

Abstract
Retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal degenerations caused by mutations in at least 50 genes. Using homozygosity mapping in Ashkenazi Jewish (AJ) patients with autosomal-recessive RP (arRP), we identified a shared 1.7 Mb homozygous region on chromosome 1p36.11. Sequence analysis revealed a founder homozygous missense mutation, c.124A>G (p.Lys42Glu), in the dehydrodolichyl diphosphate synthase gene (DHDDS) in 20 AJ patients with RP of 15 unrelated families. The mutation was not identified in an additional set of 109 AJ patients with RP, in 20 AJ patients with other inherited retinal diseases, or in 70 patients with retinal degeneration of other ethnic origins. The mutation was found heterozygously in 1 out of 322 ethnically matched normal control individuals. RT-PCR analysis in 21 human tissues revealed ubiquitous expression of DHDDS. Immunohistochemical analysis of the human retina with anti-DHDDS antibodies revealed intense labeling of the cone and rod photoreceptor inner segments. Clinical manifestations of patients who are homozygous for the c.124A>G mutation were within the spectrum associated with arRP. Most patients had symptoms of night and peripheral vision loss, nondetectable electroretinographic responses, constriction of visual fields, and funduscopic hallmarks of retinal degeneration. DHDDS is a key enzyme in the pathway of dolichol, which plays an important role in N-glycosylation of many glycoproteins, including rhodopsin. Our results support a pivotal role of DHDDS in retinal function and may allow for new therapeutic interventions for RP.
Main Text
Retinitis pigmentosa (RP; MIM 268000) is the most common inherited retinal degeneration, with an estimated worldwide prevalence of 1:4000.1–3 The disease is highly heterogeneous and has several patterns of inheritance. At present, 35 genetic loci have been implicated in nonsyndromic autosomal-recessive RP (arRP), most of which account for a few percent of RP cases each.
Although many of the early identified arRP genes were excellent candidates for the disease when mutated, mainly because of the function of the encoded protein (e.g., PDE6A4 [MIM 180071] and PDE6B5 [MIM 180072]), a large proportion of the recently identified genes were not a priori considered as candidates and were identified through whole-genome linkage or homozygosity mapping followed by mutation screening of a large number of genes in the linked intervals (e.g., EYS6 [MIM 612424] and SPATA77 [MIM 609868]). Through those studies, a new class of genes encoding proteins with housekeeping-like function (e.g., IDH3B [MIM 604526] for arRP8 and splicing factors for autosomal-dominant RP9,10) have been identified and provided new insight into processes that result in retinal degeneration. The reason for the retina-specific phenotype caused by mutations in these genes is still unclear.
The Ashkenazi Jewish (AJ) population was established by Jews who originated in the Middle East and migrated to Europe, initially settling in Germany (the “Ashkenaz” region) at or before the 4th century. The AJ lived in closed communities in European countries and developed a unique culture and language (named Yiddish, which is based on a few different languages, including German, Hebrew, and Aramaic). After the Holocaust, the population size dropped from about 8.8 million individuals to only 2.8 million, and AJ immigrated out of Europe, mainly to the United States and the emerging state of Israel. AJ currently constitute the largest Jewish ethnic group in both countries. A large amount of effort was directed to study the genetic structure of the AJ population, in the context of other Jewish ethnic groups and Middle Eastern populations, at the Y chromosome,11,12 mitochondrial,13 and genomic14,15 levels. Although consanguineous marriages are relatively uncommon among AJ (1.5% and rapidly declining),16,17 most individuals who are affected by a rare AR disease in this ethnic group are homozygous for the disease-causing mutation, mainly because of a high rate of intracommunity marriages.17 Therefore, genetic analysis of hereditary diseases in the AJ population, via homozygosity mapping, can be highly efficient.
In the present study, we used homozygosity mapping to identify the cause of disease in AJ families with arRP. The tenets of the Declaration of Helsinki were followed, and prior to donation of a blood sample, informed consent was obtained from all patients who participated in this study. DNA was extracted from the index patient, as well as from other affected and unaffected family members, with the FlexiGene DNA kit (QIAGEN). Whole-genome SNP analysis was initially performed on 11 patients with isolate or arRP who belong to eight different AJ families with either the Affymetrix 250K or 6.0 microarrays, and data analysis was performed with HomozygosityMapper. Patients from three of the families (MOL0400, MOL0565, and MOL0884; Table 1) had a shared homozygous region on chromosome 1p36.11 encompassing ∼2.32 Mb (Figure 1A). The shared homozygous region contains 56 protein coding genes, none of which is expressed exclusively in the retina. While we were analyzing candidate genes for mutations in the shared homozygous region, Zuchner and collaborators reported the identification of a missense mutation in an AJ family with arRP in a gene encoding a key enzyme in the terpenoid backbone synthesis (S.Z. et al., abstract presented at The American Society of Human Genetics annual meeting 2010). One of the genes in the linked region on 1p36.11 was the dehydrodolichyl diphosphate synthase gene (DHDDS; MIM 608172), encoding the dehydrodolichyl diphosphate synthase (Figure 1B), which is predicted to take part in the above-mentioned biochemical pathway.18 Using the Primer3 software,19 we designed primers flanking all coding exons and exon-intron boundaries of DHDDS (accession number NM_024887.2) and performed sequence analysis in the three index patients. The analysis revealed a homozygous transition, c.124A>G (Figure 1C), expected to result in an amino acid (aa) substitution, p.Lys42Glu, in all three index cases. Screening the mutation in a set of 322 ethnically matched normal controls revealed one heterozygous individual, thus indicating a carrier frequency of 0.3% (95% confidence interval 0.07%–1.7%) in the AJ population. We subsequently screened this mutation in a set of 121 AJ patients with RP, 20 AJ patients with other inherited retinal diseases, and 70 non-AJ patients with retinal degeneration of other ethnic origins. The analysis revealed 12 additional index cases with RP that were homozygous for the c.124A>G mutation, all of AJ descent. In addition, we analyzed whole-genome SNP microarray data of patients from 124 consanguineous families with RP or Leber congenital amaurosis and identified 20 index cases homozygous to the DHDDS region. DHDDS sequencing analysis in these patients, as well as in an additional set of 20 index RP patients from nonconsanguineous families, did not reveal any potential pathogenic mutation. Statistical analyses with Fisher's exact test (based on allele frequency: the mutation was found in 1 out of 644 chromosomes in the control group versus 30 out of 246 chromosomes in patients) and chi-square (based on genotype frequency: the number of homozygous, heterozygous, and wild-type individuals was 0, 1, and 321, respectively, in controls versus 15, 0, and 108 in patients) showed a significant difference between patient and control groups (p < 0.001 for each comparison).
