

Abstract
Objective
The rate-limiting step in the biosynthesis of thromboxane A2 (TxA2) and 12-hydroxyeicosatetraenoic acid (12-HETE) by platelets is activation of cytosolic phospholipase A2α (cPLA2α), which releases arachidonic acid, which is the substrate for cyclooxygenase-1 (COX-1) and 12-lipoxygenase. We evaluated signaling via the human platelet thrombin receptors, protease-activated receptor (PAR) 1 and PAR4, to the activation of cPLA2α, which provides a substrate for the biosynthesis of TxA2 and 12-HETE.
Methods and Results
Stimulating washed human platelets resulted in delayed biosynthesis of 12-HETE, which continues after maximal formation of TxA2 is completed, suggesting that 12-HETE is not formed by the same pool of arachidonic acid that provides a substrate to COX-1. PAR1-induced formation of TxA2 was inhibited by the phosphatidylinositol kinase inhibitor LY294002, whereas this inhibitor did not block 12-HETE biosynthesis. Both 1-butanol and propranolol also blocked TxA2 biosynthesis but did not inhibit 12-HETE formation.
Conclusion
The concerted evidence indicates that the platelet thrombin receptors signal activation of cPLA2α coupled to COX-1 by a pathway different from that signaling activation of the cPLA2α coupled to 12-lipoxygenase.
Keywords: aspirin, eicosanoids, thrombosis, 12-lipoxygenase, cyclooxygenase-1, cPLA2
The importance of thromboxane A2 (TxA2) in arterial thrombosis is indicated by the reduction in myocardial infarction and stroke that results from inhibiting TxA2 biosynthesis with aspirin. In patients with known occlusive vascular disease, aspirin treatment decreases serious vascular events (nonfatal myocardial infarction, nonfatal stroke, and vascular death) by about a quarter.1,2 TxA2 released by activated platelets acts to recruit additional platelets to the thrombus, and it contributes to stabilizing the thrombus. TxA2 biosynthesis is known to be activated by signaling from both of the platelet thrombin receptors, protease-activated receptor (PAR) 1 and PAR4.3–6
The rate-limiting step in TxA2 biosynthesis by platelets is activation of cytosolic phospholipase A2α (cPLA2α) with production of free arachidonic acid (AA) as substrate for cyclooxygenase-1 (COX-1). The product of COX-1, prostaglandin H2, is then metabolized to TxA2 by thromboxane synthase. Our investigation of a patient with compound heterozygous null mutations of the cPLA2α gene (PLA2G4A) demonstrated unequivocally that virtually all (≥95%) of TxA2 generated by platelet activation is derived from cPLA2α.7
Another major eicosanoid released by platelet activation is 12-hydroxyeicosatetraenoic acid (12-HETE), a product of the oxygenation of AA by 12-lipoxygenase (12-LOX). A number of investigations have suggested autocoid effects of 12-HETE on the platelet, as well as actions on the endothelium and vascular smooth muscle.8–16 As is the case with TxA2 biosynthesis, virtually all of the AA substrate for platelet 12-LOX is derived from activation of cPLA2α.7 12-HETE is released by collagen and also by higher concentrations of thrombin.12,17 To better understand the effect of thrombin on 12-HETE release, we have characterized and compared signaling of its 2 receptors, PAR1 and PAR4, with the activation of cPLA2α coupled to 12-LOX.
Here we present evidence that the platelet thrombin receptors signal activation of the cPLA2α coupled to COX-1 by a pathway different from that signaling activation of cPLA2α coupled to 12-LOX. These findings indicate that the mechanisms providing AA to COX-1 and to 12-LOX are different.
Methods
Human Platelets
Human platelets were obtained from volunteers within the Thomas Jefferson University and Vanderbilt University communities. Studies were approved by the Thomas Jefferson and Vanderbilt institutional review boards. Informed consent was obtained from all individuals before blood donation. All experiments were performed on washed platelets, prepared as described previously.18,19 Unless otherwise noted, all experiments were conducted with a platelet concentration of 2×108 platelets/mL and agonist concentrations corresponding to maximal receptor activation with thrombin, PAR1-AP (SFLLRN) or PAR4-AP (AYPGKF) (10 nmol/L, 20 μmol/L, and 200 μmol/L, respectively) based on previous studies.18,19
Platelet Aggregation and Secretion
Platelets were treated with or without 100 μmol/L aspirin for 40 minutes and 100 nmol/L SQ29548 (SQ) for 5 minutes before measurement of aggregation by light transmission (Lumi-Aggregometer model 700, Chronolog Corp) in response to thrombin, PAR1-AP, or PAR4-AP.
Time Course for Eicosanoid Formation
Activated platelets were quenched at various times by the addition of an equal volume of pH 3 water containing 0.5 ng of [2H8]12-HETE and 2 ng of [2H4]TxB2 as internal standards to the platelets. The prostanoids were extracted by adding 1 mL of ethyl acetate and vortexing. The organic phase was separated by centrifugation at 10,000g for 5 minutes at 4°C, collected, and stored at −20°C until it was analyzed by mass spectrometry.
Assay of Residual COX-1 Activity
Washed platelets were activated with either PAR1-AP (20 μmol/L) or PAR4-AP (200 μmol/L) for 1 minute. At this time, [2H8]AA (20 μmol/L) was added, and platelets were incubated at 37° for 2 minutes. The reaction was stopped by addition of an equal volume of pH 3 water containing 2 ng of [2H4]TxB2 as an internal standard to the platelets. The [2H8]TxB2 was extracted as described above for the time course experiment and analyzed by mass spectrometry as described below. For each experiment, a control was done in which the same concentration of [2H8]AA was added without preactivation of the platelets with a PAR agonist peptide. This condition was used to determine the initial activity of the enzyme.
