

Abstract
By using homozygosity mapping in a consanguineous Pakistani family, we detected linkage of nonsyndromic hearing loss to a 7.6 Mb region on chromosome 3q13.31-q21.1 within the previously reported DFNB42 locus. Subsequent candidate gene sequencing identified a homozygous nonsense mutation (c.1135G>T [p.Glu379X]) in ILDR1 as the cause of hearing impairment. By analyzing additional consanguineous families with homozygosity at this locus, we detected ILDR1 mutations in the affected individuals of 10 more families from Pakistan and Iran. The identified ILDR1 variants include missense, nonsense, frameshift, and splice-site mutations as well as a start codon mutation in the family that originally defined the DFNB42 locus. ILDR1 encodes the evolutionarily conserved immunoglobulin-like domain containing receptor 1, a putative transmembrane receptor of unknown function. In situ hybridization detected expression of Ildr1, the murine ortholog, early in development in the vestibule and in hair cells and supporting cells of the cochlea. Expression in hair cell- and supporting cell-containing neurosensory organs is conserved in the zebrafish, in which the ildr1 ortholog is prominently expressed in the developing ear and neuromasts of the lateral line. These data identify loss-of-function mutations of ILDR1, a gene with a conserved expression pattern pointing to a conserved function in hearing in vertebrates, as underlying nonsyndromic prelingual sensorineural hearing impairment.
Introduction
Hearing is mediated by highly specialized cell types within the inner ear that ultimately convert sound into electrical signals. The high degree of conservation of hearing processes in vertebrates is reflected by homologies in the anatomy of the ear as well as by the conservation of molecular pathways underlying hearing and balance.1 Sound is transduced by hair cells embedded in layers of supporting cells and the resulting electrical signal is then transmitted to specific areas of the brain for information processing. Any perturbation of the development, structure, function, or maintenance of hair cells, supporting cells, or the auditory nerve can lead to hearing impairment.2–4
In humans, hearing impairment can be due to environmental influences, but in industrialized countries most cases of early-onset hearing impairment have a genetic cause, with autosomal-recessive inheritance being observed more frequently than autosomal-dominant, X-linked, or mitochondrial inheritance patterns. More than 80 loci for nonsyndromic autosomal-recessive hearing impairment (nonsyndromic ARHI; designated as DFNB loci [MIM 220700])5 have been mapped and causative mutations have been identified at more than 30 of these loci (Hereditary Hearing Loss Homepage). Genome-wide mapping and identification of genes that are mutated in hearing impairment allows for an unbiased knowledge of proteins and molecular pathways necessary for hearing. This has led to the identification of sets of proteins important for mechanotransduction and the structure of stereocilia, maintenance of high potassium concentrations in the endolymph, and structure and function of the inner ear ribbon synapse and the auditory nerve, among others.2 Many genes associated with hearing impairment have been conserved during evolution, and targeted, chemically induced, or knockdown-mediated inactivation of their orthologs in model organisms such as mouse and zebrafish often leads to hearing loss and related phenotypes (e.g., circling behavior).6–10 Here, we report the identification of mutations of ILDR1 (MIM 609739) as the cause of autosomal-recessive hearing impairment DFNB42 (MIM 609646)11 and evaluate the expression of this gene in mouse and zebrafish.
Subjects and Methods
Family Ascertainment and Clinical Evaluations
The study was approved by the Institutional Review Boards (IRB) at the University of Cologne, Germany; BUITEMS, Quetta, Pakistan; the National Center for Excellence in Molecular Biology (NCEMB), Lahore, Pakistan; the Quaid-I-Azam University, Islamabad, Pakistan; the University of Social Welfare and Rehabilitation Sciences, Tehran, Iran; the Baylor College of Medicine and Affiliated Hospitals, Houston, TX; the University of Iowa, Iowa City, IA; and the Combined Neuroscience IRB at the National Institutes of Health, Bethesda, MD. Written informed consent was obtained from participating individuals or their parents. Some of the hearing-impaired individuals were evaluated by medical history interviews and a physical examination. In general, several affected individuals from each family underwent an otological examination and pure-tone audiometry with air and bone conduction measurements. Some individuals were further evaluated by an ophthalmologic examination with funduscopy and by serum chemistry, blood count, urinanalysis, and an electrocardiogram. Blood samples were obtained from each participating individual, and genomic DNA was extracted by standard procedures.
