

Abstract
We have leveraged a Drosophila model relevant to Alzheimer disease (AD) for functional screening of findings from a genome-wide scan for loci associated with a quantitative measure of AD pathology in humans. In six of the 15 genomic regions evaluated, we successfully identified a causal gene for the association, on the basis of in vivo interactions with the neurotoxicity of Tau, which forms neurofibrillary tangles in AD. Among the top results, rs10845990 within SLC2A14, encoding a glucose transporter, showed evidence of replication for association with AD pathology, and gain and loss of function in glut1, the Drosophila ortholog, was associated with suppression and enhancement of Tau toxicity, respectively. Our strategy of coupling genome-wide association in humans with functional screening in a model organism is likely to be a powerful approach for gene discovery in AD and other complex genetic disorders.
Main Text
Genome-wide association studies (GWAS) have emerged as powerful tools for the dissection of complex genetic traits, such as susceptibility to Alzheimer disease (AD, MIM 104300);1 however, efficient methods are needed to enhance follow-up of association signals in order to accelerate the identification and functional validation of genes affected by causal variants.2 On the basis of recent analyses, the top of GWAS-results distributions (10−3 < p < 10−7), though falling short of genome-wide significance (p < 5 × 10−8), are likely enriched for true associations, but these signals are obscured by a substantial number of chance observations with comparable statistical evidence.3–5 New strategies are therefore needed, not only to validate associations with the best evidence, but also to facilitate identification of true signals of association in circumstances where statistical power is limited and increased sample size is not feasible. One potential solution is to couple the GWAS with a functional screen that evaluates candidate genes for participation in a relevant pathological cascade, a two-stage strategy that might effectively increase overall study power. Here, we leverage a model system relevant to AD in the fruit fly, Drosophila melanogaster, to perform functional testing of 19 genes from 15 distinct genomic regions identified in a GWAS for loci influencing the burden of AD pathology in humans.
AD is the most common cause of dementia, and it is characterized at autopsy by widespread neuronal loss in association with extracellular amyloid plaques and intracellular neurofibrillary tangles, predominantly comprising the amyloid-β peptide (Aß) and Tau, respectively.6 Both rare mutations and common polymorphisms have been found to influence susceptibility for AD, and GWAS have recently been successful at discovering such loci.1,7–9 Most GWAS conducted to date have relied on the dichotomous outcome of AD clinical diagnosis; however, this study design is potentially confounded by genetic heterogeneity of dementia in cases and subclinical disease in controls. In a complementary approach, we have based our analysis on a relevant AD intermediate phenotype: a quantitative measure of global AD pathology from postmortem counts of amyloid plaques and neurofibrillary tangles. Although this approach potentially offers more statistical power than a case-control study of comparable size,10,11 it is limited by the difficulty in obtaining neuropathologic data on large numbers of older individuals. Thus, we anticipated a challenge in meeting the statistical burden of proof for gene discovery, and therefore we coupled our association analysis with a functional screening paradigm in order to validate our results.
A GWAS was performed in an autopsy cohort consisting of 227 participants from the Religious Orders Study and the Rush Memory and Aging Project, two longitudinal, epidemiologic studies of aging and AD that include brain donation at death.12–14 Written informed consent was given and an Anatomic Gift Act signed by all study participants after the procedures were fully explained, and both studies were approved by the institutional review board of Rush University Medical Center. Subjects were nondemented at recruitment and were followed prospectively with annual clinical evaluations. Proximate to death, 40% of subjects had normal cognition, 22% had mild cognitive impairment, and 38% met clinical criteria for AD (Table S1 available online). After quality control, 334,575 SNP genotypes were available for analysis (Figure S1). The outcome was a continuous measure of global AD pathology, based on averaged counts of neuritic plaques, diffuse plaques, and neurofibrillary tangles on silver-stained tissue sections from five brain regions (midfrontal, middle temporal, inferior parietal, and entorhinal cortices and the hippocampal CA1 sector).15,16 Linear regression was used to evaluate SNP associations with the continuous AD pathological trait, adjusting for both age at death and APOE ɛ4 (MIM 107741) genotype. The top independently associated regions (p < 1 × 10−3) containing candidate genes are presented in Table 1 (for full results, see Table S2). Of note, the subjects in the study cohort were also part of a larger autopsy collection used for a recent candidate-based analysis of associations with AD pathology intermediate phenotypes;11 however, none of the loci examined in that study exceeded the significance threshold applied here, and many of those SNPs were not captured by the Illumina genotyping platform used in this genome scan.