Measurement of 12-HETE
Secretion of 12-HETE was measured from platelet supernatants by liquid chromatography LC/APCI/MS/MS following addition of an internal standard (2 ng of [2H8]12-HETE) as described previously.20 The concentration of 12-HETE was determined by isotopic dilution.
Measurement of TxB2
Because TxA2 is a relatively unstable metabolite, the stable form, thromboxane B2 (TxB2), was measured as a surrogate for TxA2 formation in human platelets. [2H4]TxB2 (2 ng) was added to samples as an internal standard. Prostaglandins were measured by GC/NICI/MS as described previously.21 The concentration of TxB2 was determined by isotopic dilution.
Measurement of p38–Mitogen-Activated Protein Kinase
Platelets were stimulated with thrombin or PAR1-AP for the indicated times (from 0 to 10 minutes), lysed with 3× sample buffer (93.72 mmol/L Tris-HCl pH 6.8, 3% SDS, 37.45% glycerol, 0.01% bromophenol blue, 7.5% β-mercaptoethanol), boiled for 10 minutes, and analyzed for total and phosphorylated p38–mitogen-activated protein kinase (MAPK) by Western blot analysis using a LI-COR Odyssey system.
Statistical Analysis
Comparison between experimental groups was made using a t test from the Prism software package. Differences in mean values were considered significant at P<0.05. For comparisons of eicosanoid formation in the different tables, normal distribution was determined by the Shapiro-Wilk test. If data were found to be normally distributed, analyses were done by 1-way ANOVA followed by post-test analyses by the Bonferroni multiple comparison. If the data were found not to be normally distributed, statistical significance was analyzed by 1-way ANOVA with the Kruskal-Wallis test followed by post-test analysis by the Dunn multiple comparison.
Results
TxB2 and 12-HETE Biosynthesis Proceed at Different Rates Following PAR Activation
The kinetics of formation of TxB2 and 12-HETE were measured following activation with either the endogenous agonist thrombin or direct activation of the PARs with PAR1-activating peptide (PAR1-AP) or PAR4-AP19 (Figure 1).
Figure 1.

TxB2 and 12-HETE biosynthesis following PAR activation. Platelets were stimulated for various times with 20 μmol/L PAR1-AP (a), 200 μmol/L PAR4-AP (b), or 10 nmol/L thrombin (c). Following stimulation, platelet activation was quenched at various time points from 0 to 600 seconds. TxB2 (n=6) and 12-HETE (n=5) released into the suspension were measured via mass spectrometry. The 2 curves were found to be statistically significantly different by 2-way ANOVA (P<0.0001 for PAR1; P<0.0005 for PAR4; P<0.005 for thrombin). At 15 seconds, the production of TxB2 was significantly higher than the production of 12-HETE in response to PAR1 and thrombin activation (P<0.001, Bonferroni post test) but were not significantly different in response to PAR4 (P>0.05, Bonferroni post test).
Formation of 12-HETE was considerably and significantly delayed compared with that of TxB2 following activation of platelets with PAR1 (P<0.0001), PAR4 (P<0.0005), and thrombin (P<0.005). Importantly, whereas PAR1-induced TxB2 formation achieved maximal levels within 15 seconds, 12-HETE biosynthesis continued long after this, reaching its maximum only at 120 seconds. Moreover, the residual COX-1 activity at 1 minute after activation with PAR1-AP or PAR4-AP was 55.5±6.6% and 88.7±7.5%, respectively, indicating that the enzyme was still active even at this later time (Supplemental Figure I, available online at http://atvb.ahajournals.org). This evidence in conjunction with the fact that 12-HETE production continues well after TxB2 production is maximal indicates that the pool of cPLA2α-derived AA that was the substrate for COX-1 had been depleted at a point when AA continued to be provided by cPLA2α coupled to 12-LOX.
PAR1-AP-mediated TxB2 formation reached 100% within the first 15 seconds following agonist stimulation, whereas PAR4-AP required more than 75 seconds to achieve maximal levels. The kinetics for thrombin-mediated TxB2 formation was biphasic, with a steep early phase similar to that observed with PAR1-AP and a shallow secondary phase similar to the kinetics observed with PAR4-AP (Figure 1), supporting the hypothesis that in humans the effect of thrombin is mediated by activation of both receptors. The time of maximum formation of 12-HETE did not differ between PAR1 and PAR4 (Figure 1). Interestingly, there was a lag in the formation of 12-HETE induced by PAR4-AP in contrast to the immediate formation of the eicosanoid mediated by PAR1-AP, suggesting an initial fast response of the platelets through PAR1 activation with a delayed response mediated by PAR4.
TxB2 formation mediated by PAR1-AP and that mediated by PAR4-AP each represent ≈50% of TxB2 levels following platelet activation by thrombin, suggesting that production of TxB2 by thrombin is mediated by the additive activation of the 2 PARs. In contrast, PAR4-AP induced a significantly higher level of 12-HETE formation compared with PAR1-AP (Table 1). The amount of 12-HETE formed by thrombin exceeded the sum of that formed by PAR1 plus PAR4, suggesting a synergistic effect between the 2 receptors.
Table 1.
Eicosanoid Formation in Human Platelets
| Thrombin | PAR1-AP | PAR4-AP | PMA | |
|---|---|---|---|---|
| TxB2, ng/mL (n) | 289.8±41.4 (19) | 124.0±25.4*† (19) | 134.6±25.2*† (19) | 9.3±6.1 (4) |
| 12-HETE, ng/mL (n) | 200.6±32.1 (16) | 33.3±6.2*‡ (16) | 82.9±10.7*‡ (16) | 2.4±0.3 (4) |
Levels of eicosanoid formation were measured in human platelets following stimulation with 10 nmol/L thrombin, 20 μmol/L PAR1-AP, 200 μmol/L PAR4-AP, or 1 μmol/L PMA for 10 minutes. Eicosanoid levels at baseline (without stimulation) were subtracted from each eicosanoid value. Statistical significance was analyzed by 1-way ANOVA with Kruskal-Wallis test followed by post-test analysis by Dunn’s multiple comparison.