SNP Genotyping and Linkage Analyses
We performed a genome-wide linkage analysis with homozygosity mapping by using Affymetrix GeneChip Human Mapping 250K Sty arrays and genomic DNA samples from consanguineous family PKDF637, which was ascertained in Balochistan province of Western Pakistan. Relationship errors were evaluated with the help of the program Graphical Relationship Representation.12 The program PedCheck was applied to detect Mendelian errors13 and data for SNPs with such errors were removed from the data set. Non-Mendelian errors were identified with the program MERLIN14 and unlikely genotypes for related samples were deleted. Linkage analysis was performed assuming autosomal-recessive inheritance, full penetrance, and a disease gene frequency of 0.0001. Multipoint LOD scores were calculated with ALLEGRO,15 which was also used for haplotype reconstruction. All data handling was performed with the graphical user interface ALOHOMORA.16 In family PKDF637 and in several other families, we performed fine mapping with short tandem repeat (STR) markers from the chromosome 3q candidate region.
Results of linkage analysis and fine mapping in family DEM4012, the family in which the DFNB42 locus was originally mapped, have been reported previously.11 A total of 388 STR markers were genotyped in families PKDF223, DEM4089, DEM4098, and DEM4207. Family DEM4430 was genotyped with the Illumina Infinium HumanLinkage-12 Panel, which contains 6090 single-nucleotide polymorphism (SNP) markers. Linkage analysis in families L-867 and L-1621 was performed with 412 STR markers of the Applied Biosystems Linkage mapping set v2.5 and approximately 50,000 SNPs of the Affymetrix 50K XBA GeneChip, respectively. Screening of additional Pakistani families with STR markers from the chromosome 3q13.31-q21.1 region revealed two additional DFNB42-linked families, PKDF790 and PKDF899.
Candidate Gene Screening
For candidate gene screening in the linkage interval in family PKDF637, we designed intronic primers to PCR amplify coding exons and the respective exon-intron boundaries by using genomic DNA of an affected individual. Primer pairs for amplification of the eight ILDR1 coding exons and their approximately 50 base pairs (bp) of flanking intronic sequences (RefSeq accession NM_001199799.1 and GenBank transcript AY672838.1) are listed in Table S1, available online. PCR products were sequenced on an ABI 3730 DNA Analyzer with BigDye chemistry v1.1 or v3.1 (Applied Biosystems). Sequence traces were assembled, aligned, and analyzed with the Seqman software (DNASTAR Lasergene). Mutation nomenclature is based on transcript NM_001199799.1.
Cosegregation of the mutation with deafness in each family was tested by sequencing the respective PCR product amplified from genomic DNA of all participating family members. Depending on the ethnic background of the family for which the candidate mutation was identified, we also sequenced PCR products of 250–500 Pakistani or 60 Iranian individuals.
RT-PCR and In Situ Hybridization Studies in Mouse
RNA was isolated from six organs (cochlea, brain, heart, liver, kidney, and lung) of postnatal day 2 (P2) mice via Trizol (Invitrogen, Darmstadt, Germany).17 RT-PCR was then performed with primers located in mouse Ildr1 exons 3 and 7 (primer sequences are listed in Table S1).