Table 1.
GWAS Results and Functional Screening
| SNP | Locus | Alleles | MAF | Beta (95% CI) | p Value | Human Gene(s) |
Functional Screen |
||
|---|---|---|---|---|---|---|---|---|---|
| Fly Ortholog | LOF | GOF | |||||||
| rs393569 | 19q13 | C/T | 0.49 | 0.15 (0.09 to 0.21) | 1.64 × 10−6 | SPTBN4 | B-spec | Enh | - |
| SHKBP1 | CG9467 | - | N/A | ||||||
| LTBP4 | |||||||||
| rs1941526 | 18q12 | A/G | 0.28 | 0.15 (0.09 to 0.22) | 6.46 × 10−6 | PIK3C3 | Pi3K59F | - | N/A |
| rs17468071 | 9p21 | C/T | 0.11 | 0.22 (0.12 to 0.31) | 7.87 × 10−6 | ELAVL2 | fne | Sup | Enh |
| rs2280861 | 8p21 | C/T | 0.25 | −0.16 (−0.23 to −0.09) | 1.40 × 10−5 | ENTPD4 | NTPase | - | - |
| SLC25A37 | mfrn | - | - | ||||||
| rs10065260 | 5q14 | C/A | 0.49 | 0.13 (0.07 to 0.19) | 2.38 × 10−5 | SCAMP1 | Scamp | - | - |
| LHFPL2 | CG3770 | - | N/A | ||||||
| rs1935502 | 10p12 | A/G | 0.30 | 0.15 (0.08 to 0.21) | 2.66 × 10−5 | SLC39A12 | CG10006 | - | N/A |
| rs3824982 | 11p14 | T/C | 0.22 | 0.15 (0.08 to 0.22) | 3.22 × 10−5 | MPPED2 | CG16717 | - | N/A |
| rs12378647 | 9q33 | G/A | 0.35 | 0.14 (0.08 to 0.21) | 3.44 × 10−5 | DBC1 | |||
| rs16898 | 5q14 | T/C | 0.31 | −0.13 (−0.19 to −0.07) | 4.64 × 10−5 | HAPLN1 | |||
| rs2108720 | 7p14 | T/C | 0.22 | −0.16 (−0.23 to −0.08) | 5.23 × 10−5 | POU6F2 | pdm3 | - | N/A |
| rs527346 | 12p13 | G/A | 0.45 | −0.12 (−0.18 to −0.06) | 5.72 × 10−5 | TSPAN9 | tsp5D | - | N/A |
| rs10845990 | 12p13 | T/G | 0.39 | 0.13 (0.06 to 0.19) | 6.93 × 10−5 | SLC2A14 | Glut1 | Enh | Sup |
| NANOG | bsh | - | - | ||||||
| rs9513122 | 13q32 | G/A | 0.43 | −0.12 (−0.18 to −0.06) | 1.70 × 10−4 | HS6ST3 | hs6st | Enh | - |
| rs7591708 | 2p15 | T/C | 0.35 | 0.12 (0.06 to 0.18) | 1.93 × 10−4 | EHBP1 | CG15609 | - | N/A |
| rs7128063 | 11q14 | A/G | 0.25 | −0.13 (−0.20 to −0.06) | 5.93 × 10−4 | DLG2 | dlg | Enh | - |
| rs12634690 | 3p12 | T/C | 0.33 | −0.11 (−0.17 to −0.04) | 1.32 × 10−3 | ROBO2 | robo | - | - |
| rs297808 | 5q35 | G/A | 0.36 | 0.09 (0.03 to 0.15) | 2.60 × 10−3 | SLIT3 | slit | Enh | Sup |
Alleles are denoted as minor/major. Beta is calculated per copy of minor allele under the additive genetic model with adjustment for age at death and APOE ɛ4 genotype. CI, confidence interval. Functional Screen shows screening results based on testing of gain or loss of function (GOF and LOF, respectively) in orthologous fly genes for enhancement (Enh) or suppression (Sup) of Tau toxicity. MAF, minor allele frequency; -, no interaction observed; N/A, genetic reagent not available. Fly orthologs were identified on the basis of implementation of the tBLASTn algorithm50 within the annotated Drosophila genome. All orthologs had highly significant BLAST results: E value < 10−10 and mean score = 398 (range: 67–1462). Fly genes with evidence of functional interactions with Tau toxicity are shown in boldface type.
