

Abstract
Epigenetic modifications of cytosine residues in the DNA play a critical role for cellular differentiation and potentially also for aging. In mesenchymal stromal cells (MSC) from human bone marrow we have previously demonstrated age-associated methylation changes at specific CpG-sites of developmental genes. In continuation of this work, we have now isolated human dermal fibroblasts from young (<23 years) and elderly donors (>60 years) for comparison of their DNA methylation profiles using the Infinium HumanMethylation27 assay. In contrast to MSC, fibroblasts could not be induced towards adipogenic, osteogenic and chondrogenic lineage and this is reflected by highly significant differences between the two cell types: 766 CpG sites were hyper-methylated and 752 CpG sites were hypo-methylated in fibroblasts in comparison to MSC. Strikingly, global DNA methylation profiles of fibroblasts from the same dermal region clustered closely together indicating that fibroblasts maintain positional memory even after in vitro culture. 75 CpG sites were more than 15% differentially methylated in fibroblasts upon aging. Very high hyper-methylation was observed in the aged group within the INK4A/ARF/INK4b locus and this was validated by pyrosequencing. Age-associated DNA methylation changes were related in fibroblasts and MSC but they were often regulated in opposite directions between the two cell types. In contrast, long-term culture associated changes were very consistent in fibroblasts and MSC. Epigenetic modifications at specific CpG sites support the notion that aging represents a coordinated developmental mechanism that seems to be regulated in a cell type specific manner.
Introduction
There is a growing perception that epigenetic modifications, such as DNA methylation and histone modification, play an important role for cellular senescence and aging of the organism [1]–[3]. CpG dinucleotides in the genomic DNA can be methylated at cytosine moieties. Upon replication the same methylation pattern is established on the newly synthesized DNA strand by DNA methyltransferase 1 (DNMT1) and thereby, the methylation pattern is inherited to both daughter cells. This inheritance of epigenetic modifications might provide an ideal mechanism for the regulation of progressive alterations in the course of aging [4].
Various studies have indicated, that the global DNA methylation level decreases upon aging in murine, rat and human tissues [5]–[7]. It was also shown that the 5-methylcytosine content decreased upon long-term culture of fibroblasts [8]. This led to the suggestion that the global loss of DNA methylation might be a result of passive demethylation as a consequence of a progressive loss of DNMT1a efficiency [9]. However, a number of specific loci become hyper-methylated during aging, such as the INK4A/ARF/INK4b locus, the ribosomal gene cluster, estrogen receptor (ER), runt-related transcription factor 3 (RUNX3), insulin growth factor II (IGF2), E-cadherin, c-fos and others [1], [10]–[13]. These specific changes indicate that age-associated methylation changes are not simply based on random deterioration during ontogenic development although it is yet unknown how site-specific methylation changes are regulated.
We have recently analyzed age-associated DNA methylation changes in human mesenchymal stromal cells (MSC) [14]. These cells comprise multipotent precursors for mesodermal cell lineages such as osteocytes, chondrocytes and adipocytes and have therefore been coined as “mesenchymal stem cells”. Global methylation profiles were analyzed using the HumanMethylation27 BeadChip microarray allowing the determination of DNA methylation levels at 27,578 unique CpG sites within more than 14,000 promoter regions. Overall, methylation patterns of MSC were maintained throughout both, long-term culture and aging, whereas highly significant differences were observed at specific CpG sites. Notably, methylation changes as well as gene expression changes in MSC were overlapping in long-term culture in vitro and aging in vivo [14]–[17]. This supports the notion of replicative senescence and aging to represent related developmental processes, regulated by specific epigenetic modifications.
Distinct age-related phenotypes, such as wrinkle formation, hair graying and impaired wound healing, as well as the accessibility of samples from differently aged healthy donors make human skin an ideal model system for the analysis of age-related epigenetic changes [18], [19]. Dermal fibroblasts are important for skin architecture and extracellular matrix (ECM) synthesis and they are morphologically indistinguishable from MSC preparations. It is controversially discussed if immunomodulatory capacities and differentiation potential vary between the two cell types - some studies indicated that dermal fibroblasts display similar in vitro differentiation potential as MSC [20]–[22]. This might be explained by the finding that human dermal fibroblasts are composed of different subtypes with distinct gene expression profiles according to the anatomical site of origin [23]. With this in mind, we have isolated fibroblasts from different dermal regions to investigate age-associated changes in their DNA methylation profiles in comparison to MSC.
Results
Comparison of dermal fibroblasts with bone marrow MSC
Human dermal fibroblasts were isolated from young (6–23 years old; 9 samples) and elderly donors (60–73 years old; 6 samples). Mesenchymal stromal cells were isolated from bone marrow of young (21–50 years old; 4 samples) and elderly donors (53–85 years old; 4 samples) as described in our previous work [14], [16]. Both cell types revealed a very similar spindle-shaped morphology and growth pattern. Furthermore, fibroblasts and MSC displayed the same and uniform immunophenotype for surface markers that are commonly used for the definition of MSC (CD14−, CD29+, CD31−, CD34−, CD45−, CD73+, CD90+, CD105+; Figure 1A). Simultaneous in vitro differentiation revealed that adipogenic differentiation could be induced in about 40% of the cells within MSC preparations, whereas hardly any fat droplet formation was observed in fibroblasts (n = 5, Figure 1B,E). Osteogenic differentiation induced calcific deposition only in MSC but not in fibroblasts (Figure 1C,F) and chondrogenic differentiation in micromass culture induced sulfated glycosaminoglycan rich matrix only in MSC but not in fibroblasts (Figure 1D). These results support the notion, that MSC but not fibroblasts comprise a multipotent subset of progenitor cells.
Figure 1. Molecular characterization of fibroblasts and MSC.