Part of the Ildr1 mRNA (NM_134109) was amplified from murine embryonic day (E) 12.5 otic vesicle with Phusion polymerase (New England Biolabs) via primers located in exons 5 and 7 to yield a 898 bp PCR fragment (primer sequences are listed in Table S1). The PCR product was then cloned into pGEM-T (Promega) and in situ hybridizations were carried out on cochlear sections of P1, P4, and P10 mice as previously described.18,19
In Situ Hybridization in Zebrafish
For zebrafish in situ hybridization, the 918 bp ildr1 EST EE699102.1 (Danio rerio cDNA clone IMAGE:8817233) derived from zebrafish whole-body cDNA and cloned into the pExpress-1 vector was purchased from imaGenes (Berlin, Germany). ildr1 in situ hybridization antisense and control sense probes were generated by in vitro transcription with the SP6 and T7 promoters of the pExpress-1 vector. In situ hybridizations on zebrafish of different stages of development were performed as previously described.20
Results
Identification of a Nonsense Mutation of ILDR1
In family PKDF637, four hearing-impaired and seven normal-hearing children were born to three normal-hearing couples who are first or second cousins within a large consanguineous family (Figure 1A), consistent with ARHI. By sequence analysis we found no mutations of GJB2 (encoding connexin 26; MIM 121011)21 and SLC26A4 (encoding pendrin; MIM 605646),22 two genes commonly associated with ARHI. We then performed a genome-wide linkage analysis with 250k SNP arrays followed by homozygosity mapping. By using a reduced marker panel of approximately 20,000 SNPs, we identified linkage to a single genomic region on chromosome 3q13.31-q21.1 with a maximum LOD score of 5.0 (Table 1). This LOD score was the maximum expected from simulation analysis, and the region on chromosome 3 was the only region within the genome with a LOD score that exceeded 2.0. With consideration of all SNPs from the 250K SNP array and after haplotype reconstruction, the region of shared homozygosity was determined to be flanked by SNP markers rs16823850 and rs2717225 (data not shown), defining a critical region of 7.6 Mb. The mapping results were confirmed by STR marker genotyping (Figure 1A). The linked region contains 52 annotated known and predicted coding genes and 1 miRNA gene (UCSC Genome Bioinformatics, build hg19). We sequenced miRNA-198 and 41 coding genes in one affected individual (Table S2), representing approximately 69% of the coding exons, and identified a homozygous nonsynonymous mutation not listed in dbSNP build 131. This mutation is a transversion (c.1135G>T) located in exon 7 of ILDR1 and is predicted to lead to a premature stop codon (p.Glu379X; Figure 1B; Table 1). This nonsense mutation cosegregated with ARHI in the family as expected from the haplotype analysis (Figure 1A) and was not likely to be a polymorphism because it is absent not only from dbSNP but also from the 1000 Genomes database. We also did not find it in 1000 Pakistani control chromosomes (Table 1).
Figure 1.
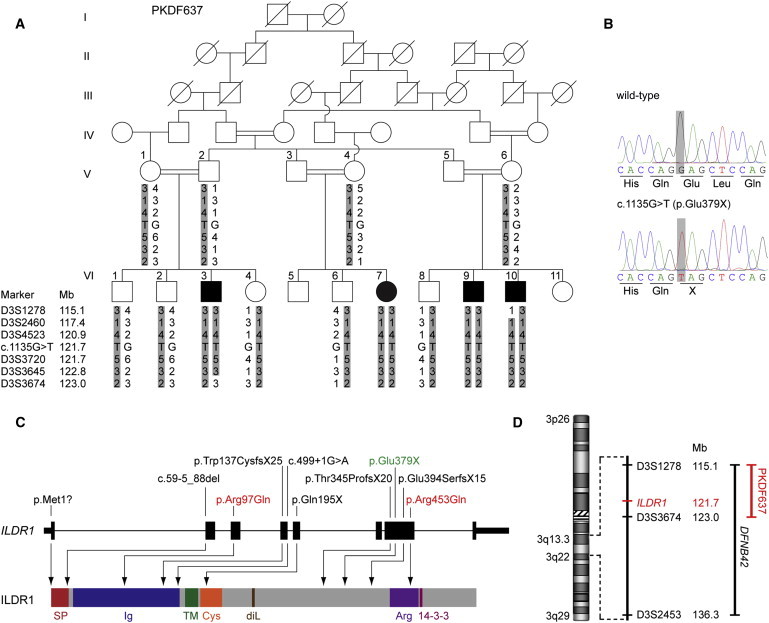
Identification of an ILDR1 Nonsense Mutation Causing Autosomal-Recessive Hearing Impairment
(A) Pedigree of family PKDF637 and haplotype analysis showing homozygous haplotypes on chromosome 3q13.3-q21.1 in affected individuals. c.1135G>T (NM_001199799.1) denotes an ILDR1 nonsense mutation leading to p.Glu379X. Mb, megabases from human genome reference sequence build hg19.