As expected for our small study, no variant achieved genome-wide significance, and we therefore implemented our functional screening strategy. Candidate genes in the vicinity of top-scoring SNPs were identified on the basis of linkage disequilibrium criteria (Table 1 and Table S2), and in each case, all such genes were included for further evaluation in an unbiased fashion. In nine out of 24 cases, no candidate genes were identified in the target genomic region around an index SNP, and these association signals were not pursued further. We additionally chose to evaluate two genomic regions that were identified by SNP associations of more modest significance but contained genes (SLIT3 [MIM 603745] and ROBO2 [MIM 602431]) that function as ligand and receptor, respectively, in a common neuronal signaling pathway. Nineteen out of the 22 candidate genes had conserved orthologs in Drosophila and were promoted to functional testing.
A variety of Drosophila experimental models relevant to AD have been developed, including transgenic systems based on the neurotoxicity of both Aß and Tau.17–19 For functional screening of GWAS results, we selected the Tau transgenic model because (1) it has previously been successfully employed for rapid genetic screening20 and (2) there is growing consensus that Tau is a downstream mediator of Aß toxicity in AD.6,21–23 Expression of human Tau (MAPT [MIM 157140]) in the Drosophila nervous system recapitulates several features of AD, including age-dependent neurodegeneration, decreased lifespan, and abnormally phosphorylated and misfolded Tau.19 We used transgenic animals, allowing tissue-specific expression of TauV337M, a mutant form of Tau associated with familial frontotemporal dementia (FTD [MIM 600274]). Importantly, wild-type and mutant forms of human Tau demonstrate similar mechanisms of toxicity when expressed in the Drosophila nervous system and show consistent interactions with known genetic modifiers.19,24,25 Therefore, similar to transgenic mouse models based on FTD mutant Tau,26,27 the fly model selected for our study is relevant to understanding the mechanisms of Tau toxicity in AD.28
TauV337M expression in the fly eye causes a moderately reduced eye size and roughened surface (Figure 1B), a phenotype that is amenable to rapid screening for second-site genetic modifiers.20 Specifically, by scoring for lines that either exacerbate or rescue the eye phenotype, genes can be characterized as enhancers or suppressors of Tau toxicity, respectively. For loss-of-function analysis, transgenic RNA-interference (RNAi) lines were tested for all 19 target genes,29,30 and classical Drosophila mutant alleles were also available in most cases.31,32 In addition, we evaluated lines known or predicted to activate gene expression, allowing assessment for gain-of-function interactions for many loci.33 Genetic modifier effects were scored with the use of a semiquantitative rating scale of rough-eye severity, allowing statistical comparison with Tau transgenic controls (Figure S4).
Figure 1.