Table 1.
Ashkenazi Jewish RP Families with the Homozygous p.Lys42Glu Mutation in DHDDS
| Family | Affected Siblings | Level of Consanguinitya | Size of Homozygous Region (Mb) |
|---|---|---|---|
| MOL0397 | 1 | None | NP |
| MOL0400 | 1 | 2:2 | 15.1 |
| MOL0565 | 2 | None | 3.8 |
| MOL0718 | 1 | None | 3.0 |
| MOL0735 | 1 | None | 3.0 |
| MOL0779 | 2 | None | 3.0 |
| MOL0884 | 3 | None | 3.7 |
| TB31/R54 | 1 | None | NP |
| TB61/R226 | 1 | None | 3.4 |
| CHRD0262 | 2 | None | NP |
| CHRD0677 | 1 | None | 3.0 |
| CHRD3323 | 1 | None | 3.2 |
| CHRD3458 | 1 | None | 4.9 |
| CHRD4047 | 2 | None | 3.8 |
| CHRD5151 | 1 | None | 4.0 |
NP denotes not performed.
Level of consanguinity is measured by the number of generations separating the spouse from the common ancestor (e.g., 2:2 denotes first cousins).
Figure 1.

Homozygosity Mapping, Gene Structure, and the Mutation Identified in DHDDS
(A) The chromosomal region harboring the homozygous haplotype at chromosome 1 (top). Rulers are based on the February 2009 USCS genome build (hg19). The set of horizontal bars represents the homozygous region in the three different families, as identified by whole-genome SNP analysis. The family number is shown on the left. The region flanked by SNP markers rs3924468 and rs11247703, covering 2.32 Mb between 25.8 and 28.1 Mb on chromosome 1, was considered the shared homozygous region.
(B) Intron-exon structure of DHDDS and known or predicted protein domains. The structure of DHDDS is depicted (top) with exons marked as black rectangles (wider for the coding region). The location of the c.124A>G (p.Lys42Glu) missense mutation is marked below exon 3. Known or putative domains of the DHDDS enzyme are depicted (bottom), with an asterisk pointing to the mutation position.
(C) Sequence chromatograms of part of DHDDS exon 3 of a homozygous mutant and a wild-type individual. The c.124A>G mutation (boxed) changes an AAG codon to GAG (p.Lys42Glu).
(D) An amino acid sequence alignment of DHDDS including position 42 (boxed). The aa types are color-coded: small aa in red, acidic in blue, basic in magenta, and hydroxyl + amine + basic in green.
To better characterize the shared homozygous region, we performed whole-genome SNP analysis (with Affymetrix 6.0 arrays) on nine additional homozygous index cases (see Table 1 and Table S1 available online; whole-genome SNP array genotyping data are available by request). This allowed us to analyze the homozygous region in a total of 15 homozygous patients and 1 unaffected sibling representing 12 different AJ families. The analysis revealed an identical shared region of 1.68 Mb (26.4–28.08) on chromosome 1, harboring 41 protein-coding genes, including DHDDS. The shared haplotype contains 244 homozygous SNPs representing a founder RP-associated haplotype in the AJ population. No other shared homozygous regions (over 1 Mb) could be detected in this set of patients, nor in the original three families used for homozygosity mapping.
The c.124A>G mutation affects a conserved amino acid residue (Lys42; Figure 1D) located in close proximity to a binding site of farnesyl diphosphate. The mutation substitutes the wild-type basic aa lysine with an acidic one, glutamic acid. The predicted effect of this substitution, with different tools designed to predict functional effects of missense changes, is not conclusive. Some tools did not predict a functional effect (e.g., PMUT: a neutral score of 0.4429; SAP: a neutral score of 0.53), whereas other tools did predict a functional effect (e.g., PolyPhen2: possibly damaging with a score of 0.206; SIFT: an intolerant score of 0.03; MutationTaster: a disease-causing prediction with a probability of 0.998).
In summary, the p.Lys42Glu DHDDS mutation was identified in 15 (out of 123, 12%) AJ RP index patients in our cohort, and it cosegregated with the disease when additional family members were available for the study (Figure 2).
Figure 2.