Dermal fibroblast and mesenchymal stromal cells from bone marrow display a very similar immunophenotype (A). Adipogenic differentiation could not be induced in fibroblasts whereas fat droplet formation was observed in about 40% of the MSC upon differentiation (B,E; blue: DAPI; green: BODIPY; scale bar: 100 µm). Osteogenic differentiation was monitored by Alizarin Red staining of calcium phosphate precipitates and this could only be induced in MSC but not in fibroblasts (C,F; scale bar: 100 µm). Chondrogenic differentiation was analyzed by Alcian Blue staining of glucosaminoglycans in micromass culture and this was again only observed in MSC but not in fibroblasts (D; scale bar: 50 µm).
DNA methylation changes between fibroblasts and MSC
DNA methylation profiles of fibroblasts and MSC of early passage (passage 3 and passage 2 respectively) were compared using the HumanMethylation27 BeadChip microarray. Hierarchical cluster analysis clearly separated fibroblasts and MSC into two different groups (Figure 2A) and histograms of P-values demonstrate a skew towards 0 which supports the notion of a high number of significant differences (Figure S1A). Various CpG sites revealed differences in average beta greater than 0.15 (referred to as more than 15% difference in methylation level) between fibroblasts and MSC and additionally passed a second filter criterion of highly significant changes (P<0.001 with limma moderated T-test): 766 CpG sites were hyper-methylated in fibroblasts whereas 752 CpG sites were hypo-methylated. These CpG sites corresponded to 521 and 499 non-redundant genes that were categorized by Gene Onthology classification. Hyper-methylated CpG sites in fibroblasts were significantly over-represented in genes of categories that mediate development, immune response and hormone secretion. Hypo-methylated genes were rather involved in immune response, defense response and wound healing (Figure S2). Taken together, fibroblasts and MSC differ in their epigenetic profiles and this may result in their dissimilar differentiation potential and immunomodulatory function.
Figure 2. Relationship of DNA methylation profiles.

DNA methylation profiles of fibroblasts and MSC were analyzed with the HumanMethylation27 BeadChip that facilitates simultaneous analysis of 27,578 unique CpG sites. Unsupervised hierarchical clustering (Euclidian distance) of all CpG sites separated fibroblasts and MSC in two different groups. Furthermore, cell preparations from the same anatomical site clustered together (A). Alternatively, we used principal components analysis (PCA) to visualize the relationship of fibroblasts from different dermal regions (B).
Epigenetic positional memory of fibroblasts
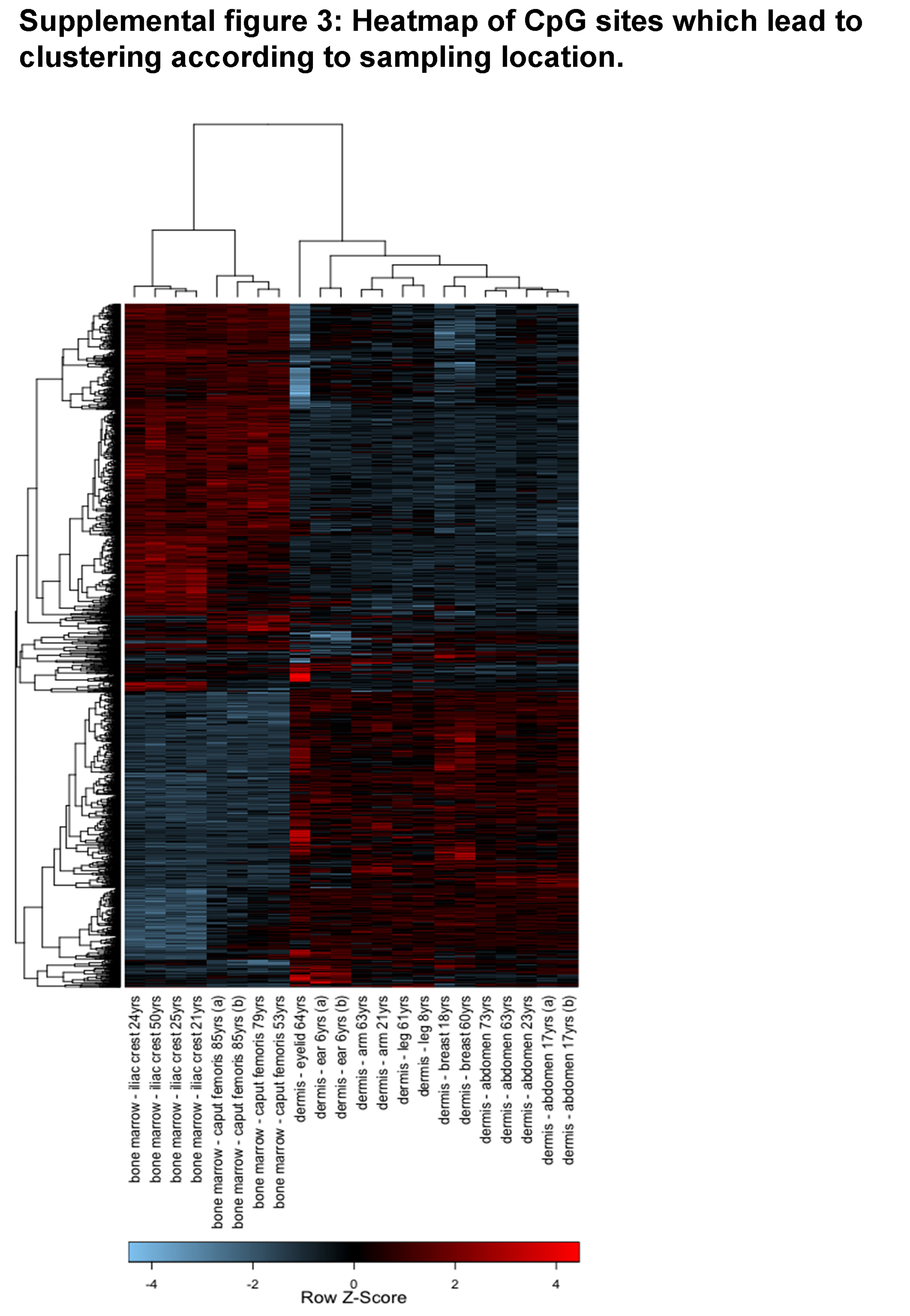
It was striking that DNA methylation profiles of fibroblasts from the same dermal region such as ear, arm, breast, abdomen or leg clustered always closely together in hierarchical cluster analysis (Figure 2A). To depict differences that lead to the clustering, we selected 3182 CpG sites that might be differentially methylated among anatomical sites (P<0.001; limma moderated F-statistic). These CpG sites were then subjected to a heatmap (Figure S3). Differences in fibroblasts from different anatomical sites are further supported by principal components analysis (PCA; Figure 2B). Such positional memory might be governed by epigenetic modification of homeobox containing or HOX-genes that have been shown to be important for patterning the primary and secondary axes [24], [25]. Hierarchical clustering of 85 CpG sites represented on the microarray that are associated with the four HOX clusters revealed methylation changes that correlate with different anatomical sites (Figure S4). Thus, fibroblasts and MSC maintain positional information in their DNA methylation profile even after culture expansion for three passages.