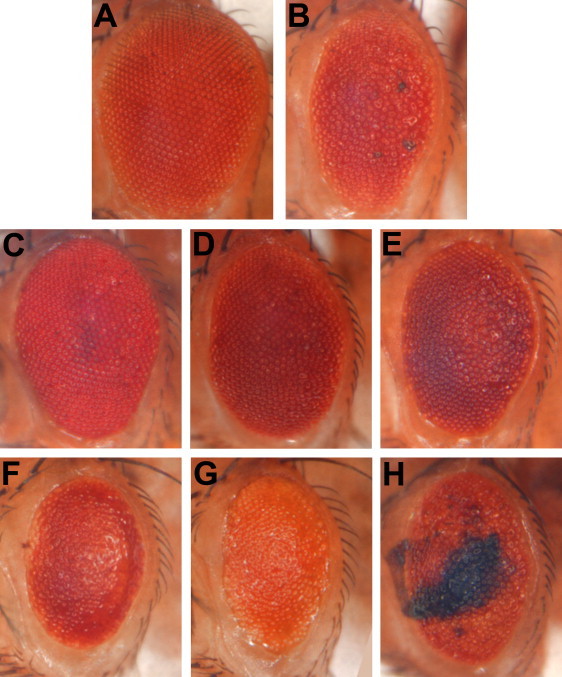
Functional Screening of GWAS Results, Based on Interactions of Gene Orthologs with Tau Toxicity In Vivo
Compared to control animals (A, GMR-Gal4/+), expression of human Tau generates a reduced eye size and moderate roughened appearance (B, UAS-TauV337M/+; GMR-Gal4/+).19 Lines predicted to increase the expression of glut1 (C, UAS-TauV337M/+; GMR-Gal4/+; Glut1d05758/+)33 and slit (D, UAS-TauV337M/+; GMR-Gal4/+;UAS-sli.B/+)47 or RNAi directed against fne (E, UAS-TauV337M/+; GMR-Gal4/UAS-fne.IR.v101508) suppressed Tau toxicity, restoring a near-wild-type eye. Reciprocally, RNAi directed against glut1 (F, UAS-TauV337M/+; GMR-Gal4,UAS-Dcr2/UAS-glut1.IR.v13326) and slit (G, UAS-TauV337M/+; GMR-Gal4,UAS-Dcr2/UAS-slit.IR.v38233) or increasing expression of fne (H, UAS-TauV337M/+; GMR-Gal4/+;UAS-fne.4-10B/+)48 enhanced Tau toxicity, exacerbating the rough-eye phenotype. Spatially and temporally defined expression of the yeast GAL4 transcription factor within the Drosophila retina, via the GMR-GAL4 driver line, directs Tau transgene expression from upstream activating sequence (UAS) sites. In the case of activating gain-of-function and RNAi lines for candidate genes, coexpression is also directed to the eye via the GAL4/UAS system.49 All photographed animals are female so as to facilitate comparisons, but consistent modifier effects were observed in both sexes. All crosses used a w1118 genetic background and were conducted at 25°C, with the exception of UAS-fne.4-10B, which was lethal in combination with UAS-TauV337M at this temperature and was therefore tested at 23°C. All genetic enhancer lines were also tested in the absence of Tau to confirm that there was no significant toxicity in isolation (Figure S3). Immunoblot analysis was performed to confirm that reagents identified as modifiers of the Tau eye phenotype did not alter Tau expression levels. All genetic modifier effects were scored with the use of a semiquantitative scale and were shown to be significantly different (p < 0.0001) from Tau controls (Figure S4).