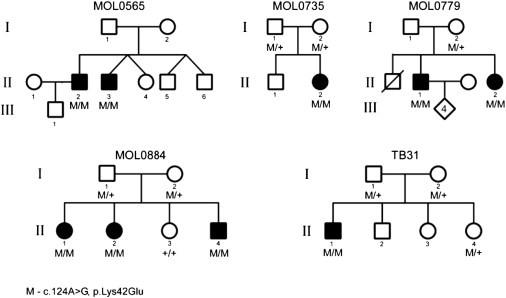
Cosegregation of Mutation and Phenotype in Identified Families
The family number is indicated above the corresponding family tree. Filled symbols represent affected individuals, whereas clear symbols represent unaffected individuals. Generation numbers are depicted on the left. The DHDDS genotype is shown below recruited individual symbols: M/M denotes homozygous for the mutation; M/+ denotes heterozygous for the mutation; +/+ denotes homozygous for the wild-type allele.
Aiming to clinically characterize patients with the p.Lys42Glu DHDDS mutation, we performed full ophthalmologic examinations, electroretinography (ERG), kinetic visual fields, color vision testing, optical coherence tomography (OCT), and autofluoresence (AF) imaging, as previously described.20–22 Clinical evaluation of 18 patients showed a spectrum of findings (Table S2). The patients had visual acuity that ranged from light perception to 20/20 (LogMAR 0.0; Table S2). Funduscopic findings at various disease stages included waxy appearance of the optic nerve head, attenuation of retinal blood vessels, and bone spicule-like pigmentation (Figure S1). OCT imaging in early disease showed preserved central retinal photoreceptors but a decline in photoreceptor layer thickness with distance from the fovea (Figure 3E), and occasionally the presence of cystoid macular edema (Figure S2). Kinetic visual fields revealed reduced peripheral function in the youngest patients studied and only small central islands of vision remaining later in life (Figures 3A and 3B and Figure S3). ERG responses were nondetectable in most patients (Table S2).
Figure 3.
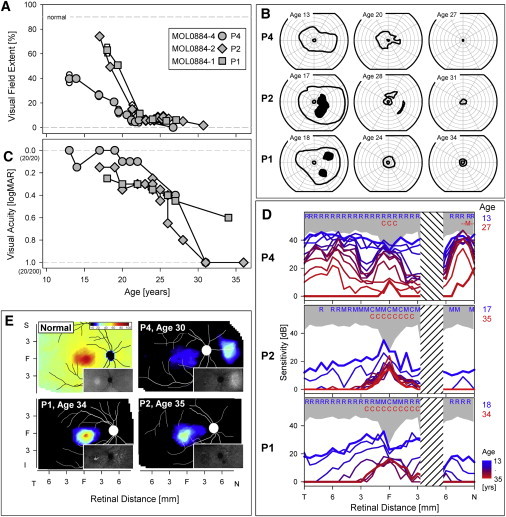
Ocular Phenotype of Three Siblings Who Are Homozygous for the DHDDS c.124A>G Mutation
(A) Visual field extent (kinetic perimetry with V-4e test target) expressed as a percentage of normal mean and plotted as a function of age in the three siblings of family MOL0884.
(B) Visual fields (V-4e, I-4e test targets) at different ages showing progressive peripheral vision loss.
(C) Visual acuity (expressed in logMAR units) as a function of age in the patients.
(D) Dark-adapted static perimetric profiles across the horizontal meridian with a 500 nm stimulus in the three siblings. Data span a minimum of 1.5 decades in each patient. Based on two-color (500 nm and 650 nm) dark-adapted perimetry, photoreceptor mediation was determined for each test locus at the earliest (blue letters) and latest (red letters) ages studied. The following abbreviations are used: R, rod-mediated; M, mixed rod- and cone-mediated; C, cone-mediated; N, nasal; T, temporal visual field. Gray represents normal limits (mean ± 2 standard deviation). Hashed band is the location of the physiological blind spot.
(E) Photoreceptor layer thickness topography mapped to a pseudocolor scale (shown above normal map) in a normal subject and in the three affected siblings. Insets: near-infrared excited autofluorescence imaging of melanin fluorophore distribution to estimate retinal pigment epithelium (RPE) layer health.
The natural history of the disease in family MOL0884 can be gleaned from serial ocular data acquired from the second to the fourth decades of life in the three affected siblings (Figure 3). Symptoms occurred during the latter part of the second decade of life in all three siblings, and these related to decreasing night and side vision; loss of reading (central) vision was not an early complaint. Visual field extent was quantified via kinetic perimetry to a large target (Figures 3A and 3B); fields became progressively reduced with only a small central island remaining by ages 21–24. Best-corrected visual acuity had a more prolonged time course of loss but was 20/200 or worse in two siblings by age 30–31 (Figure 3C). Rod-mediated vision was measurable via dark-adapted chromatic sensitivities in all three patients at younger ages, but this progressively diminished until only cone-mediated function was detectable (Figure 3D). Photoreceptor topography in a wide expanse of central retina (Figure 3E) revealed that by the fourth decade of life, there was only a markedly thinned photoreceptor layer remaining in and around the fovea; in two of three siblings, photoreceptors were not detectable surrounding the foveal region. Patient MOL0884-4 (II:4 in Figure 2) also had a locus of detectable photoreceptors nasal to the optic nerve head (Figure 3E, P4), and this correlated with an area of limited rod sensitivity by dark-adapted psychophysical testing (Figure 3D). Near-infrared autofluoresence imaging showed islands of preserved retinal pigment epithelium corresponding in retinal location to the regions of preserved photoreceptors (Figure 3E, insets). Two of the patients were imaged with wide-angle view, and bone spicule-like pigment was visible (Figure S2). In addition, both parents and an asymptomatic sibling in family MOL0884 were examined clinically, with kinetic perimetry and standard ERGs. No abnormalities were detected, with the exception of a borderline rod ERG amplitude in the father's recording.