Age-associated DNA methylation changes in fibroblasts
Comparison of DNA methylation profiles in fibroblasts derived from young (<23 years old) or elderly donors (>60 years old) revealed that 75 CpG sites change their methylation level more than 15% upon aging (Figure 3A). However, none of these were highly significant using the stringent criteria as mentioned above (P<0.001; limma moderated T-test). This might be due to the relatively small sample number or other parameters which may impact differential methylation. Alternatively, we performed pair wise comparisons with fibroblasts from the same dermal location to exclude bias by sampling from different anatomical sites, and these results were very similar (data not shown). Only skin samples from female donors were utilized to avoid DNA methylation differences due to dose compensation of sex chromosomes [19]. On the other hand, there is a tendency towards zero in the histogram of P-values indicating that there might be specific age-associated changes (Figure S1B,C). We used rank product (RankProd) test as alternative non-parametric method to generate adjusted p-values and 257 CpG sites reached a significance level below 0.05. These included 31 CpG sites of the 75 CpG sites with methylation changes of more than 15% (Table S1).
Figure 3. Age-associated methylation changes in fibroblasts and MSC.

Mean DNA methylation of 27,578 CpG sites was compared between fibroblasts from young donors (<23 years, eight samples) and from elderly donors (>60 years; six samples) by scatter plot analysis. 75 CpG sites revealed more than 15% age-associated differential methylation (indicated as black spots; A). Scatterplot analysis of MSC from young (<50 years, four samples) and elderly donors (>50 years, four samples) demonstrated that 1060 CpG have more than 15% age-associated DNA methylation changes (B). Comparison of fibroblasts revealed that 30 CpG sites were overlapping differentially methylated upon aging in MSC and fibroblasts (black spots; C). This is highly significantly more than expected by chance alone but differential methylation often occurred in opposite directions.
Genes with CpG sites that revealed the highest age-associated hyper-methylation included sperm associated antigen 7 (SPAG7; 26%), cyclin-dependent kinase inhibitor 2B (CDKN2B; 26%) and Stannin (SNN; 20%). In contrast, genes with hypo-methylated CpG sites included aldehyde dehydrogenase 1A1 (ALDH1A1; −30%); complement factor D (CFD; −22%); WNT1 inducible signaling pathway protein 2 (WISP2; −18%) and cyclin-dependent kinase 5, regulatory subunit 1 (CDK5R1; −17%). Subsequently, we have tested how differential DNA methylation is reflected on gene expression level: quantitative real time PCR of 7 genes demonstrated that in tendency, hypomethylation upon aging is associated with higher gene expression and this was significant for WISP2, ALDH1A1 and CFD (Figure S5).
Other authors indicated that the global methylation level decays upon aging. Therefore, we reanalyzed non-quantil normalized raw-data and there was a slight decline of the methylation level upon aging in fibroblasts (Figure S6). The 90-quantil of methylation level in this boxplot was 80.5% in fibroblasts from younger donors whereas it was 78.5% in fibroblasts from elderly donors (P = 0.02). In contrast, global age-associated hypomethylation was not significant in MSC (P = 0.93). 1060 CpG sites were more than 15% differentially methylated in MSC of young and elderly donors [14] (Figure 3B). There was no linear correlation in age-associated changes in fibroblasts and MSC. Strikingly, 30 CpG sites were in the overlap of CpG sites with age-associated changes in fibroblasts and MSC (Figure 3C, Table S2). Thus, 40% of age-associated changes in fibroblast CpG sites were also differentially methylated in MSC and hypergeometric distribution revealed that this association is highly significant (P<10−25). Several CpG sites that are hyper-methylated upon aging of fibroblasts were hypo-methylated upon aging of MSC and vice versa.
Age-associated methylation changes were analyzed in more detail for CDKN2B that is part of the INK4A/ARF/INK4b locus. Notably, all 6 CpG sites within the second exon of CDKN2B represented on the HumanMethylation27 BeadChip microarray were hyper-methylated upon aging in fibroblasts, whereas they were hypo-methylated in MSC. These age-associated DNA methylation changes were exemplarily validated by pyrosequencing and this method revealed very similar age-associated methylation changes in five neighboring CpG sites (Figure 4). Taken together, there are some age-associated DNA methylation changes at specific CpG sites in fibroblasts and MSC but they are often regulated in opposite directions in a cell type dependent manner.
Figure 4. Age-associated DNA methylation changes in CDKN2B.

Cyclin-dependent kinase inhibitor 2B (CDKN2B) belongs to the INK4A/ARF/INK4B locus and six CpG sites within the second exon are represented by the microarray (A). All of these were hyper-methylated upon aging in fibroblasts (B) and they were all hypo-methylated in MSC (C). Pyrosequencing of five neighbouring CpG sites in this region verified significant hyper-methylation upon aging in fibroblasts (D) and hypo-methylation in MSC (E). * P<0.05; ** P<0.01; *** P<0.001).