(B) Sequence chromatograms of a part of ILDR1 exon 7 of an individual homozygous for the reference sequence (wild-type) and an affected individual from family PKDF637 homozygous for the c.1135G>T (p.Glu379X) mutation.
(C) Genomic structure of ILDR1 based on the longest open reading frame (NM_001199799.1) containing eight coding exons (black rectangles). The positions of the ten ILDR1 mutations are shown both at the gene (top) and the protein level (bottom). The protein diagram depicts the predicted functional domains and sequence motifs. The nonsense mutation identified in family PKDF637 is highlighted in green and the two identified missense substitutions are shown in red. 14-3-3, 14-3-3 binding site; Arg, arginine-rich region; Cys, cysteine-rich region; diL, dileucine motif; Ig, immunoglobulin superfamily domain; SP, signal peptide; TM, transmembrane domain.
(D) Overview of the overlapping linkage intervals on chromosome 3q in family PKDF637 and in the original DFNB42 family (designated DEM4012 in this report).
Table 1.
Summary of the Results of Linkage Analysis and ILDR1 Mutations Detected in 11 Families with Autosomal-Recessive Hearing Impairment
| Family ID | Origin | Markers Genotyped | Maximum LOD Score | ILDR1 Mutation (cDNA)a | Location of the Mutation (Type of Mutation) | ILDR1 Mutation (Protein)b | Frequency in Control Chromosomesc |
|---|---|---|---|---|---|---|---|
| PKDF637 | Pakistan | genome-wide 250k SNP array | 5.0 | c.1135G>T | exon 7 (nonsense) | p.Glu379X | 0/1000 |
| DEM4012 (original DFNB42 family) | Pakistan | genome-wide 388 STR markers | 3.7 | c.3G>A | exon 1 (start codon) | p.Met1? | 0/500 |
| L-1621 | Iran | genome-wide 50k SNP array | 4.9 | c.59-5_88del | intron 1/exon 2 (splice site / deletion) | ND | 0/120 |
| PKDF790 | Pakistan | DFNB42-linked STRs | 5.7 | c.290G>A | exon 3 (missense) | p.Arg97Gln | 0/692 |
| PKDF223 | Pakistan | genome-wide 388 STR markers | 2.6 | c.411delGd | exon 4 (frameshift) | p.Trp137CysfsX25 | 0/688 |
| c.1387C>Td | exon 7 (missense) | p.Arg463Cys | 0/690 | ||||
| DEM4098 | Pakistan | genome-wide 388 STR markers | 4.6 | c.499+1G>A | intron 4 (splice site) | ND | 0/688 |
| L-867 | Iran | genome-wide 412 STR markers | 2.5 | c.583C>T | exon 5 (nonsense) | p.Gln195X | 0/120 |
| DEM4089 | Pakistan | genome-wide 388 STR markers | 3.1 | c.1032delG | exon 7 (frameshift) | p.Thr345ProfsX20 | 0/500 |
| DEM4207 | Pakistan | genome-wide 388 STR markers | 4.0 | c.1032delG | exon 7 (frameshift) | p.Thr345ProfsX20 | 0/500 |
| DEM4430 | Pakistan | genome-wide 6k SNP array | 4.5 | c.1180delG | exon 7 (frameshift) | p.Glu394SerfsX15 | 0/690 |
| PKDF899 | Pakistan | DFNB42-linked STRs | 2.1 | c.1358G>A | exon 7 (missense) | p.Arg453Gln | 0/690 |
ND, not determined
Nucleotide numbering starts from A of the translation initiation ATG codon of transcript NM_001199799.1. All mutations were present in homozygous state in all affected individuals of the respective families.
Accession number NP_001186728.1.
Ethnically matched controls (Pakistani or Iranian).
Two homozygous mutations of ILDR1 completely cosegregate with deafness in family PKDF223.