DHDDS contains eight coding exons along a genomic region of ∼37 kb18 and is known to produce a few splice variants.23 No data, however, are available on its expression or function in the retina. To verify the retinal expression of DHDDS, we isolated RNA from the human retina with TRI-reagent (Sigma-Aldrich) and synthesized cDNA with the Verso cDNA kit (Thermo) in accordance with the manufacturer's protocol. PCR-specific primers (Table S3) were designed with Primer3, and RT-PCR analysis was performed with three primer sets that were designed to amplify different parts of the cDNA molecules (Figure 4A). The analysis confirmed the expression of the full-length transcript in the retina, as well as three alternatively spliced variants, two of which are in-frame and are likely to encode a protein (Figure 4A): a NAGNAG sequence24 in the acceptor splice site of intron 8 and a transcript in which exon 6 is skipped. The third transcript had a frameshift addition of 23 nucleotides because of a cryptic acceptor splice site in intron 3, resulting in a premature stop codon (Figure 4A) within exon 4, and is therefore likely to be degraded by the nonsense-mediated mRNA decay or, alternatively, to produce a nonfunctional protein. Expressed sequence tags (ESTs) representing these variants were previously deposited in GenBank. To better characterize the distribution of DHDDS in human tissues, we performed RT-PCR analysis on cDNAs that were synthesized from RNA derived from 20 different human tissues (Clontech; category 636643, lot 8101369A), as well as the human retina (Figure 4B). Human PGM1 (MIM 171900) was used as a control. Ubiquitous expression of DHDDS was observed, with a band of higher intensity in the retinal sample as compared to other tissues (Figure 4B, right lane). The ubiquitous expression supports data obtained with RNA blot in a set of tissues that did not include ocular ones.18
Figure 4.

DHDDS Transcripts and RT-PCR Analysis in Human Tissues
(A) DHDDS can produce different transcripts through alternative splicing. The scheme represents the full-length open reading frame (ORF) of DHDDS (top), as well as alternatively spliced transcripts identified in this study: skipping of exon 6 (middle) and recognition of a cryptic acceptor site within intron 3 (bottom, blue box), leading to the addition of 23 nucleotides. Arrows represent the location and direction of primers used for RT-PCR (see also Table S3). The NAGNAG sequence in exon 9 represents an alternative usage of either the first or second NAG sequence as an acceptor splice site of intron 8. Ex denotes exon, Int denotes intron, PTC denotes premature termination codon. It should be noted that a transcript in which exon 5 is skipped has been deposited in GenBank, but we were not able to verify it in any of the analyzed tissues.
(B) The expression level of DHDDS in 21 tissues is depicted. Two sets of primers were used to amplify DHDDS: set 1 amplified exons 4 trough 6 (F4-R6), and set 2 primers amplified exons 5 through 7 (F5-R7). PGM1 amplification served as a control for cDNA amount used in the PCR reaction (set 3). The lower faint band in set 2 could be observed in all tissues, and sequence analysis revealed skipping of exon 6.
To study the localization of the DHDDS enzyme in the human retina, we performed immunohistological studies with two commercially available anti-DHDDS antibodies (HPA026721 and HPA026727 [rabbit polyclonal, Sigma Life Science] at a final concentration of 1:15 and 1:175, respectively; secondary antibody Cy™2-conjugated donkey anti-rabbit IgG [1:200, Jackson ImmunoResearch Laboratories]). Prominent staining of the inner segments of photoreceptors was identified, which differed between rod and cone photoreceptors (Figure 5B, compared to the hematoxylin and eosin staining in Figure 5A and the negative control section in Figure 5C). Although the cone inner segments demonstrated a uniform signal, only the ellipsoid (distal region) and myoid (proximal region) of the rod photoreceptor inner segments were labeled, whereas the central part had much lower fluorescence (Figures 5D and 5E). The labeling of the cone inner segments is in accordance with a uniform distribution of cone inner segment organelles. In the rod inner segments, the myoid region contains the cellular protein synthesis machinery (including the Golgi apparatus and the endoplasmatic reticulum) in which N-glycosylation takes part, and the ellipsoid region contains mainly the mitochondria, which was shown to contain dolichol and dolichyl phosphate.25,26 The subcellular localization of DHDDS in rod photoreceptors is therefore in accordance with the expected distribution of the enzyme. DHDDS-positive staining was also evident in the basal aspect of retinal pigment epithelium cells (Figure 5E), and a weak staining was evident in other retinal layers (Figure 5B). The labeling pattern of DHDDS in the human retina is compatible with its function in dolichol metabolism (expected to occur in the photoreceptor inner segments), as well as the phenotype obtained (photoreceptor degeneration) as a result of the mutation identified in DHDDS.
Figure 5.

DHDDS Expression in the Human Retina
(A) Hematoxylin-Eosin stained section of a normal human retina, with identification of the different retinal layers. The following abbreviations are used: GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer, containing the photoreceptor nuclei; IS, inner segments of photoreceptors; OS, outer segments of photoreceptors; RPE, retinal pigment epithelium layer.
(B and C) Low-magnification images of anti-DHDDS antibody-labeled (B, in green fluorescence) and control (C, no primary antibody applied) retinal sections. Note prominent labeling of the rod and cone inner segments. Yellow color of the RPE layer is due to autofluorescence of these cells (original magnification 20×). Scale bar represents 100 μm.