Replicative senescence affects DNA methylation patterns
Long-term culture of MSC has impact on proliferation, cell size and differentiation potential [26] and we have recently demonstrated that this is associated with reproducible and highly significant DNA methylation changes at specific CpG sites [14]. To determine if similar epigenetic modifications are induced by long-term culture of fibroblasts, we have compared DNA methylation profiles of passage 3 and passage 21 of fibroblasts (6 year old donor, othoplastic). Comparison of long-term culture associated DNA methylation changes in fibroblasts and MSC revealed a striking correlation in CpG sites that were more than 15% differentially methylated in both cell types (P<10−31, chi-square analysis of 161 CpG sites; Figure 5A). Thus, replicative senescence induces similar hyper-methylation and hypo-methylation at specific CpG sites in fibroblasts and MSC. Furthermore, there was a significant correlation between DNA methylation changes in fibroblasts upon long-term culture in vitro and aging in vivo (P = 0.001, chi-square analysis of 32 CpG sites with more than 15% methylation changes in both comparisons; Figure 5B).
Figure 5. Long-term culture-associated methylation changes in fibroblasts and MSC.

Differential methylation between passage 3 and passage 21 of fibroblasts (6 year old donor) was plotted against differential DNA methylation data upon long-term culture of MSC that has been described before [14] (A). Black spots demonstrate CpG sites with differential methylation of more than 15% upon long-term culture of both cell types and these showed a very good correlation in long-term culture associated changes in fibroblasts and MSC (R = 0.764). We have also compared long-term culture-associated DNA methylation changes and age-associated changes in fibroblasts and they display only moderate correlation (R = 0.337; B).
Discussion
Fibroblasts are diverse with various functions in different dermal regions [23]. This diversity is also reflected in global DNA methylation patterns: epigenetic modifications at specific CpG sites provide a positional memory of the sampling location and there are age-associated DNA methylation changes that may contribute to aging of the skin.
The question, how to distinguish between fibroblasts and MSC, is not trivial. MSC raise high hopes for cellular therapy and regenerative medicine and they are concurrently tested in various clinical trials [27]. Despite intensive research over the last decade, there is still no molecular definition of multipotent subsets. MSC and fibroblasts are very similar in cell size, morphology, growth pattern and immunophenotype [28]–[30]. Traditionally, MSC have been defined by their multipotent differentiation potential [31], [32] but this demarcation has lost its clarity since it has been shown, that human dermal fibroblasts may also comprise multipotent cells with adipogenic, osteogenic and chondrogenic differentiation potential [33], [34]. However, for the samples used in this study, adiopogenic and osteogenic differentiation could only be induced in MSC but not fibroblasts. Hence, the difference in DNA methylation profiles between fibroblasts and MSC may also arise from a specific epigenetic make up of the multipotent subset within MSC. Further MSC preparations isolated under different culture conditions and other cell types need to be considered to determine epigenetic modifications that are characteristic for adult stem cells [32].
Reciprocal epithelial-mesenchymal interactions shape site-specific development of the skin. Recombination experiments in chick suggested that dermal signals dictate positional identity of epidermal differentiation: for example transplantation of wing epithelium to leg mesenchyme transforms the development of feathers to the development of scales [35], [36]. Various studies have addressed the anatomic diversity of human fibroblasts on the gene expression level [23], [36], [37]. These datasets demonstrated the association of global gene expression profiles with the site of fibroblast origin and they were related to three anatomic divisions: anterior - posterior, proximal - distal and dermal versus non-dermal. It has also been shown, that site-specific HOX expression in fibroblasts maintains features of the embryonic HOX pattern over decades in vivo and over numerous cell divisions in vitro [36]. In this study, we demonstrate that fibroblasts also maintain an epigenetic positional memory even after culture expansion for three passages. Hierarchical clustering of CpG sites related to HOX genes revealed a separation of fibroblasts from the top half (anterior) versus the bottom half (posterior) of the human body. Recently it has been shown, that variably methylated regions (VMRs) exist in the genome which are highly variable in DNA-methylation among individuals [38]. Several of these were in developmental genes including HOX genes and therefore inter-individual differences in fibroblast CpG methylation may act as a potential contributor to the site-dependent structure. Thus, a larger number of samples from various regions will be necessary to gain further insight into the systematics of positional determination by epigenetic modifications. This might also be useful for forensic analysis of tissue samples of unknown origin.
Various molecular mechanisms have been implicated in aging including telomere-shortening, accumulation of mutations, oxidative stress and alteration of molecular pathways [39]. Decreases in the global level of methylation along with a concomitant increase in promoter methylation has become a hallmark of age-associated epigenetic changes [5]–[7], [40]. Decrease of the 5-methylcytosine content upon aging has been demonstrated almost 40 years ago in the brain, heart and spleen of rats [6] and there is even evidence, that the global methylation level changes upon aging in plants [41]. In this study, we observed only a moderate age-associated hypo-methylation in fibroblasts. This might be due to the bias for promoter regions within the 27,578 CpG sites represented on the microarray whereas it has been shown, that hypo-methylation upon aging occurs predominantly in specific interspersed repetitive sequences (IRSs) [42]. Age-associated DNA methylation changes appear to be more complex as methylation of specific CpG sites reveal directional perturbations [12], [43], [44]. For example, Christensen et al. provided clear evidence of tissue–specific methylation changes related to aging [43].
The INK4a/ARF/INK4b locus (also known as CDKN2A and CDKN2B) plays a pivotal role in aging and cancer [45]. Gene expression of p16INK4A (CDKN2A) increases with age and it has even been proposed as a biomarker of aging [46]. Furthermore, functional studies in hematopoietic stem cells, neuronal stem cells and pancreatic islets have suggested, that p16INK4A is one cause of aging by down-regulation of self-renewal potential of adult stem cells [46]–[48] and reprogramming towards induced pluripotent stem cells is critically dependent on the repression of p19ARF in mice and p16INK4A in humans [49]. In this study, we observed the second-highest hyper-methylation upon aging of fibroblasts in CDKN2B. However, it is intriguing that the same CpG sites are hypo-methylated upon aging in MSC.