P<0.05 compared with thrombin;
PAR1-AP vs PAR4-AP not significantly different;
P<0.05 for PAR1-AP vs PAR4-AP.
Phosphatidylinositol Kinase Products Mediate PAR-Induced Thromboxane Biosynthesis But Not Formation of 12-HETE
To investigate the participation of the phosphatidylinositol (PI) kinases in activating cPLA2α coupled to COX-1 and 12-LOX,22,23 platelets were pretreated with increasing concentrations of the PI kinase inhibitor LY294002 (LY), which was shown previously to completely inhibit PAR-mediated formation of both PI(4,5)P2 and PI(3,4,5)P3 in platelets.24 Increasing the concentration of LY resulted in a complete inhibition of TxB2 formation, with an IC50 of approximately 25 μmol/L LY for both PAR1-AP and PAR4-AP (Figure 2a and 2b). In contrast, LY increased 12-HETE formation by 2-fold in a dose-dependent fashion following platelet activation with both PAR1-AP and PAR4-AP. To ascertain that the effects of LY on thromboxane production were not due to direct inhibition of COX-1, we monitored TxB2 production when [2H8]AA was provided to the platelets (Figure 2c). In these conditions, the cPLA2α is bypassed, and the production of eicosanoids is directly proportional to the residual activities of the enzymes catalyzing the production of TxB2 (COX-1 and TxA2 synthase). Our results clearly demonstrated that LY at a concentration of 100 μmol/L had no significant effect on either enzyme. Taken together, these results indicate that following PAR signaling, products of the PI kinases are required for activation of the cPLA2α coupled to COX-1 but not for activation of the cPLA2α that provides substrate for 12-LOX.
Figure 2.

Regulation of PAR-mediated eicosanoid formation and aggregation by lipid signaling pathways. Platelets were stimulated with 10 nmol/L thrombin, 20 μmol/L PAR1-AP, or 200 μmol/L PAR4-AP, and subsequent eicosanoid formation, platelet aggregation and dense granule secretion were measured. a, Aggregation, TxB2, and 12-HETE levels were measured following stimulation with PAR1-AP or PAR4-AP in platelets treated with increasing concentrations of LY from 0 to 250 μmol/L. b, Washed platelets were treated with 100 μmol/L LY, 0.4% 1-butanol (1-ButOH), 100 μmol/L propranolol, or vehicle followed by stimulation for 10 minutes with thrombin, PAR1-AP, or PAR4-AP. Eicosanoid formation was then measured in each condition tested. TxB2 and 12-HETE: Bars represent mean±SEM (P<0.001 and P<0.05 for TxB2 and 12-HETE, respectively, by repeated-measures ANOVA; *P<0.05, ***P<0.001 compared with control condition by the Dunnett multiple comparison test). NS indicates not significant.
1-Butanol and Propranolol Inhibit PAR1-Induced Thromboxane Biosynthesis But Not Formation of 12-HETE
Treatment with the lipid signaling inhibitors 1-butanol or propranolol each produced a marked and significant inhibition of TxA2 formation following stimulation with thrombin, PAR1-AP, or PAR4-AP (P<0.001) (Figure 2b). By contrast, PAR1-induced 12-HETE formation was unaffected by either propranolol or 1-butanol. Only with thrombin signaling did propranolol induce a slight but significant change in 12-HETE levels (P<0.05), and 1-butanol caused a decrease in 12-HETE only with PAR4-AP (P<0.001). This evidence, particularly the differences seen with PAR1 signaling, further supports the concept that separate signaling pathways may regulate the activity of the cPLA2α that is coupled to formation of these 2 eicosanoids.
Protein Kinase C Stimulates Platelet Activation But Does Not Contribute to PAR-Mediated Activation of cPLA2α
Protein kinase C (PKC) is activated by PARs and has been reported to directly phosphorylate cPLA2.25 Aggregation and eicosanoid formation were measured by direct addition of PMA (a diacylglycerol mimetic known to activate PKC) to the washed platelets (Supplemental Figure II). Although addition of PMA was sufficient to induce platelet aggregation, it was unable to induce either calcium mobilization or downstream eicosanoid formation even at high concentrations26 (Figure 3; Table 1).
Figure 3.

TP receptor signaling contributes to PAR-mediated eicosanoid formation. Platelets were treated with or without 100 μmol/L aspirin (ASA) for 40 minutes or 100 nmol/L SQ for 5 minutes followed by stimulation under stirring conditions with 10 nmol/L thrombin, 20 μmol/L PAR1-AP, or 200 μmol/L PAR4-AP. Following stimulation, eicosanoids released into the suspension were measured via mass spectrometry. a, TxB2 was measured following stimulation with the different agonists (n=6). Treatment with ASA resulted in complete inhibition of TxB2 formation relative to control conditions (***P<0.001). Treatment with SQ resulted in partial inhibition of TxB2 formation with a higher degree of inhibition following PAR1-AP (***P<0.001) relative to thrombin or PAR4-AP-treated conditions (*P<0.05) as compared with control. b, 12-HETE was measured following stimulation with the different agonists (n=6). Treatment with ASA resulted in a complete increase of 12-HETE formation following stimulation with thrombin or PAR4-AP and a significant decrease in 12-HETE formation following stimulation with PAR1-AP (***P<0.001). Treatment with SQ resulted in partial inhibition of TxB2 formation following stimulation with thrombin (not significant) or PAR4-AP (*P<0.05) but a high level of inhibition following treatment with PAR1-AP (***P<0.001) as compared with control.