ILDR1, a gene of unknown function, encodes the immunoglobulin-like domain containing receptor 1, a predicted type 1 transmembrane protein.23 Alternative splicing produces up to five different transcripts, four of which have been reported previously,23,24 an additional mRNA being annotated in the UCSC Genome Browser (AY134857.1; Figure S1). The longest ILDR1 open reading frame consists of eight coding exons and encodes a 546 amino acid protein. ILDR1 is predicted to contain a signal peptide, an extracellular immunoglobulin (Ig) superfamily domain, and a transmembrane domain as well as other predicted functional domains such as a cysteine-rich and an arginine-rich domain, an LSR (lipolysis stimulated lipoprotein receptor) domain, a dileucine motif, and a 14-3-3 binding site (Figure 1C).23 Isoforms lacking the dileucine motif or the transmembrane domain have been identified but the tissue distribution, and functional significance of the putative membrane-bound and soluble ILDR1 isoforms are unknown. The p.Glu379X alteration located in exon 7 affects four ILDR1 isoforms (Figure S1) and might render the transcripts susceptible to nonsense-mediated mRNA decay (NMD). No RNA from an affected individual of family PKDF637 was available to test this hypothesis. If the protein is expressed, it would lack the arginine-rich region and the predicted 14-3-3 binding site (Figure 1C).
ILDR1 Mutation in the Original DFNB42 Family
ILDR1 is contained within the 21.1 Mb DFNB42 interval defined by linkage analysis in an unrelated Pakistani family segregating ARHI (family DEM4012; Figure 1D; Table 1).11 To determine whether an ILDR1 mutation causes deafness also in the original DFNB42 family DEM4012, we sequenced the ILDR1 coding exons and splice sites in an affected family member. We identified a homozygous start codon mutation (c.3G>A [p.Met1?]; Table 1; Figure S2). This variant cosegregated with autosomal-recessive/pseudodominant hearing impairment (Figure 2A) and was not found in 500 Pakistani control chromosomes. Therefore, we conclude that ILDR1 is the gene mutated in DFNB42 hearing impairment.
Figure 2.
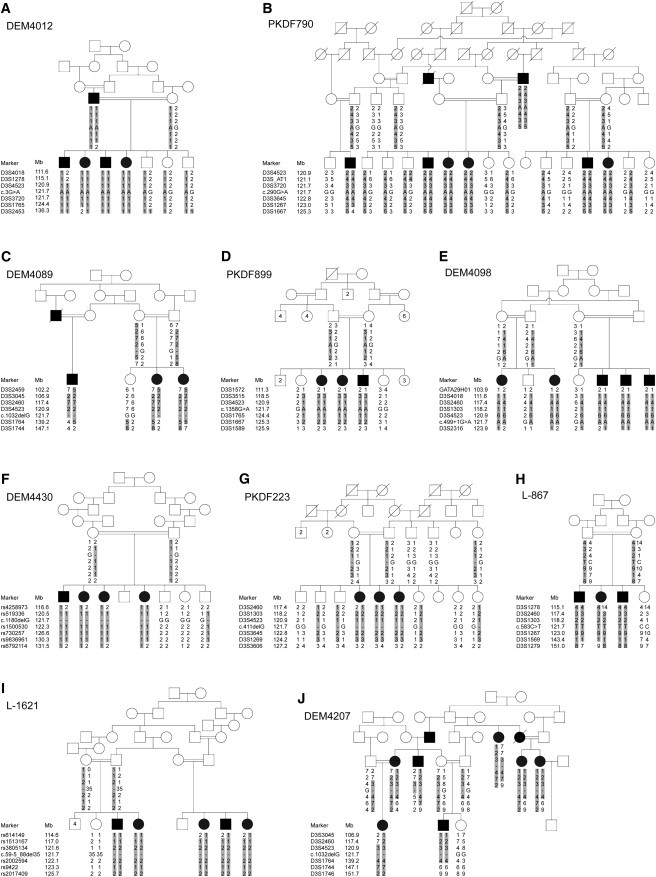
Haplotype Analysis at the DFNB42 Locus and Cosegregation of ILDR1 Mutations with Autosomal-Recessive Hearing Impairment in Ten Families
(A) Pedigree of the original DFNB42 family (DEM4012).