Overall, age-associated DNA methylation changes occurred at similar CpG sites upon aging in fibroblasts and MSC, but they were often regulated in different directions. This may underline the functional difference between the two cell types and the involvement of different mechanisms for cellular aging. It is also conceivable, that epigenetic modifications are differently regulated by the diverse cellular microenvironments in dermis and bone marrow. In our analysis, ages in fibroblasts and MSC were not matched and aging in MSC might be biased by the different available sources of bone marrow in young and elderly donors as described before [14]. All the more, the significant interrelation of age-associated methylation changes in fibroblasts and MSC is noteworthy. Recently, polycomb group protein targets have been shown to become far more likely methylated with age [13]. Furthermore, hyper-methylation occurs predominantly at bivalent chromatin domain promoters in CD4+ T-cells and CD14+ monocytes [12]. These are developmentally regulated genes harboring both, activating (H3K4me3) and inactivating (H3K27me3) histone marks in embryonic stem cells [50]–[52]. The bivalent epigenetic modifications raise the possibility of age-associated genes being poised in a bipotential state, which may be resolved differently in various cell lineages. This might explain the opposing age-associated DNA methylation changes in different tissues [43] and between fibroblasts and MSC.
Replicative senescence upon long-term culture is not an inevitable fate of all cells. Embryonic stem cells as well as induced pluripotent stem cells do not show signs of replicative senescence in long-term culture [53]. Loss of pluripotency seems to be accompanied with limitation of replicative live span and this might be governed by specific epigenetic modifications. We have previously demonstrated continuous changes in gene expression profiles of MSC upon long-term culture [15], [54] and observed a moderate but significant concordance in the expression profiles upon long-term culture in vitro and aging in vivo [16]. Furthermore, we have shown that long-term culture of MSC induces hyper-methylation and hypo-methylation at specific CpG sites and that these epigenetic modifications are also related to those between MSC from young and elderly donors [14]. Here, we have exemplarily analyzed DNA methylation changes upon long-term culture of fibroblasts and there was a striking concordance of senescence-associated methylation changes in fibroblasts and MSC. Additional fibroblast samples are currently culture-expanded to further discern long-term culture associated DNA methylation changes. This might facilitate identification of specific CpG sites for monitoring the replicative lifespan in cellular products for therapeutic application [17]. Notably, there was also a significant association of methylation changes upon long-term culture and aging in fibroblasts. This indicates that both mechanisms might be regulated by similar epigenetic modifications.
Two fundamental hypothesis are currently discussed to account for aging: it might be evoked by stochastic or random, accidental events, or it is a result of a purposeful program driven as developmental process [55]. Most likely, interplay of both mechanisms promotes aging at various levels. However, the highly reproducible epigenetic modification at specific CpG sites in developmental genes argues for a coordinated developmental mechanism. This study demonstrates, that fibroblasts and MSC maintain their positional memory as well as age-associated epigenetic modifications even after in vitro culture. Age-associated modifications seem to be regulated in a cell type specific manner. Further research on other characterized cell preparations and larger numbers of samples are important to gain insight into the mechanisms that control DNA methylation at specific sites in the genome.
Materials and Methods
Isolation of primary cells
Human dermal fibroblasts were isolated from skin samples donated by young (6–23 years) and elderly (60–73 years) female patients undergoing surgical interventions. All samples were taken after written consent, using guidelines approved by the Ethic Committee on the Use of Human Subjects at the University of Aachen. Skin samples were treated with dispase II (Roche Diagnostics, Mannheim, Germany) for 12 hours at 4°C to separate the dermis from the epidermis. The dermis was digested with 0,2% collagenase and 1,5% BSA in collagenase buffer (100 mM HEPES, 120 mM NaCl, 50 mM KCl, 1 mM CaCl2, 5 mM Glucose) for 45 minutes. Dermal remnants were removed by filtering the digest through a 100 µm nylon strainer (Falcon, Becton Dickinson [BD], San Jose, USA). The cells were subsequently washed and expanded in standard medium consisting of DMEM (PAA; 1 g/L glucose) supplemented with glutamine (PAA), penicillin/streptomycin (PAA) and 10% fetal calf serum (Biochrom, Berlin, Germany) in a humidified atmosphere at 5% CO2.
Mesenchymal stromal cells were isolated from human bone marrow after written consent using guidelines approved by the Ethic Committee on the Use of Human Subjects at the University of Heidelberg as described before [16]. In brief, MSC were isolated from bone marrow aspirates from the iliac crest of young healthy donors for allogeneic transplantation or from the caput femoris after hip fracture of elderly donors. MSC were culture-expanded in culture medium M1 as previously described [16], [30].
Immunophenotypic analysis
Surface marker expression was analyzed on a FACS canto II (BD) upon staining with the following antibodies as described before [16]: CD14-allophycocyanin (APC, clone M5E2, BD), CD29-phycoerythrin (PE, clone MAR4, BD), CD31-PE (clone WM59, BD), CD34-APC (clone 8G12, BD), CD45-APC (clone HI30, BD), CD73-PE (clone AD2, BD), CD90-APC (clone 5E10, BD), CD105-fluorescein isothiocyanate (FITC, clone MEM-226, ImmunoTools, Friesoythe, Germany).
In vitro differentiation
Osteogenic, adipogenic and chondrogenic differentiation of fibroblasts and MSC was simultaneously performed under the same differentiation conditions as described before [15], [56]. After three weeks, osteogenic differentiation was analyzed by Alizarin Red staining and quantified with a method based on acetic acid extraction and neutralization with ammonium hydroxide [57]. A Tecan infinite M200 plate reader was employed to measure the absorbance at 405 nm.
Adipogenic differentiation was stained with the green fluorescent dye BODIPY (4,4-difluoro-1,2,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene) and counter-stained with DAPI (4′,6-Diamidin-2-phenylindol; both Molecular Probes, Eugene, Oregon, USA). Fluorescence microscopic pictures were always taken from four randomly chosen areas with a Leica DM IL HC microscope (Leica, Wetzlar, Germany). Cell nuclei and BODIPY positive cells were counted with ImageJ software (rsbweb.nih.gov/ij/) to determine the percentage of cells that differentiated towards adipogenic lineage as described before [58].