Intracellular Calcium Contributes to PAR-Induced Eicosanoid Formation
Reduction of cytosolic calcium with BAPTA markedly inhibited the TxA2 biosynthesis that was induced by thrombin, and by the PAR1 and PAR4 agonists (Table 2). BAPTA also inhibited the production of 12-HETE by these agonists, but to a lesser extent.
Table 2.
Calcium-Dependent Eicosanoid Biosynthesis by Human Platelets
| Thrombin | PAR1-AP | PAR4-AP | |
|---|---|---|---|
| TxB2, % inhibition by BAPTA (n) | 89 (9)* | 93 (9)* | 95 (9)* |
| 12-HETE, % inhibition by PABTA (n) | 81 (9)* | 57 (9)*‡ | 79 (9)* |
Eicosanoids were measured in human platelets following stimulation with 10 nmol/L thrombin, 20 μmol/L PAR1-AP, or 200 μmol/L PAR4-AP for 10 minutes. Eicosanoids produced in presence of 20 μmol/L BAPTA are expressed as percentage inhibition compared with control conditions (no BAPTA present) and represent the calcium-dependent eicosanoid biosynthesis. Statistical significance was analyzed by repeated ANOVA (P<0.0001) followed by post-test analysis by Bonferroni’s multiple comparison.
P<0.001 vs control;
PAR1-AP vs thrombin or PAR4-AP.
TP Receptor Signaling Contributes to PAR-Induced Eicosanoid Formation
We have previously shown that TxA2 formed during the first phase of activation stimulates a secondary wave of TxA2 biosynthesis through autocrine activation of the TP receptor.7 To investigate the role of the initial biosynthesis of TxA2 on eicosanoid formation and platelet function, we pretreated washed platelets with or without aspirin (a cyclooxygenase inhibitor) or SQ (a TP antagonist) before stimulation with thrombin, PAR1-AP or PAR4-AP (Figure 3). As expected, treatment with aspirin completely inhibited TxB2 formation by thrombin, PAR1-AP and PAR4-AP. SQ-treated platelets, which are able to directly form TxB2 following PAR activation but are unable to initiate a second wave of activation through the TP receptor, show a significant decrease in total TxB2 formed; thrombin and PAR4-AP-mediated TxB2 formation was decreased by 40%, whereas PAR1-AP-mediated TxB2 formation was inhibited by approximately 80% (Figure 3a). 12-HETE formation in the presence of aspirin and SQ also showed a differential effect based on type of activation (Figure 3b). In both thrombin and PAR4-AP conditions, treatment with SQ had only a partial inhibitory effect on 12-HETE formation, and aspirin treatment resulted in an increase in 12-HETE formation. In contrast, PAR1-AP activated platelets showed a significant decrease in 12-HETE formation in the presence of either SQ or aspirin, indicating that TxA2 feedback on TP mediates the production of 12-HETE following PAR1 (Table 3).
Table 3.
TP-Dependent Eicosanoid Biosynthesis by Human Platelets
| Thrombin | PAR1-AP | PAR4-AP | |
|---|---|---|---|
| TxB2, % inhibition by SQ (n) | 40 (8)* | 79 (8)† | 40 (8)* |
| 12-HETE, % inhibition by SQ (n) | NS (8) | 60 (8)‡ | 38 (8)* |
Eicosanoids were measured in human platelets following stimulation with 10 nmol/L thrombin, 20 μmol/L PAR1-AP, or 200 μmol/L PAR4-AP for 10 minutes. Eicosanoids produced in presence of the TP receptor antagonist were expressed as percent inhibition compared to control condition (no antagonist present) and represent the TP-dependent eicosanoid biosynthesis. Data were analyzed by repeated ANOVA (P<0.005) followed by post-test analysis by Dunn’s multiple comparison.
Difference with eicosanoid in control was significant:
P<0.05,
P<0.0005,
P<0.0001.
NS indicates not significant.
To determine whether the observed differences in regulation of eicosanoid formation might be related to the phases of platelet activation, platelet aggregation was measured in the presence of aspirin or SQ following stimulation with increasing concentrations of nmol/L thrombin, PAR1-AP, or PAR4-AP (Figure 4). Treatment with aspirin or SQ did not alter either maximal or stable platelet aggregation induced by PAR4-AP, whereas thrombin and PAR1-AP-stimulated platelet aggregation was significantly attenuated by aspirin or SQ at lower concentrations of each agonist.
Figure 4.

Inhibition of TP signaling significantly attenuates PAR1-mediated platelet aggregation. Platelets were treated with or without 100 μmol/L aspirin (ASA) for 40 minutes or 100 nmol/L SQ for 5 minutes. Following treatment, platelet aggregation was measured following stimulation with increasing concentrations of thrombin (0 to 10 nmol/L), PAR1-AP (0 to 20 μmol/L), or PAR4-AP (0 to 200 μmol/L). Treatment with ASA or SQ significantly shifted PAR1-AP- and thrombin-mediated platelet aggregation at lower concentrations but had minimal effects on thrombin or PAR4-AP-induced platelet aggregation. n=8 (***P<0.001, *P<0.05).
The Time Course of p38 MAPK Phosphorylation
cPLA2αcan be phosphorylated by isozymes of the MAPK family, such as extracellular signal–regulated kinases and p38.27 We therefore sought to determine whether the time course for PAR-mediated activation of MAPK would correlate with the time course of eicosanoid production. Our results show that detectable p38-MAPK phosphorylation is initiated after 30 seconds following thrombin or PAR stimulation and reaches maximum levels within 5 minutes post-stimulation (Figure 5). This is not concordant with the rapid release of TxA2, particularly that in response to PAR1-AP, and it is consistent with the findings of Kuliopulos et al demonstrating no effect of a p38 MAPK inhibitor on TxA2-dependent collagen-induced aggregation.28
Figure 5.