Out of the 19 genes evaluated in the fly model, six genes show interactions with Tau toxicity in vivo (Table 1, Figure 1, and Figure S2), providing functional evidence that strengthens the validity of the GWAS results. In three notable cases, both loss- and gain-of-function experiments demonstrate reciprocal interactions. Specifically, SLC2A14 (MIM 611039) was selected for evaluation on the basis of an associated intronic SNP (rs10845990), and a single ortholog (glut1) is present in the Drosophila genome.34 A line predicted to increase glut1 expression was a potent Tau suppressor, restoring the eye to nearly wild-type appearance (Figure 1C), and a glut1 RNAi line had the opposite effect, enhancing Tau toxicity and leading to a worsened eye phenotype (Figure 1F). Similarly, SLIT3 was selected for testing on the basis of an intronic SNP, rs297808. Increasing expression of the orthologous fly gene, slit, rescues the Tau-induced eye phenotype (Figure 1D), whereas slit RNAi increases Tau toxicity (Figure 1G). In addition, we find evidence to support functional validation of ELAVL2 (MIM 601673), a gene found in the vicinity of rs17468071. Transgene-mediated expression of found in neurons (fne), an ortholog of ELAVL2, strongly increased Tau toxicity in the fly eye (Figure 1H), and at higher levels, fne caused pupal lethality when coexpressed with Tau. Reciprocally, an fne RNAi line attenuated Tau toxicity (Figure 1E). The Drosophila genome contains two other ELAVL2 orthologs, including the founding family member, elav, and Rbp9; however, manipulating the expression of these genes in the absence of Tau was associated with substantial toxicity, limiting further evaluation using our screening strategy. Finally, RNAi directed against three other fly genes, β-spectrin, heparan sulfate 6-O-sulfotransferase, and discs large 1, each enhance Tau toxicity, supporting functional validation of the orthologous loci implicated by our GWAS (Table 1 and Figure S2).
For the six loci highlighted by the Drosophila functional screen, we genotyped the index SNP in an additional 305 deceased study participants with completed neuropathological evaluation (Table S3). rs10845990, within the SLC2A14 locus, showed suggestive evidence of replication (p = 0.03), and the association was improved in a pooled analysis of 532 subjects, including both the discovery and the replication cohorts (pDISC = 6.9 × 10−5, pJOINT = 8.1 × 10−6). SLC2A14, encoding a glucose transporter (GLUT14), is an attractive biological candidate given the well-known dysregulation of glucose metabolism in the AD brain and likely pathogenic role of oxidative stress.6 Although predominantly expressed in the testes,35 less abundant SLC2A14 transcripts are also detected in the central nervous system, on the basis of publically available transcriptome data (see Web Resources).36–38 Glucose transporter expression has been reported to be reduced in brains affected by AD, correlated with both Tau phosphorylation and neurofibrillary tangle burden.39 Interestingly, genetic and pharmacological manipulation of oxidative stress has previously been shown to modulate Tau-induced toxicity in flies,40 potentially consistent with this mechanism of action for the observed interaction with glut1.
In summary, on the basis of genetic association in humans and functional screening in a pertinent model organism, we have identified six candidate loci that influence the accumulation of AD neuropathology. Our strategy of integrating human GWAS with a Drosophila genetic screen builds on similar successful cross-species studies in which fly models of neurodegenerative disease enabled secondary screens to reinforce findings from mammalian systems, including transcriptome analysis41 and drug discovery.42 The Drosophila Tau transgenic model selected for our functional screening pipeline has been used in prior successful genetic screens and numerous other investigations,20,24,25,43 and many results have been consistent with findings in mouse models and other AD experimental paradigms.28,44 In current hypotheses about the mechanisms of AD pathogenesis, supported by a large body of work, Tau-induced neurotoxicity defines a key pathway mediating the effects of Aß.6,21–23 Therefore, our functional screen may be relevant to many susceptibility loci that influence downstream mechanisms of Aß toxicity. Nevertheless, our approach would not be expected to detect genes that directly influence the processing of amyloid precursor protein (APP), Aß aggregation, or other proximal events in the pathologic cascade. In the future, such loci might be functionally screened with the use of either APP or Aß transgenic flies or Aß/Tau dual transgenic flies.17,24,45
Additional strengths of our approach include the substantial genomic conservation between flies and mammals46 and the availability of reagents to manipulate the function of nearly all Drosophila genes.31 The success rate of our strategy exceeds the returns of unbiased Drosophila genetic screens using the same transgenic model,20 suggesting that the list of 19 loci tested was enriched for genes influencing the development of AD pathology. Although a negative result in our screen does not exclude a gene as potentially associated with AD, the six validated loci highlight pathways of potential relevance to disease pathogenesis. Future functional investigation in Drosophila, and in other experimental systems, may reveal the mechanisms by which these genes modulate Tau-induced neurodegeneration, and these loci are also excellent targets for further replication analysis in human cohorts. Importantly, our functional screening strategy highlights genes that are likely responsible for association signals, and in two cases, rs393569 and rs10845990, we are able to nominate causal genes (SPTBN4 and SLC2A14, respectively) for which more than one candidate was initially found on the basis of linkage disequilibrium with the index SNP, a commonly encountered problem in following up GWAS results.