Chondrogenic differentiation was induced in micromass culture for three weeks. Subsequently the pellets were fixed with 10% formalin and paraffin embedded. 1 µm sections were stained with Alcian blue in combination with PAS (Periodic acid-Schiff) in an automated slide stainer and photo-documented.
DNA isolation
Genomic DNA was isolated from 106 cells using the QIAGEN DNA Blood Midi-Kit. DNA quality was assessed with a NanoDrop ND-1000 spectrometer (NanoDrop Technologies, Wilmigton, USA) and gel electrophoresis. 600 ng DNA were subsequently bisulfite converted using the EpiTect Bisulfite Kit (Qiagen, Hilden, Germany).
DNA methylation profiling
DNA methylation profiles were analyzed using the HumanMethylation27 BeadChip (Illumina, San Diego, USA) as described before [14]. About 200 ng of bisulfite converted DNA were applied per BeadChip according to the manufacturer's instructions. During hybridization, the DNA molecules anneal to two different bead types with locus-specific DNA oligomers - one corresponds to the methylated (C) and the other to the unmethylated (T) state. Allele-specific primer annealing is followed by single-base extension using DNP- and Biotin-labeled ddNTPs. After extension, the array is fluorescently stained, scanned, and the intensities of the unmethylated and methylated bead types measured. Hybridization and initial data analysis with the BeadStudio Methylation Module was performed at the DKFZ Gene Core Facility in Heidelberg. The precise distribution of samples across the beadchips is presented in Table S3. The complete CpG methylation values have been deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE22595. (Hyperlink for review: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=rbsjfkqoqoukstc&acc=GSE22595 ).
DNA methylation values, described as beta values, are recorded for each locus in each sample. Background normalized raw data were only used to compare the overall methylation level in samples from young and elderly donors. For all other analysis, quantile normalization of the raw data was performed to minimize the chip effect [59]. Hierarchical clustering by Euclidian distance was performed using the MultiExperiment Viewer (MeV, TM4) [60]. Principal components analysis (PCA) was calculated with prcomp in R package stats. For analysis of age-associated DNA methylation changes we have calculated mean methylation of samples from young and elderly donors. To focus on biologically relevant CpG sites we have selected those with differences in average beta value greater than 0.15 (more than 15% difference in methylation level). Standard deviation in DNA-methylation measurements are not uniformly distributed across the range of beta values – it has a parabolic shape with the maximum peak around 0.5 [61]. Smaller changes at the two extremes of the beta-scale might also be important but they are less likely to be biologically relevant, more difficult to validate and using the 15% methylation threshold reduces beadchip effects which might exist despite quantile normalization.
To further select for significantly differentially methylated CpG sites we used a false discovery (FDR) approach using limma moderated T-test, which is based on an empirical Bayes moderation approach [62]. For comparison of fibroblasts and MSC we used very stringent parameters with 15% methylation changes and adjusted p-value (q-value) <0.001 [61]. However, these criteria were too stringent for analysis of aging in fibroblast because of the relatively small number of samples and therefore, we used an additional non-parametric method named RankProd with q-value <0.05. This method shared many CpG sites with the above mentioned cut-off of 15% methylation change. Genes associated with the differentially methylated CpG sites were classified by GeneOntology analysis using GoMiner software (http://discover.nci.nih.gov/gominer/).
Pyrosequencing
Methylation changes in CDKN2B were validated by pyrosequencing as described before [14]. Pyrosequencing was performed at Varionostic GmbH (Ulm, Germany).
Quantitative real-time PCR analysis
Expression of 7 differentially methylated genes was analyzed by real-time quantitative PCR (qRT-PCR) using the StepOne™ Instrument (Applied Biosystems, Applera Deutschland GmbH, Darmstadt, Germany). Total RNA was isolated and reversely transcribed as described in our previous work [16]. Primers were obtained from Metabion (Martinsried, Germany) (Table S4). QRT-PCR reactions were performed with the Fast SYBR® green PCR master mix in a MicroAmp optical 96-well reaction plate according to the manufacturer's instructions (Applied Biosystems). Gene expression levels were normalized to GAPDH expression.
Statistics
All results are expressed as mean ± standard deviation. To estimate the probability of differences we have adopted the two-sided Student's T-test. Probability value of P<0.05 denoted statistical significance. Probability that age-associated methylation changes are related in different datasets was estimated by hypergeometric distribution, and linear correlation was determined by Pearson correlation coefficient. Representation of differentially methylated genes in functional categories was analyzed by Fischer's Exact p-value.
Supporting Information
Histograms of limma moderated T-test raw p-values. For comparison of fibroblasts of young versus elderly donors, we performed nonspecific filtering to exclude the CpG sites with low variation by applying nsFilter function in genefilter package in R/Bioconductor. We then compared the histograms of raw p-values before and after filtering. This shows that the non-specific step can improve our analysis.
(JPG)
{kind=link}
Gene Onthology analysis of genes with differentially methylated CpG sites between fibroblasts and MSC.
(TIF)
Heat map of CpG sites which lead to clustering according to sampling location. Samples were classified according to their sampling location (iliac crest, caput femoris, eyelid, ear, arm, leg, breast and abdomen). 3182 CpG sites were selected that might be differentially methylated (P<0,001; limma moderated F-statistic). These CpG sites were then subjected to a heatmap. Each CpG was normalized to zero and unit standard deviation.
(JPG)
{kind=link}
Hierarchical clustering of 85 CpG sites associated with the four HOX clusters.
(TIF)
qRT-PCR of differentially methylated genes in fibroblasts of young and elderly donors.
(TIF)
Boxplot of methylation level in raw data of beadchips.
(TIF)
Age-associated methylation changes in fibroblasts.
(DOC)
Age-associated changes in fibroblasts and MSC.
(DOC)
Distribution of all the samples across the beadchips.
(DOC)
Primer sequences for qRT-PCR.