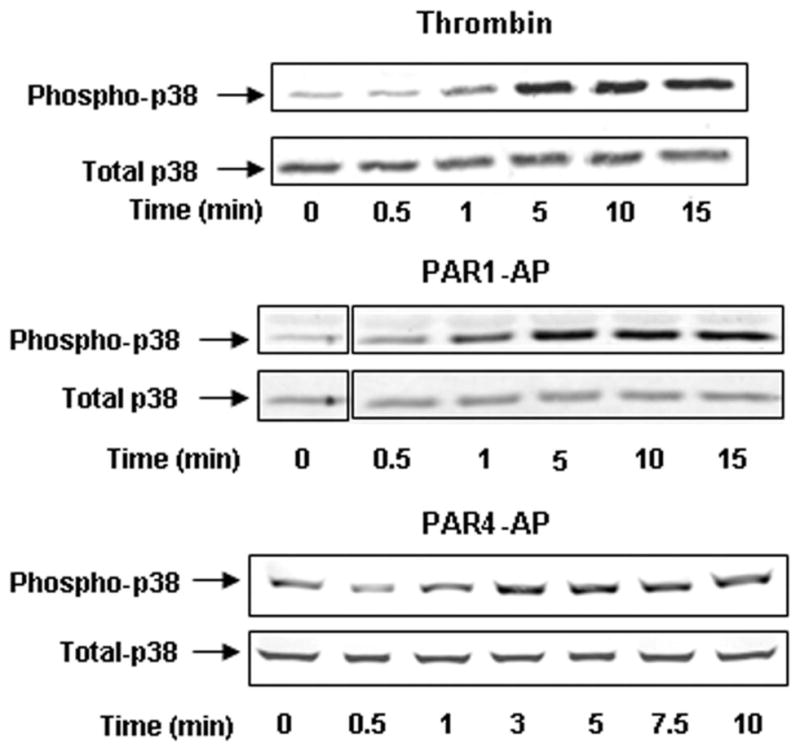
PKC does not play a role in PAR-mediated eicosanoid formation. Platelets were stimulated with 10 nmol/L thrombin, 20 μmol/L PAR1-AP, or 200 μmol/L PAR4-AP and subsequently measured for p38 phosphorylation. Platelets were stimulated from 0 to 15 minutes, and phosphorylation of p38 MAPK was measured. p38 was phosphorylated within 1 minute, with maximum phosphorylation occurring at 5 minutes following stimulation. Total cPLA2 was also measured to ensure that equal protein was loaded for each condition (n=2).
Discussion
In the platelet, the same enzyme, phospholipase, cPLA2α (PLA2G4A), delivers AA both to COX-1 for formation of TxA2 and to 12-LOX for formation of 12-HETE, as was demonstrated by the virtual absence of agonist-induced biosynthesis of both of these eicosanoids in a patient with null mutations of cPLA2α.7 Thus, it was of considerable interest to find that PAR-induced signaling to activate the cPLA2α coupled to COX-1 is quite different from PAR-induced signaling to the cPLA2α coupled to 12-LOX. Three lines of evidence delineate the difference between these 2 cPLA2α activation pathways in the platelet: (1) 12-HETE formation continues well after TxB2 biosynthesis has ceased, (2) PI kinases participate in the lipid signaling pathway for TxA2 production but not for 12-HETE, and (3) 1-butanol and propranolol inhibit PAR1-induced formation of TxB2 but do not block 12-HETE production.
The rate of PAR1-induced biosynthesis of TxA2 is markedly greater than that of 12-HETE. Maximal formation of TxA2 is reached in ≤15 seconds after the PAR1 peptide, whereas formation of 12-HETE increases gradually over 120 seconds. These differences between the rates of formation and the time at which maximal formation is achieved are very similar to those previously reported for the formation of TxA2 and 12-HETE following collagen-induced platelet activation.29 It is clear from the persisting 12-HETE formation long after TxA2 production has ceased that the AA substrate is not provided by the same mechanisms to COX-1 and to 12-LOX after PAR1 activation.
LY249002, a PI kinase inhibitor that blocks formation of both PI(4,5)P2 and PI(3,4,5)P3,24 markedly inhibits PAR1 and PAR4-induced biosynthesis of TxA2. In contrast, LY does not inhibit PAR1 or PAR4-induced formation of 12-HETE. Indeed, LY actually increases PAR1-induced 12-HETE formation. Also, our results indicate that LY does not have direct effects on the enzymatic activities of either COX-1 or TxA2 synthase. Thus, the results of the studies with LY also indicate that the PAR signaling that activates cPLA2α coupled to COX-1 is different from the PAR signaling to the cPLA2α coupled to 12-LOX.
Further evidence that the PAR signaling pathway activating the cPLA2α coupled to COX-1 is discrete from that signaling to 12-LOX coupled cPLA2α is derived from the results of the studies with 1-butanol and propranolol. Neither 1-butanol nor propranolol inhibits PI kinases.30,31 Thus, inhibition of the activation of the cPLA2 coupled to COX-1 by these agents occurs at a site in the signaling pathway different from the PI kinases. Importantly, neither 1-butanol nor propranolol inhibits PAR1-induced activation of the cPLA2 coupled to 12-LOX, thus providing additional evidence that this signaling pathway differs from that leading to activation of the cPLA2 coupled to COX-1.