(D and E) Higher-magnification images of the photoreceptor-RPE region. Note segmental staining of the ellipsoid (red arrow) and myoid (white arrow) of the rod photoreceptor inner segments (original magnification 40× with digital zoom). Scale bar represents 25 μm. A representative cone inner segment is marked by a yellow star.
Dehydrodolichyl diphosphate (Dedol-PP) synthase is a highly conserved enzyme that can be found in all animal species, as well as in plants and bacteria. It catalyzes cis-prenyl chain elongation to produce the polyprenyl backbone of dolichol (Figure 6A). The human Dedol-PP enzyme was reported to be able to complement for the lack of the orthologous enzyme in a yeast mutant strain.18 In mammals, dolichol is present in most tissues, with relatively high levels in human tissues compared to other animals,27,28 and the levels of dolichol compounds are reported to increase substantially with age.29 Dolichyl pyrophosphate plays an important role as a glycosyl carrier in the biosynthesis of the N-linked oligosaccharide chains of freshly synthesized glycoproteins (Figure 6B).
Figure 6.
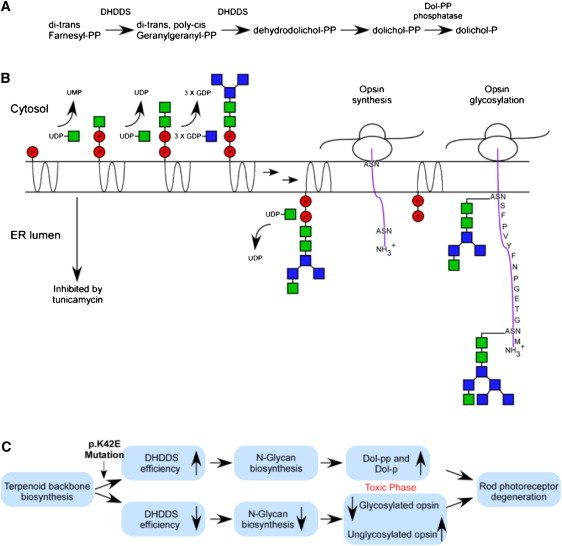
The Function of the DHDDS Enzyme and Possible Mechanisms Underlying DHDDS-Related Retinal Degeneration
(A) DHDDS is a key enzyme in dolichol biosynthesis, which catalyzes cis-prenyl chain elongation to produce the polyprenyl backbone of dolichol. It is required for two enzymatic steps (arrows) along the dolichol pathway.
(B) Schematic representation of the role of dolichol in N-glycosylation. Dolichol is a membrane-bound glycosyl carrier and plays an important role in the biosynthesis of the N-linked oligosaccharide chains of glycoproteins. The two arrows represent multiple steps that are not shown here. N-glycosylation of the opsin molecule is schematically represented on the right. The ribosome (oval shape) synthesizes fresh opsin molecules with two sites of N-glycosylation (ASN) at the N terminus (positions 2 and 15). Dol-PP transfers the oligosaccharide chain to these sites to glycosylate the opsin molecule (based on 49).
(C) Scheme of putative mechanisms by which the DHDDS mutation could lead to retinal degeneration. In the top panel, the possibility that the mutation causes a hyperactivity in the enzymatic function of DHDDS leading to excessive accumulation of Dol-P and/or Dol-PP, because of “overflow” of the N-glycosylation pathway, is shown. These substances might be toxic to the photoreceptor cell, causing photoreceptor degeneration. Alternatively, the mutation can by hypormorph, leading to reduced enzymatic function of DHDDS. This might in turn reduce N-glycosylation levels, producing a certain amount of nonglycosylated, and therefore nonfunctional, opsin molecules, leading to photoreceptor degeneration. Either of the proposed two mechanisms might mimic the retinal degeneration phenotype cause by the inhibitor of N-glycosylation followed by exposure to tunicamycin (panel B).
Direct proof for the activity of the dolichol pathway in the retina has been obtained by classical biochemical methods (reviewed by 30), as well as by measuring the absolute rate of dolichyl phosphate (Dol-P) synthesis in the retina.31 Protein N-glycosylation is a fundamental biological process (reviewed by 32–34) and is a requisite for maintaining normal structure and function of the retina (reviewed by 35,36). The most important and abundant rod photoreceptor protein, rhodopsin, is composed of a single polypeptide chain to which two unusually short N-linked oligosaccharide chains are covalently coupled37–39 at positions N-2 and N-15 (Figure 6B). Experiments in the 1980s with tunicamycin (a specific inhibitor of the N-glycosylation pathway; Figure 6B)32 demonstrated aberrant photoreceptor outer segment membrane morphogenesis in vitro in isolated Xenopus retinas maintained in short-term organ culture,40 as well as retinal degeneration in living frogs.41 In the former study, intraocular injection of tunicamycin produced a progressive RP-like phenotype in which shortening of rod outer segments occurred, accompanied by decreased ERG amplitudes and disruption of the rod outer segment membrane assembly.41 It should be noted that the ocular phenotype in these animals was restricted to the photoreceptors, although the concentration of tunicamycin was expected to be the lowest in this layer.41 The photoreceptor-specific phenotype might be explained by the observation that the highest activity of the lipid-intermediate pathway of glycoprotein synthesis in the retina is in the photoreceptor layer.42 Within the eye, the phenotype obtained in these animals is therefore in concordance with the RP phenotype in patients with the DHDDS mutation.