The association signals uncovered in our GWAS are comparable to that of numerous published reports in larger case-control cohorts that have identified candidate risk loci with suggestive but not definitive statistical evidence of association to AD or other relevant intermediate traits.1 Evidence is emerging in support of a polygenic model of inheritance for complex genetic disorders, particularly neuropsychiatric diseases, in which hundreds or even thousands of common variants collectively contribute to disease risk.3–5 Given the very small effect sizes, it is unrealistic that the majority of such loci can be validated individually by statistical evidence alone. Our strategy of coupling GWAS in humans to functional genetic screening in a model organism will therefore likely be a powerful strategy for follow-up of such signals in the future for the prioritization of genes and pathways for further investigation.
Acknowledgments
We are grateful to our colleagues, Lei Yu and Sue Leurgans, for assistance with statistical analyses. We thank Christian Klambt, Jimena Sierralta, and Hiroshi Nakato for generously providing Drosophila stocks. We are grateful Dr. Bradley Hyman and Chris Cotsapas for comments on the manuscript and valuable discussion. We also thank the Bloomington Drosophila stock center, the Vienna Drosophila RNAi Center (VDRC), and the Harvard Transgenic RNAi Project (TRiP, NIH/NIGMS R01GM084947) for providing fly stocks. J.M.S. is supported by NIH grant K08AG034290 and by the Clinical Investigator Training Program: Beth Israel Deaconess Medical Center – Harvard/MIT Health Sciences and Technology, in collaboration with Pfizer Inc. and Merck & Co. M.B.F. is supported by the Ellison Medical Foundation. The authors also thank the participants of the Religious Orders Study and the Rush Memory and Aging Project, which were supported by NIH grants P30AG10161, R01AG15819, and R01AG17917.
Contributor Information
Mel B. Feany, Email: mel_feany@hms.harvard.edu.
Philip L. De Jager, Email: pdejager@rics.bwh.harvard.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BioGPS, http://biogps.gnf.org
FlyBase, http://flybase.org/
Harvard Transgenic RNAi Project (TRiP), http://www.flyrnai.org/TRiP-HOME.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
Vienna Drosophila RNAi Center (VDRC), http://stockcenter.vdrc.at/control/main
References
- 1.Bertram L., Tanzi R.E. Genome-wide association studies in Alzheimer's disease. Hum. Mol. Genet. 2009;18:R137–R145. doi: 10.1093/hmg/ddp406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ioannidis J.P.A., Thomas G., Daly M.J. Validating, augmenting and refining genome-wide association signals. Nat. Rev. Genet. 2009;10:318–329. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International Schizophrenia Consortium. Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F., Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J., Benyamin B., McEvoy B.P., Gordon S., Henders A.K., Nyholt D.R., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International Multiple Sclerosis Genetics Consortium. Bush W.S., Sawcer S.J., de Jager P.L., Oksenberg J.R., McCauley J.L., Pericak-Vance M.A., Haines J.L. Evidence for polygenic susceptibility to multiple sclerosis–the shape of things to come. Am. J. Hum. Genet. 2010;86:621–625. doi: 10.1016/j.ajhg.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Querfurth H.W., LaFerla F.M. Alzheimer's disease. N. Engl. J. Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 7.Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M.L., Pahwa J.S., Moskvina V., Dowzell K., Williams A. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambert J.-C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M., Combarros O., Zelenika D., Bullido M.J., Tavernier B. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 9.Seshadri S., Fitzpatrick A.L., Ikram M.A., DeStefano A.L., Gudnason V., Boada M., Bis J.C., Smith A.V., Carassquillo M.M., Lambert J.C. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett D.A., De Jager P.L., Leurgans S.E., Schneider J.A. Neuropathologic intermediate phenotypes enhance association to Alzheimer susceptibility alleles. Neurology. 2009;72:1495–1503. doi: 10.1212/WNL.0b013e3181a2e87d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shulman J.M., Chibnik L.B., Aubin C., Schneider J., De Jager P., Bennett D. Intermediate phenotypes identify divergent pathways to Alzheimer's disease. PLoS ONE. 2010;5:e1124. doi: 10.1371/journal.pone.0011244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett D.A., Schneider J., Arvanitakis Z., Kelly J., Aggarwal N., Shah R., Wilson R. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 13.Bennett D.A., Wilson R., Schneider J., Evans D., Beckett L., Aggarwal N., Barnes L., Fox J., Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- 14.Bennett D.A., Schneider J.A., Buchman A.S., Mendes de Leon C., Bienias J.L., Wilson R.S. The Rush Memory and Aging Project: study design and baseline characteristics of the study cohort. Neuroepidemiology. 2005;25:163–175. doi: 10.1159/000087446. [DOI] [PubMed] [Google Scholar]
- 15.Bennett D.A., Schneider J.A., Tang Y., Arnold S.E., Wilson R.S. The effect of social networks on the relation between Alzheimer's disease pathology and level of cognitive function in old people: a longitudinal cohort study. Lancet Neurol. 2006;5:406–412. doi: 10.1016/S1474-4422(06)70417-3. [DOI] [PubMed] [Google Scholar]
- 16.Bennett D.A., Wilson R.S., Schneider J.A., Evans D.A., Aggarwal N.T., Arnold S.E., Cochran E.J., Berry-Kravis E., Bienias J.L. Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer's disease. Neurology. 2003;60:246–252. doi: 10.1212/01.wnl.0000042478.08543.f7. [DOI] [PubMed] [Google Scholar]
- 17.Finelli A., Kelkar A., Song H.J., Yang H., Konsolaki M. A model for studying Alzheimer's Abeta42-induced toxicity in Drosophila melanogaster. Mol. Cell. Neurosci. 2004;26:365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Moloney A., Sattelle D.B., Lomas D.A., Crowther D.C. Alzheimer's disease: insights from Drosophila melanogaster models. Trends Biochem. Sci. 2010;35:228–235. doi: 10.1016/j.tibs.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wittmann C.W., Wszolek M., Shulman J., Salvaterra P., Lewis J., Hutton M., Feany M. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 20.Shulman J.M., Feany M. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165:1233–1242. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ittner L.M., Ke Y.D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B.C., Christie M.J., Napier I.A. Dendritic Function of Tau Mediates Amyloid-beta Toxicity in Alzheimer's Disease Mouse Models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 22.Roberson E.D., Scearce-Levie K., Palop J., Yan F., Cheng I., Wu T., Gerstein H., Yu G., Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 23.Vossel K.A., Zhang K., Brodbeck J., Daub A.C., Sharma P., Finkbeiner S., Cui B., Mucke L. Tau Reduction Prevents Abeta-Induced Defects in Axonal Transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fulga T.A., Elson-Schwab I., Khurana V., Steinhilb M.L., Spires T.L., Hyman B.T., Feany M.B. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- 25.Khurana V., Lu Y., Steinhilb M., Oldham S., Shulman J., Feany M. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol. 2006;16:230–241. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 26.Gotz J., Chen F., van Dorpe J., Nitsch R. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 27.Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 28.Götz J., Ittner L.M. Animal models of Alzheimer's disease and frontotemporal dementia. Nat. Rev. Neurosci. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- 29.Dietzl G., Chen D., Schnorrer F., Su K.C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 30.Ni J.Q., Markstein M., Binari R., Pfeiffer B., Liu L.P., Villalta C., Booker M., Perkins L., Perrimon N. Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat. Methods. 2008;5:49–51. doi: 10.1038/nmeth1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matthews K.A., Kaufman T.C., Gelbart W.M. Research resources for Drosophila: the expanding universe. Nat. Rev. Genet. 2005;6:179–193. doi: 10.1038/nrg1554. [DOI] [PubMed] [Google Scholar]