The intracellular localization of COX-1 and 12-LOX is pertinent to a consideration of differential regulation of the cPLA2α coupled to each of the enzymes. Platelet COX-1 is localized to the intracellular membrane complex with characteristics of the endoplasmic reticulum that is referred to as the dense tubular system.32 On the other hand, the 12-LOX is predominantly localized in the cytosol of rat platelets that have been isolated so as to minimize ex vivo activation, and it undergoes calcium- and thrombin-dependent translocation to a membrane fraction,33,34 from which it can be coimmunoprecipitated with cPLA2α.17 Thus, the differences between PAR-induced signaling to the cPLA2α providing substrate to COX-1 and that coupled to 12-LOX may occur in the context of localization of the 2 oxygenases at separate sites in the platelet. cPLA2α activity is regulated by several factors in cells, including interaction with membrane lipids and phosphorylation.35–40 Although these mechanisms have been well described in vitro, their relevance in vivo is still debated. Our results suggest that, in human platelets, a product of the PI kinases (likely PI[4,5]P2) participates in the activation of the phospholipase coupled to COX-1 (Figure 6). By contrast, activation of the cPLA2α coupled to 12-LOX does not appear to require signaling through a PI kinase. Together, our results support the hypothesis that regulation of cPLA2α in human platelets determines the metabolic pathway of AA by translocating the lipase to separate pools of phospholipids, specifically coupling the lipase to either COX-1 or 12-LOX, or both.
Figure 6.
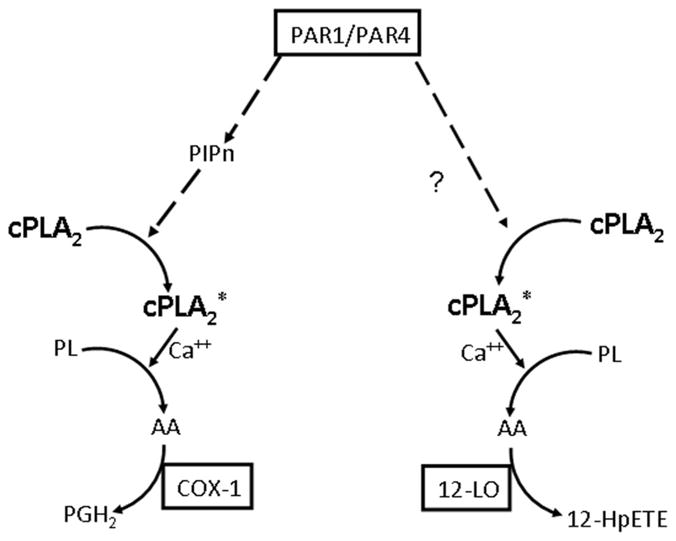
Hypothetical model for PAR1-mediated formation of eicosanoids. cPLA2* indicates activated cPLA2; cPLA2, nonactivated cPLA2; PIP, phosphatidyl inositols; PL, phospholipids; PGH2, prostaglandin H2; 12-HpETE, 12-hydroperoxyeicosatetraenoic acid.
In contrast to the difference in the lipid signaling pathways that activate COX-1-coupled cPLA2α but not 12-LOX-coupled cPLA2α, calcium is required for the PAR-induced activation of the cPLA2α coupled to both COX-1 and 12-LOX. Also, secondary signaling via the TP receptor contributes to the ultimate PAR1-induced activation of both COX-1-coupled cPLA2α and 12-LOX-coupled cPLA2α. TP-induced augmentation of the initial calcium response to PAR1 likely participates in this positive feedback loop regulating the extent of cPLA2α activation. Previous research has shown that aspirin inhibits 12-HETE production by platelets activated by collagen41 and thrombin,42 and from that evidence, it was suggested that inhibition of the conversion of 12-hydroperoxyeicosatetraenoic acid to 12-HETE by a peroxidase accounted for this effect of aspirin. The present results indicate that TP receptor signaling contributes substantially to 12-HETE formation in response to PAR1. PAR4-induced 12-HETE formation is not affected by aspirin and is inhibited to a lesser extent by the TP receptor antagonist.
Both the TP receptor and PAR1 are Gq-coupled receptors that signal calcium release via the PLCβ pathway, which also leads to activation of PKC. Activation of PKC with PMA, however, produced platelet aggregation but did not lead to formation of either TxA2 or 12-HETE, thus excluding the PKC pathway as a mechanism for activation of cPLA2α.
We found that activation of the PAR4 receptor generated TxA2 more slowly than activation of PAR1 in this direct comparison of the agonists for the 2 receptors. These findings are consistent with previous evaluations in which the effects of PAR14 and PAR46 agonists were carried out in separate studies. Following stimulation of PAR4, the rate of 12-HETE formation is also less than that resulting from PAR1.
In conclusion, the PAR-induced signaling pathway that activates the cPLA2α that provides AA to COX-1 is different from that activating the cPLA2α that liberates substrate for 12-LOX. Thus, both COX-1 and 12-LOX are functionally coupled to a discrete subset of platelet cPLA2α that provides AA separately to each of these oxygenases.
Supplementary Material
Acknowledgments
We thank W. James Hudson, Nancy Colowick, Megha Patel, Taneem Amin, Audra Judd, and Joseph Chambers for performing a number of the assays used in this study.
Sources of Funding
This work was supported in part by the National Institutes of Health Grants R00-HL089457 (to M.H.) and P50-HL081009 (to J.A.O. and H.E.H.).
Footnotes
Disclosures
None.