It is unclear, however, why the phenotype in the human patients is restricted to the retina and why no dysfunction is evident in other organs. Disruption of the N-glycosylation pathway in humans, as a result of loss-of-function mutations in several genes, is known to cause a set of syndromes known as congenital disorders of N-glycosylation.43 It is logical to assume that the mutation we identified in DHDDS alters, rather than abolishes, enzymatic function, perhaps either by reducing the level of DHDDS protein or by preventing requisite interactions between DHDDS and a photoreceptor-specific protein. Reduced protein levels might have a mild effect on other organs but might severely affect rhodopsin function and/or intracellular trafficking, leading to progressive photoreceptor degeneration over time (Figure 6C). Alternatively, the photoreceptor degeneration might not be related to alteration in opsin N-glycosylation, but rather to a toxic accumulation of isoprenoid compounds (Figure 6C). Dolichol accumulation has been reported in neuronal ceroid lipofuscinosis,44 in which retinal degeneration occurs, and alteration of dolichol/Dol-P levels has been reported in patients with Alzheimer disease.45–48
Our finding of a prevalent arRP-associated mutation in a specific ethnic group has practical implications for genetic testing in AJ RP patients. In this genetically heterogeneous condition in which each gene accounts for a small fraction of cases, the relatively high frequency of this DHDDS mutation (over 10% of AJ RP patients) points to a useful modification of the testing strategy for RP. In simplex and multiplex RP patients with AJ ancestry, the p.Lys42Glu DHDDS mutation should be one of the screening targets. Moreover, the identification of a DHDDS mutation may become the first step toward recommendation of specific therapeutic options for this currently incurable progressive disease leading to blindness.
Acknowledgments
This study was financially supported by the Foundation Fighting Blindness USA (BR-GE-0510-0490-HUJ to D.S.; C-CL-07-0411,0412 to A.V.C. and S.G.J.), by the Legacy Heritage Biomedical Program of the Israeli Science Foundation (grant 612/09 to T.B.-Y. and D.S.), Hope for Vision (to A.V.C. and S.G.J.), and the Yedidut 1 research grant (to E.B.). We thank the patients and their families for their participation in the study. The authors thank Michal Ben-Hur, Rob W. Collin, Sandro Banfi, Robert K. Koenekoop, Israel Barzel, Alejandro Roman, Malgorzata Swider, Alexander Sumaroka, Yelena Piontek, and Michael Kogan for excellent and generous assistance. The authors would also like to thank Steven J. Fliesler for helpful discussions.
Contributor Information
Samuel G. Jacobson, Email: jacobsos@mail.med.upenn.edu.
Dror Sharon, Email: dror.sharon1@gmail.com.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ClustalW2 multiple alignment tool at The European Bioinformatics Institute, http://www.ebi.ac.uk/Tools/clustalw2/index.html
HomozygosityMapper, http://www.homozygositymapper.org/
MutationTaster, http://www.mutationtaster.org
NCBI Basic Local Alignment Search Tool (BLAST), http://blast.ncbi.nlm.nih.gov/
NCBI UniGene, http://www.ncbi.nlm.nih.gov/unigene/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/index.shtml
Primer3, http://frodo.wi.mit.edu/primer3/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Rosenberg T. Epidemiology of hereditary ocular disorders. Dev. Ophthalmol. 2003;37:16–33. doi: 10.1159/000072036. [DOI] [PubMed] [Google Scholar]
- 2.Bundey S., Crews S.J. A study of retinitis pigmentosa in the City of Birmingham. II Clinical and genetic heterogeneity. J. Med. Genet. 1984;21:421–428. doi: 10.1136/jmg.21.6.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bunker C.H., Berson E.L., Bromley W.C., Hayes R.P., Roderick T.H. Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol. 1984;97:357–365. doi: 10.1016/0002-9394(84)90636-6. [DOI] [PubMed] [Google Scholar]
- 4.Huang S.H., Pittler S.J., Huang X., Oliveira L., Berson E.L., Dryja T.P. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat. Genet. 1995;11:468–471. doi: 10.1038/ng1295-468. [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin M.E., Sandberg M.A., Berson E.L., Dryja T.P. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat. Genet. 1993;4:130–134. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- 6.Abd El-Aziz M.M., Barragan I., O'Driscoll C.A., Goodstadt L., Prigmore E., Borrego S., Mena M., Pieras J.I., El-Ashry M.F., Safieh L.A. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat. Genet. 2008;40:1285–1287. doi: 10.1038/ng.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H., den Hollander A.I., Moayedi Y., Abulimiti A., Li Y., Collin R.W., Hoyng C.B., Lopez I., Abboud E.B., Al-Rajhi A.A. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am. J. Hum. Genet. 2009;84:380–387. doi: 10.1016/j.ajhg.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartong D.T., Dange M., McGee T.L., Berson E.L., Dryja T.P., Colman R.F. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat. Genet. 2008;40:1230–1234. doi: 10.1038/ng.