(DOC)
Acknowledgments
We want to thank Andrea Fritz, Christine Volkmar and Stephanie Brosig for excellent technical assistance and Dipl. Ing. Matthias Schick (DKFZ, Heidelberg, Germany) for hybridization and preliminary analysis of DNA methylation data.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by the Heidelberg Academy of Sciences (WIN-Kolleg), by the German Ministry of Education and Research (BMBF) within the supporting program "cell based regenerative medicine" (CB-HERMES), by the excellence initiative of the German federal and state governments, by the START-Program of the Faculty of Medicine, RWTH Aachen and by the Stem Cell Network North Rhine Westphalia. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23:413–418. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Fraga MF, Agrelo R, Esteller M. Cross-talk between aging and cancer: the epigenetic language. Ann N Y Acad Sci. 2007;1100:60–74. doi: 10.1196/annals.1395.005. [DOI] [PubMed] [Google Scholar]
- 3.Munoz-Najar UM, Sedivy JM. Antioxid Redox Signal; 2010. Epigenetic Control of Aging. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 5.Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem. 1987;262:9948–9951. [PubMed] [Google Scholar]
- 6.Vanyushin BF, Nemirovsky LE, Klimenko VV, Vasiliev VK, Belozersky AN. The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents. Gerontologia. 1973;19:138–152. [PubMed] [Google Scholar]
- 7.Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, et al. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Ann Hum Genet. 2004;68:196–204. doi: 10.1046/j.1529-8817.2004.00081.x. [DOI] [PubMed] [Google Scholar]
- 8.Wilson VL, Jones PA. DNA methylation decreases in aging but not in immortal cells. Science. 1983;220:1055–1057. doi: 10.1126/science.6844925. [DOI] [PubMed] [Google Scholar]
- 9.Casillas MA, Jr, Lopatina N, Andrews LG, Tollefsbol TO. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol Cell Biochem. 2003;252:33–43. doi: 10.1023/a:1025548623524. [DOI] [PubMed] [Google Scholar]
- 10.Murgatroyd C, Wu Y, Bockmuhl Y, Spengler D. The Janus face of DNA methylation in aging. Aging (Albany NY) 2010;2:107–110. doi: 10.18632/aging.100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.So K, Tamura G, Honda T, Homma N, Waki T, et al. Multiple tumor suppressor genes are increasingly methylated with age in non-neoplastic gastric epithelia. Cancer Sci. 2006;97:1155–1158. doi: 10.1111/j.1349-7006.2006.00302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bork S, Pfister S, Witt H, Horn P, Korn B, et al. DNA Methylation Pattern Changes upon Long-Term Culture and Aging of Human Mesenchymal Stromal Cells. Aging Cell. 2010;9:54–63. doi: 10.1111/j.1474-9726.2009.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner W, Horn P, Castoldi M, Diehlmann A, Bork S, et al. Replicative Senescence of Mesenchymal Stem Cells - a Continuous and Organized Process. PLoS ONE. 2008;5:e2213. doi: 10.1371/journal.pone.0002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagner W, Bork S, Horn P, Krunic D, Walenda T, et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS ONE. 2009;4:e5846. doi: 10.1371/journal.pone.0005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner W, Bork S, Lepperdinger G, Joussen S, Ma N, et al. How to track cellular aging of mesenchymal stromal cells. Aging (Albany NY) 2010;2:224–230. doi: 10.18632/aging.100136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boukamp P. Skin aging: a role for telomerase and telomere dynamics? Curr Mol Med. 2005;5:171–177. doi: 10.2174/1566524053586644. [DOI] [PubMed] [Google Scholar]
- 19.Gronniger E, Weber B, Heil O, Peters N, Stab F, et al. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genet. 2010;6:e1000971. doi: 10.1371/journal.pgen.1000971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haniffa MA, Collin MP, Buckley CD, Dazzi F. Mesenchymal stem cells: the fibroblasts' new clothes? Haematologica. 2009;94:258–263. doi: 10.3324/haematol.13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Junker JP, Sommar P, Skog M, Johnson H, Kratz G. Adipogenic, chondrogenic and osteogenic differentiation of clonally derived human dermal fibroblasts. Cells Tissues Organs. 2010;191:105–118. doi: 10.1159/000232157. [DOI] [PubMed] [Google Scholar]
- 22.Cappellesso-Fleury S, Puissant-Lubrano B, Apoil PA, Titeux M, Winterton P, et al. Human Fibroblasts Share Immunosuppressive Properties with Bone Marrow Mesenchymal Stem Cells. J Clin Immunol. 2010;30:607–619. doi: 10.1007/s10875-010-9415-4. [DOI] [PubMed] [Google Scholar]
- 23.Chang HY, Chi JT, Dudoit S, Bondre C, van de RM, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tabin CJ. Why we have (only) five fingers per hand: hox genes and the evolution of paired limbs. Development. 1992;116:289–296. doi: 10.1242/dev.116.2.289. [DOI] [PubMed] [Google Scholar]
- 25.Nelson CE, Morgan BA, Burke AC, Laufer E, DiMambro E, et al. Analysis of Hox gene expression in the chick limb bud. Development. 1996;122:1449–1466. doi: 10.1242/dev.122.5.1449. [DOI] [PubMed] [Google Scholar]
- 26.Wagner W, Ho AD, Zenke M. Different Facets of Aging in Human Mesenchymal Stem Cells. Tissue Eng Part B Rev. 2010;16:445–453. doi: 10.1089/ten.TEB.2009.0825. [DOI] [PubMed] [Google Scholar]
- 27.Sensebe L, Krampera M, Schrezenmeier H, Bourin P, Giordano R. Mesenchymal stem cells for clinical application. Vox Sang. 2009;98:93–107. doi: 10.1111/j.1423-0410.2009.01227.x. [DOI] [PubMed] [Google Scholar]
- 28.Bae S, Ahn JH, Park CW, Son HK, Kim KS, et al. Gene and microRNA expression signatures of human mesenchymal stromal cells in comparison to fibroblasts. Cell Tissue Res. 2009;335:565–573. doi: 10.1007/s00441-008-0729-y. [DOI] [PubMed] [Google Scholar]
- 29.Haniffa MA, Wang XN, Holtick U, Rae M, Isaacs JD, et al. Adult human fibroblasts are potent immunoregulatory cells and functionally equivalent to mesenchymal stem cells. J Immunol. 2007;179:1595–1604. doi: 10.4049/jimmunol.179.3.1595. [DOI] [PubMed] [Google Scholar]