References
- 1.Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, Buring J, Hennekens C, Kearney P, Meade T, Patrono C, Roncaglioni MC, Zanchetti A. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849–1860. doi: 10.1016/S0140-6736(09)60503-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henriksen RA, Hanks VK. PAR-4 agonist AYPGKF stimulates thromboxane production by human platelets. Arterioscler Thromb Vasc Biol. 2002;22:861–866. doi: 10.1161/01.atv.0000014742.56572.25. [DOI] [PubMed] [Google Scholar]
- 4.Kramer RM, Roberts EF, Hyslop PA, Utterback BG, Hui KY, Jakubowski JA. Differential activation of cytosolic phospholipase A2 (CPLA2) by thrombin and thrombin receptor agonist peptide in human platelets: evidence for activation of CPLA2 independent of the mitogen-activated protein kinases ERK1/2. J Biol Chem. 1995;270:14816–14823. doi: 10.1074/jbc.270.24.14816. [DOI] [PubMed] [Google Scholar]
- 5.Shankar H, Garcia A, Prabhakar J, Kim S, Kunapuli SP. P2Y12 receptor-mediated potentiation of thrombin-induced thromboxane A2 generation in platelets occurs through regulation of Erk1/2 activation. J Thromb Haemost. 2006;4:638–647. doi: 10.1111/j.1538-7836.2006.01789.x. [DOI] [PubMed] [Google Scholar]
- 6.Wu CC, Hwang TL, Liao CH, Kuo SC, Lee FY, Teng CM. The role of PAR4 in thrombin-induced thromboxane production in human platelets. Thromb Haemost. 2003;90:299–308. doi: 10.1160/TH03-02-0103. [DOI] [PubMed] [Google Scholar]
- 7.Adler DH, Cogan JD, Phillips JA, III, Schnetz-Boutaud N, Milne GL, Iverson T, Stein JA, Brenner DA, Morrow JD, Boutaud O, Oates JA. Inherited human CPLA(2α) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J Clin Invest. 2008;118:2121–2131. doi: 10.1172/JCI30473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbertsson H, Kuhme T, Hammarstrom S. A 12(S)-HETE receptor in Lewis lung carcinoma cells. Adv Exp Med Biol. 1999;447:193–198. doi: 10.1007/978-1-4615-4861-4_18. [DOI] [PubMed] [Google Scholar]
- 9.Fonlupt P, Croset M, Lagarde M. 12-HETE inhibits the binding of PGH2/TXA2 receptor ligands in human platelets. Thromb Res. 1991;63:239–248. doi: 10.1016/0049-3848(91)90287-7. [DOI] [PubMed] [Google Scholar]
- 10.Johnson EN, Brass LF, Funk CD. Increased platelet sensitivity to ADP in mice lacking platelet-type 12-lipoxygenase. Proc Natl Acad Sci U S A. 1998;95:3100–3105. doi: 10.1073/pnas.95.6.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller AW, Katakam PV, Lee HC, Tulbert CD, Busija DW, Weintraub NL. Arachidonic acid-induced vasodilation of rat small mesenteric arteries is lipoxygenase-dependent. J Pharmacol Exp Ther. 2003;304:139–144. doi: 10.1124/jpet.102.041780. [DOI] [PubMed] [Google Scholar]
- 12.Nyby MD, Sasaki M, Ideguchi Y, Wynne HE, Hori MT, Berger ME, Golub MS, Brickman AS, Tuck ML. Platelet lipoxygenase inhibitors attenuate thrombin- and thromboxane mimetic-induced intracellular calcium mobilization and platelet aggregation. J Pharmacol Exp Ther. 1996;278:503–509. [PubMed] [Google Scholar]
- 13.Ozeki Y, Ito H, Nagamura Y, Unemi F, Igawa T. 12(S)-HETE plays a role as a mediator of expression of platelet CD62 (P-selectin) Platelets. 1998;9:297–302. doi: 10.1080/09537109876537. [DOI] [PubMed] [Google Scholar]
- 14.Pidgeon GP, Lysaght J, Krishnamoorthy S, Reynolds JV, O’Byrne K, Nie D, Honn KV. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Rev. 2007;26:503–524. doi: 10.1007/s10555-007-9098-3. [DOI] [PubMed] [Google Scholar]
- 15.Quintana LF, Guzman B, Collado S, Claria J, Poch E. A coding polymorphism in the 12-lipoxygenase gene is associated to essential hypertension and urinary 12(S)-HETE. Kidney Int. 2006;69:526–530. doi: 10.1038/sj.ki.5000147. [DOI] [PubMed] [Google Scholar]
- 16.Zink MH, Oltman CL, Lu T, Katakam PV, Kaduce TL, Lee H, Dell-sperger KC, Spector AA, Myers PR, Weintraub NL. 12-Lipoxygenase in porcine coronary microcirculation: implications for coronary vasoregulation. Am J Physiol Heart Circ Physiol. 2001;280:H693–H704. doi: 10.1152/ajpheart.2001.280.2.H693. [DOI] [PubMed] [Google Scholar]
- 17.Coffey MJ, Coles B, Locke M, Bermudez-Fajardo A, Williams PC, Jarvis GE, O’Donnell VB. Interactions of 12-lipoxygenase with phospholipase A2 isoforms following platelet activation through the glycoprotein VI collagen receptor. FEBS Lett. 2004;576:165–168. doi: 10.1016/j.febslet.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 18.Holinstat M, Voss B, Bilodeau ML, Hamm HE. Protease-activated receptors differentially regulate human platelet activation through a phosphatidic acid-dependent pathway. Mol Pharmacol. 2007;71:686–694. doi: 10.1124/mol.106.029371. [DOI] [PubMed] [Google Scholar]
- 19.Holinstat M, Voss B, Bilodeau ML, McLaughlin JN, Cleator J, Hamm HE. PAR4, but not PAR1, signals human platelet aggregation via CA2+ mobilization and synergistic P2Y12 receptor activation. J Biol Chem. 2006;281:26665–26674. doi: 10.1074/jbc.M602174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SH, Williams MV, DuBois RN, Blair IA. Targeted lipidomics using electron capture atmospheric pressure chemical ionization mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:2168–2176. doi: 10.1002/rcm.1170. [DOI] [PubMed] [Google Scholar]