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKie A.B., McHale J.C., Keen T.J., Tarttelin E.E., Goliath R., van Lith-Verhoeven J.J., Greenberg J., Ramesar R.S., Hoyng C.B., Cremers F.P. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum. Mol. Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- 10.Chakarova C.F., Hims M.M., Bolz H., Abu-Safieh L., Patel R.J., Papaioannou M.G., Inglehearn C.F., Keen T.J., Willis C., Moore A.T. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- 11.Nebel A., Filon D., Brinkmann B., Majumder P.P., Faerman M., Oppenheim A. The Y chromosome pool of Jews as part of the genetic landscape of the Middle East. Am. J. Hum. Genet. 2001;69:1095–1112. doi: 10.1086/324070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behar D.M., Thomas M.G., Skorecki K., Hammer M.F., Bulygina E., Rosengarten D., Jones A.L., Held K., Moses V., Goldstein D. Multiple origins of Ashkenazi Levites: Y chromosome evidence for both Near Eastern and European ancestries. Am. J. Hum. Genet. 2003;73:768–779. doi: 10.1086/378506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Behar D.M., Metspalu E., Kivisild T., Achilli A., Hadid Y., Tzur S., Pereira L., Amorim A., Quintana-Murci L., Majamaa K. The matrilineal ancestry of Ashkenazi Jewry: Portrait of a recent founder event. Am. J. Hum. Genet. 2006;78:487–497. doi: 10.1086/500307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bray S.M., Mulle J.G., Dodd A.F., Pulver A.E., Wooding S., Warren S.T. Signatures of founder effects, admixture, and selection in the Ashkenazi Jewish population. Proc. Natl. Acad. Sci. USA. 2010;107:16222–16227. doi: 10.1073/pnas.1004381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atzmon G., Hao L., Pe'er I., Velez C., Pearlman A., Palamara P.F., Morrow B., Friedman E., Oddoux C., Burns E., Ostrer H. Abraham's children in the genome era: Major Jewish diaspora populations comprise distinct genetic clusters with shared Middle Eastern Ancestry. Am. J. Hum. Genet. 2010;86:850–859. doi: 10.1016/j.ajhg.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zlotogora J., Bach G., Munnich A. Molecular basis of mendelian disorders among Jews. Mol. Genet. Metab. 2000;69:169–180. doi: 10.1006/mgme.2000.2969. [DOI] [PubMed] [Google Scholar]
- 17.Cohen T., Vardi-Saliternik R., Friedlander Y. Consanguinity, intracommunity and intercommunity marriages in a population sample of Israeli Jews. Ann. Hum. Biol. 2004;31:38–48. doi: 10.1080/0301446032000159255. [DOI] [PubMed] [Google Scholar]
- 18.Endo S., Zhang Y.W., Takahashi S., Koyama T. Identification of human dehydrodolichyl diphosphate synthase gene. Biochim. Biophys. Acta. 2003;1625:291–295. doi: 10.1016/s0167-4781(02)00628-0. [DOI] [PubMed] [Google Scholar]
- 19.Rozen S., Skaletsky H.J. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S., Misener S., editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press; Totowa, NJ: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 20.Beit-Ya'acov A., Mizrahi-Meissonnier L., Obolensky A., Landau C., Blumenfeld A., Rosenmann A., Banin E., Sharon D. Homozygosity for a novel ABCA4 founder splicing mutation is associated with progressive and severe Stargardt-like disease. Invest. Ophthalmol. Vis. Sci. 2007;48:4308–4314. doi: 10.1167/iovs.07-0244. [DOI] [PubMed] [Google Scholar]
- 21.Jacobson S.G., Cideciyan A.V., Aleman T.S., Sumaroka A., Roman A.J., Gardner L.M., Prosser H.M., Mishra M., Bech-Hansen N.T., Herrera W. Usher syndromes due to MYO7A, PCDH15, USH2A or GPR98 mutations share retinal disease mechanism. Hum. Mol. Genet. 2008;17:2405–2415. doi: 10.1093/hmg/ddn140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cideciyan A.V., Aleman T.S., Jacobson S.G., Khanna H., Sumaroka A., Aguirre G.K., Schwartz S.B., Windsor E.A., He S., Chang B. Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: Implications for therapy of Leber congenital amaurosis. Hum. Mutat. 2007;28:1074–1083. doi: 10.1002/humu.20565. [DOI] [PubMed] [Google Scholar]
- 23.Rebl A., Anders E., Wimmers K., Goldammer T. Characterization of dehydrodolichyl diphosphate synthase gene in rainbow trout (Oncorhynchus mykiss) Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2009;152:260–265. doi: 10.1016/j.cbpb.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Hiller M., Huse K., Szafranski K., Jahn N., Hampe J., Schreiber S., Backofen R., Platzer M. Widespread occurrence of alternative splicing at NAGNAG acceptors contributes to proteome plasticity. Nat. Genet. 2004;36:1255–1257. doi: 10.1038/ng1469. [DOI] [PubMed] [Google Scholar]
- 25.Ardail D., Lermé F., Louisot P. Dolichol kinase activity: A key factor in the control of N-glycosylation in inner mitochondrial membranes. Biochim. Biophys. Acta. 1990;1024:131–138. doi: 10.1016/0005-2736(90)90216-b. [DOI] [PubMed] [Google Scholar]
- 26.Eggens I., Chojnacki T., Kenne L., Dallner G. Separation, quantitation and distribution of dolichol and dolichyl phosphate in rat and human tissues. Biochim. Biophys. Acta. 1983;751:355–368. doi: 10.1016/0005-2760(83)90294-1. [DOI] [PubMed] [Google Scholar]