- 32.Tweedie S., Ashburner M., Falls K., Leyland P., McQuilton P., Marygold S., Millburn G., Osumi-Sutherland D., Schroeder A., Seal R. FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 2009;37:D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bellen H.J., Levis R.W., Liao G., He Y., Carlson J.W., Tsang G., Evans-Holm M., Hiesinger P.R., Schulze K.L., Rubin G.M. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Escher S.A., Rasmuson-Lestander A. The Drosophila glucose transporter gene: cDNA sequence, phylogenetic comparisons, analysis of functional sites and secondary structures. Hereditas. 1999;130:95–103. doi: 10.1111/j.1601-5223.1999.00095.x. [DOI] [PubMed] [Google Scholar]
- 35.Wu X., Freeze H.H. GLUT14, a duplicon of GLUT3, is specifically expressed in testis as alternative splice forms. Genomics. 2002;80:553–557. doi: 10.1006/geno.2002.7010. [DOI] [PubMed] [Google Scholar]
- 36.Rhead B., Karolchik D., Kuhn R.M., Hinrichs A.S., Zweig A.S., Fujita P.A., Diekhans M., Smith K.E., Rosenbloom K.R., Raney B.J. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38:D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su A.I., Cooke M.P., Ching K.A., Hakak Y., Walker J.R., Wiltshire T., Orth A.P., Vega R.G., Sapinoso L.M., Moqrich A. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. USA. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C.L., Haase J., Janes J., Huss J.W. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y., Liu F., Iqbal K., Grundke-Iqbal I., Gong C.-X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008;582:359–364. doi: 10.1016/j.febslet.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dias-Santagata D., Fulga T.A., Duttaroy A., Feany M.B. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J. Clin. Invest. 2007;117:236–245. doi: 10.1172/JCI28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desai U.A., Pallos J., Ma A.A.K., Stockwell B.R., Thompson L.M., Marsh J.L., Diamond M.I. Biologically active molecules that reduce polyglutamine aggregation and toxicity. Hum. Mol. Genet. 2006;15:2114–2124. doi: 10.1093/hmg/ddl135. [DOI] [PubMed] [Google Scholar]
- 42.Karsten S.L., Sang T.-K., Gehman L.T., Chatterjee S., Liu J., Lawless G.M., Sengupta S., Berry R.W., Pomakian J., Oh H.S. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron. 2006;51:549–560. doi: 10.1016/j.neuron.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 43.Blard O., Feuillette S., Bou J., Chaumette B., Frebourg T., Campion D., Lecourtois M. Cytoskeleton proteins are modulators of mutant tau-induced neurodegeneration in Drosophila. Hum. Mol. Genet. 2007;16:555–566. doi: 10.1093/hmg/ddm011. [DOI] [PubMed] [Google Scholar]
- 44.Moloney A., Sattelle D.B., Lomas D.A., Crowther D.C. Alzheimer's disease: insights from Drosophila melanogaster models. Trends Biochem Sci. 2009;35:228–235. doi: 10.1016/j.tibs.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fossgreen A., Bruckner B., Czech C., Masters C., Beyreuther K., Paro R. Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc. Natl. Acad. Sci. USA. 1998;95:13703–13708. doi: 10.1073/pnas.95.23.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rubin G.M., Yandell M., Wortman J., Gabor Miklos G., Nelson C., Hariharan I., Fortini M., Li P., Apweiler R., Fleischmann W. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Battye R., Stevens A., Jacobs J.R. Axon repulsion from the midline of the Drosophila CNS requires slit function. Development. 1999;126:2475–2481. doi: 10.1242/dev.126.11.2475. [DOI] [PubMed] [Google Scholar]
- 48.Samson M.L., Chalvet F. found in neurons, a third member of the Drosophila elav gene family, encodes a neuronal protein and interacts with elav. Mech. Dev. 2003;120:373–383. doi: 10.1016/s0925-4773(02)00444-6. [DOI] [PubMed] [Google Scholar]
- 49.Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 50.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.