- 21.Boutaud O, Brame CJ, Salomon RG, Roberts LJ, 2nd, Oates JA. Characterization of the lysyl adducts formed from prostaglandin H2 via the levuglandin pathway. Biochemistry. 1999;38:9389–9396. doi: 10.1021/bi990470+. [DOI] [PubMed] [Google Scholar]
- 22.Balsinde J, Balboa MA, Li WH, Llopis J, Dennis EA. Cellular regulation of cytosolic group iv phospholipase A2 by phosphatidylinositol bisphosphate levels. J Immunol. 2000;164:5398–5402. doi: 10.4049/jimmunol.164.10.5398. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian P, Vora M, Gentile LB, Stahelin RV, Chalfant CE. Anionic lipids activate group iva cytosolic phospholipase A2 via distinct and separate mechanisms. J Lipid Res. 2007;48:2701–2708. doi: 10.1194/jlr.M700356-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Holinstat M, Preininger AM, Milne SB, Hudson WJ, Brown HA, Hamm HE. Irreversible platelet activation requires protease-activated receptor 1-mediated signaling to phosphatidylinositol phosphates. Mol Pharmacol. 2009;76:301–313. doi: 10.1124/mol.109.056622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nemenoff RA, Winitz S, Qian NX, Van Putten V, Johnson GL, Heasley LE. Phosphorylation and activation of a high molecular weight form of phospholipase A2 by p42 microtubule-associated protein 2 kinase and protein kinase C. J Biol Chem. 1993;268:1960–1964. [PubMed] [Google Scholar]
- 26.McNicol A, Shibou TS. Translocation and phosphorylation of cytosolic phospholipase A2 in activated platelets. Thromb Res. 1998;92:19–26. doi: 10.1016/s0049-3848(98)00097-8. [DOI] [PubMed] [Google Scholar]
- 27.Gijon MA, Spencer DM, Kaiser AL, Leslie CC. Role of phosphorylation sites and the C2 domain in regulation of cytosolic phospholipase A2. J Cell Biol. 1999;145:1219–1232. doi: 10.1083/jcb.145.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuliopulos A, Mohanlal R, Covic L. Effect of selective inhibition of the p38 map kinase pathway on platelet aggregation. Thromb Haemost. 2004;92:1387–1393. doi: 10.1160/TH04-03-0187. [DOI] [PubMed] [Google Scholar]
- 29.Dutilh CE, Haddeman E, Jouvenaz GH, Ten Hoor F, Nugteren DH. Study of the two pathways for arachidonate oxygenation in blood platelets. Lipids. 1979;14:241–246. doi: 10.1007/BF02533876. [DOI] [PubMed] [Google Scholar]
- 30.He H, Genovese KJ, Nisbet DJ, Kogut MH. Phospholipase C, phosphatidylinositol 3-kinase, and intracellular [Ca(2+)] mediate the activation of chicken HD11 macrophage cells by CpG oligodeoxynucleotide. Dev Comp Immunol. 2008;32:1111–1118. doi: 10.1016/j.dci.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Husebye ES, Flatmark T. Phosphatidylinositol kinase of bovine adrenal chromaffin granules: modulation by hydrophilic and amphiphilic cations. Biochem Pharmacol. 1988;37:4149–4156. doi: 10.1016/0006-2952(88)90109-8. [DOI] [PubMed] [Google Scholar]
- 32.Gerrard JM, White JG, Rao GH, Townsend D. Localization of platelet prostaglandin production in the platelet dense tubular system. Am J Pathol. 1976;83:283–298. [PMC free article] [PubMed] [Google Scholar]
- 33.Baba A, Sakuma S, Okamoto H, Inoue T, Iwata H. Calcium induces membrane translocation of 12-lipoxygenase in rat platelets. J Biol Chem. 1989;264:15790–15795. [PubMed] [Google Scholar]
- 34.Chang WC, Nakao J, Orimo H, Murota S. Effects of reduced glutathione on the 12-lipoxygenase pathways in rat platelets. Biochem J. 1982;202:771–776. doi: 10.1042/bj2020771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clark JD, Schievella AR, Nalefski EA, Lin LL. Cytosolic phospholipase A2. J Lipid Mediat Cell Signal. 1995;12:83–117. doi: 10.1016/0929-7855(95)00012-f. [DOI] [PubMed] [Google Scholar]
- 36.Das S, Cho W. Roles of catalytic domain residues in interfacial binding and activation of group IV cytosolic phospholipase A2. J Biol Chem. 2002;277:23838–23846. doi: 10.1074/jbc.M202322200. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh M, Tucker DE, Burchett SA, Leslie CC. Properties of the group iv phospholipase A2 family. Prog Lipid Res. 2006;45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Leslie CC. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids. 2004;70:373–376. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 39.Six DA, Dennis EA. Essential Ca(2+)-independent role of the group iva cytosolic phospholipase A(2) C2 domain for interfacial activity. J Biol Chem. 2003;278:23842–23850. doi: 10.1074/jbc.M301386200. [DOI] [PubMed] [Google Scholar]
- 40.Tucker DE, Ghosh M, Ghomashchi F, Loper R, Suram S, John BS, Girotti M, Bollinger JG, Gelb MH, Leslie CC. Role of phosphorylation and basic residues in the catalytic domain of cytosolic phospholipase A2α in regulating interfacial kinetics and binding and cellular function. J Biol Chem. 2009;284:9596–9611. doi: 10.1074/jbc.M807299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eynard AR, Galli G, Tremoli E, Maderna P, Magni F, Paoletti R. Aspirin inhibits platelet 12-hydroxy-eicosatetraenoic acid formation. J Lab Clin Med. 1986;107:73–78. [PubMed] [Google Scholar]
- 42.Siegel MI, McConnell RT, Cuatrecasas P. Aspirin-like drugs interfere with arachidonate metabolism by inhibition of the 12-hydroperoxy-5,8,10,14-eicosatetraenoic acid peroxidase activity of the lipoxygenase pathway. Proc Natl Acad Sci U S A. 1979;76:3774–3778. doi: 10.1073/pnas.76.8.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.