- 27.Rip J.W., Rupar C.A., Ravi K., Carroll K.K. Distribution, metabolism and function of dolichol and polyprenols. Prog. Lipid Res. 1985;24:269–309. doi: 10.1016/0163-7827(85)90008-6. [DOI] [PubMed] [Google Scholar]
- 28.Carroll K.K., Guthrie N., Ravi K. Dolichol: Function, metabolism, and accumulation in human tissues. Biochem. Cell Biol. 1992;70:382–384. doi: 10.1139/o92-059. [DOI] [PubMed] [Google Scholar]
- 29.Pullarkat R.K., Reha H. Accumulation of dolichols in brains of elderly. J. Biol. Chem. 1982;257:5991–5993. [PubMed] [Google Scholar]
- 30.Kean E.L. The dolichol pathway in the retina and its involvement in the glycosylation of rhodopsin. Biochim. Biophys. Acta. 1999;1473:272–285. doi: 10.1016/s0304-4165(99)00198-1. [DOI] [PubMed] [Google Scholar]
- 31.Keller R.K., Fliesler S.J., Nellis S.W. Isoprenoid biosynthesis in the retina. Quantitation of the sterol and dolichol biosynthetic pathways. J. Biol. Chem. 1988;263:2250–2254. [PubMed] [Google Scholar]
- 32.Elbein A.D. Inhibitors of the biosynthesis and processing of N-linked oligosaccharides. CRC Crit. Rev. Biochem. 1984;16:21–49. doi: 10.3109/10409238409102805. [DOI] [PubMed] [Google Scholar]
- 33.West C.M. Current ideas on the significance of protein glycosylation. Mol. Cell. Biochem. 1986;72:3–20. doi: 10.1007/BF00230632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kukuruzinska M.A., Lennon K. Protein N-glycosylation: Molecular genetics and functional significance. Crit. Rev. Oral Biol. Med. 1998;9:415–448. doi: 10.1177/10454411980090040301. [DOI] [PubMed] [Google Scholar]
- 35.Fliesler S.J. Retinal rod outer segment membrane assembly: Studies with inhibitors of N-linked oligosaccharide biosynthesis and processing. In: Ohyama M., Muramatsu T., Furuta S., editors. Glycoconjugates in Medicine. Professional Postgraduate Services; Tokyo: 1988. pp. 316–323. [Google Scholar]
- 36.Murray A.R., Fliesler S.J., Al-Ubaidi M.R. Rhodopsin: The functional significance of asn-linked glycosylation and other post-translational modifications. Ophthalmic Genet. 2009;30:109–120. doi: 10.1080/13816810902962405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang C.J., Yamashita K., Muellenberg C.G., Shichi H., Kobata A. Structure of the carbohydrate moieties of bovine rhodopsin. J. Biol. Chem. 1979;254:6414–6418. [PubMed] [Google Scholar]
- 38.Fukuda M.N., Papermaster D.S., Hargrave P.A. Rhodopsin carbohydrate. Structure of small oligosaccharides attached at two sites near the NH2 terminus. J. Biol. Chem. 1979;254:8201–8207. [PubMed] [Google Scholar]
- 39.Duffin K.L., Lange G.W., Welply J.K., Florman R., O'Brien P.J., Dell A., Reason A.J., Morris H.R., Fliesler S.J. Identification and oligosaccharide structure analysis of rhodopsin glycoforms containing galactose and sialic acid. Glycobiology. 1993;3:365–380. doi: 10.1093/glycob/3.4.365. [DOI] [PubMed] [Google Scholar]
- 40.Fliesler S.J., Rayborn M.E., Hollyfield J.G. Membrane morphogenesis in retinal rod outer segments: Inhibition by tunicamycin. J. Cell Biol. 1985;100:574–587. doi: 10.1083/jcb.100.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fliesler S.J., Rapp L.M., Hollyfield J.G. Photoreceptor-specific degeneration caused by tunicamycin. Nature. 1984;311:575–577. doi: 10.1038/311575a0. [DOI] [PubMed] [Google Scholar]
- 42.Fliesler S.J., Tabor G.A., Hollyfield J.G. Glycoprotein synthesis in the human retina: Localization of the lipid intermediate pathway. Exp. Eye Res. 1984;39:153–173. doi: 10.1016/0014-4835(84)90005-8. [DOI] [PubMed] [Google Scholar]
- 43.Haeuptle M.A., Hennet T. Congenital disorders of glycosylation: An update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum. Mutat. 2009;30:1628–1641. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- 44.Wolfe L.S., Ng Ying Kin N.M., Palo J., Haltia M. Dolichols in brain and urinary sediment in neuronal ceroid lipofuscinosis. Neurology. 1983;33:103–106. doi: 10.1212/wnl.33.1.103. [DOI] [PubMed] [Google Scholar]
- 45.Wolfe L.S., Ng Ying Kin N.M., Palo J., Haltia M. Raised levels of cerebral cortex dolichols in Alzheimer's disease. Lancet. 1982;2:99. doi: 10.1016/s0140-6736(82)91717-2. [DOI] [PubMed] [Google Scholar]
- 46.Söderberg M., Edlund C., Alafuzoff I., Kristensson K., Dallner G. Lipid composition in different regions of the brain in Alzheimer's disease/senile dementia of Alzheimer's type. J. Neurochem. 1992;59:1646–1653. doi: 10.1111/j.1471-4159.1992.tb10994.x. [DOI] [PubMed] [Google Scholar]
- 47.Hooff G.P., Wood W.G., Müller W.E., Eckert G.P. Isoprenoids, small GTPases and Alzheimer's disease. Biochim. Biophys. Acta. 2010;1801:896–905. doi: 10.1016/j.bbalip.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edlund C., Söderberg M., Kristensson K., Dallner G. Ubiquinone, dolichol, and cholesterol metabolism in aging and Alzheimer's disease. Biochem. Cell Biol. 1992;70:422–428. doi: 10.1139/o92-065. [DOI] [PubMed] [Google Scholar]
- 49.Fujita S., Endo T., Ju J., Kean E.L., Kobata A. Structural studies of the N-linked sugar chains of human rhodopsin. Glycobiology. 1994;4:633–640. doi: 10.1093/glycob/4.5.633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.