- 30.Wagner W, Wein F, Seckinger A, Frankhauser M, Wirkner U, et al. Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp Hematol. 2005;33:1402–1416. doi: 10.1016/j.exphem.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 32.Wagner W, Ho AD. Mesenchymal stem cell preparations-comparing apples and oranges. Stem Cell Rev. 2007;3:239–248. doi: 10.1007/s12015-007-9001-1. [DOI] [PubMed] [Google Scholar]
- 33.Lorenz K, Sicker M, Schmelzer E, Rupf T, Salvetter J, et al. Multilineage differentiation potential of human dermal skin-derived fibroblasts. Exp Dermatol. 2008;17:925–932. doi: 10.1111/j.1600-0625.2008.00724.x. [DOI] [PubMed] [Google Scholar]
- 34.Chen FG, Zhang WJ, Bi D, Liu W, Wei X, et al. Clonal analysis of nestin(−) vimentin(+) multipotent fibroblasts isolated from human dermis. J Cell Sci. 2007;120:2875–2883. doi: 10.1242/jcs.03478. [DOI] [PubMed] [Google Scholar]
- 35.Brotman HF. Epidermal-dermal tissue interactions between mutant and normal embryonic back skin: site of mutant gene activity determining abnormal feathering is in the epidermis. J Exp Zool. 1977;200:243–257. doi: 10.1002/jez.1402000206. [DOI] [PubMed] [Google Scholar]
- 36.Rinn JL, Wang JK, Allen N, Brugmann SA, Mikels AJ, et al. A dermal HOX transcriptional program regulates site-specific epidermal fate. Genes Dev. 2008;22:303–307. doi: 10.1101/gad.1610508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rinn JL, Bondre C, Gladstone HB, Brown PO, Chang HY. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006;2:e119. doi: 10.1371/journal.pgen.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feinberg AP, Irizarry RA, Fradin D, Aryee MJ, Murakami P, et al. Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci Transl Med. 2010;2:49ra67. doi: 10.1126/scitranslmed.3001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vijg J, Campisi J. Puzzles, promises and a cure for ageing. Nature. 2008;454:1065–1071. doi: 10.1038/nature07216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agrawal A, Tay J, Yang GE, Agrawal S, Gupta S. Age-associated epigenetic modifications in human DNA increase its immunogenicity. Aging (Albany NY) 2010;2:93–100. doi: 10.18632/aging.100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraga MF, Rodriguez R, Canal MJ. Genomic DNA methylation-demethylation during aging and reinvigoration of Pinus radiata. Tree Physiol. 2002;22:813–816. doi: 10.1093/treephys/22.11.813. [DOI] [PubMed] [Google Scholar]
- 42.Jintaridth P, Mutirangura A. Physiol Genomics; 2010. Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. [DOI] [PubMed] [Google Scholar]
- 43.Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 47.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 48.Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Collado M, Villasante A, Strati K, Ortega S, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 51.Azuara V, Perry P, Sauer S, Spivakov M, Jorgensen HF, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 52.Zhao XD, Han X, Chew JL, Liu J, Chiu KP, et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007;1:286–298. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 53.Zeng X. Human embryonic stem cells: mechanisms to escape replicative senescence? Stem Cell Rev. 2007;3:270–279. doi: 10.1007/s12015-007-9005-x. [DOI] [PubMed] [Google Scholar]
- 54.Schallmoser K, Bartmann C, Rohde E, Bork S, Guelly C, et al. Replicative senescence-associated gene expression changes in mesenchymal stromal cells are similar under different culture conditions. Haematologica. 2010;95:867–874. doi: 10.3324/haematol.2009.011692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayflick L. Biological aging is no longer an unsolved problem. Ann N Y Acad Sci. 2007;1100:1–13. doi: 10.1196/annals.1395.001. [DOI] [PubMed] [Google Scholar]
- 56.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 57.Gregory CA, Gunn WG, Peister A, Prockop DJ. An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem. 2004;329:77–84. doi: 10.1016/j.ab.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 58.Horn P, Bokermann G, Cholewa D, Bork S, Walenda T, et al. Cytotherapy; in press; 2010. Comparison of Individual Platelet Lysates for Isolation of Human Mesenchymal Stromal Cells. [Google Scholar]
- 59.Kuan PF, Wang S, Zhou X, Chu H. Bioinformatics; in press; 2010. A statistical framework for Illumina DNA methylation arrays. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saeed AI, Sharov V, White J, Li J, Liang W, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 61.Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16:383–393. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histograms of limma moderated T-test raw p-values. For comparison of fibroblasts of young versus elderly donors, we performed nonspecific filtering to exclude the CpG sites with low variation by applying nsFilter function in genefilter package in R/Bioconductor. We then compared the histograms of raw p-values before and after filtering. This shows that the non-specific step can improve our analysis.
(JPG)
Gene Onthology analysis of genes with differentially methylated CpG sites between fibroblasts and MSC.
(TIF)
Heat map of CpG sites which lead to clustering according to sampling location. Samples were classified according to their sampling location (iliac crest, caput femoris, eyelid, ear, arm, leg, breast and abdomen). 3182 CpG sites were selected that might be differentially methylated (P<0,001; limma moderated F-statistic). These CpG sites were then subjected to a heatmap. Each CpG was normalized to zero and unit standard deviation.
(JPG)
Hierarchical clustering of 85 CpG sites associated with the four HOX clusters.
(TIF)
qRT-PCR of differentially methylated genes in fibroblasts of young and elderly donors.
(TIF)
Boxplot of methylation level in raw data of beadchips.
(TIF)
Age-associated methylation changes in fibroblasts.
(DOC)
Age-associated changes in fibroblasts and MSC.
(DOC)
Distribution of all the samples across the beadchips.
(DOC)
Primer sequences for qRT-PCR.
(DOC)