(B–J) Pedigrees of nine additional families segregating autosomal-recessive hearing impairment. The haplotypes at the DFNB42 locus indicate shared homozygosity in affected individuals in each family. Genotypes of the respective ILDR1 mutations are shown on the haplotypes. Mb, megabases.
Although the consequences of this mutation at the transcript and protein level are unknown, we note that Met136 is the next downstream in-frame methionine that might be used as a translation initiation codon, possibly leading to an N-terminal protein truncation. However, Met136 does not show a strong Kozak consensus sequence (G at position −3 but a T instead of a consensus G at position +4). Alternatively, MutationTaster25 predicts use of the next downstream ATG, which indeed shows a strong Kozak consensus sequence but is out-of-frame, potentially producing a 43 amino acid polypeptide with no similarity to ILDR1.
ILDR1 Mutations in Autosomal-Recessive Hearing Impairment
We next sequenced ILDR1 in affected individuals of nine additional unrelated ARHI families. These families were identified either by targeted screening for linkage to STR markers at the DFNB42 locus or through genome-wide homozygosity mapping with LOD scores ranging from 2.1 to 5.7 at this locus (Table 1). Seven of nine DFNB42 families originated from different regions in Pakistan and two are from Iran. We identified a homozygous ILDR1 mutation in affected individuals from all nine families. In total we identified eight different homozygous ILDR1 mutations, one mutation being present in two families. The mutant alleles included one nonsense and one canonical splice donor site mutation, two missense variants, three frameshift mutations leading to premature stop codons, and a 35 bp deletion that removes 5 bp at the exon 2 splice acceptor site and extends for 30 bp into exon 2 (Figures 2B–2J; Table 1; sequence chromatograms are shown in Figure S2). All mutations cosegregated with hearing impairment in the respective families and none of them were present in dbSNP, in the 1000 Genomes database, or in 120 to 692 chromosomes from ethnically matched controls (Figures 2B–2J; Table 1).
One frameshift mutation (c.1032delG [p.Thr345ProfsX20]) was present in two apparently unrelated Pakistani families (DEM4089 and DEM4207) and STR genotyping indicated that affected individuals from both families shared identical haplotypes at three STR markers including the ILDR1 intragenic marker D3S3720 (data not shown), suggesting that the mutation was inherited from a common ancestor of the two families. Two missense substitutions replaced arginine residues at positions 97 and 453 with glutamine. Whereas the p.Arg97Gln change (c.290G>A) is located in the Ig superfamily domain (Figure 1C) and is predicted to be “possibly damaging” by Polyphen and “not tolerated” by SIFT, p.Arg453Gln (c.1358G>A) was identified in the family with the lowest LOD score of all 11 families (2.1; Table 1) and is predicted to be benign by Polyphen and SIFT. Multiple species sequence alignments confirmed a high degree of conservation of Arg97 among ILDR1 orthologs and paralogs whereas Arg453 is moderately conserved (Figure S3). Thus, confirmation of the possible pathogenicity of p.Arg453Gln awaits future functional analyses. However, we note that p.Arg453Gln was not observed in 690 control chromosomes (Table 1), arguing for its pathogenicity. Furthermore, Arg453 is one of the arginine residues of the arginine-rich region located at amino acid positions 431–466 of the 546 residue isoform (Figure 1C). Although the function(s) of arginine-rich regions are not comprehensively documented, some can mediate protein-protein interactions26 and substitution of critical arginine residues might impede this function.
A third homozygous missense substitution, p.Arg463Cys (encoded by c.1387C>T), cosegregated with deafness in family PKDF223. However, affected family members are also homozygous for a truncating ILDR1 mutation, which we consider sufficient to cause deafness, so a possible pathogenic effect of p.Arg463Cys alone is speculative. In conclusion, multiple mutant alleles of ILDR1 are distributed across the gene (Figure 1C), and a majority of them are truncating mutations that might induce NMD and/or produce likely nonfunctional truncated proteins.
Clinical Characterization of DFNB42 Hearing Impairment
All individuals homozygous for an ILDR1 mutation had sensorineural hearing impairment. Hearing impairment was prelingual (and possibly congenital), bilateral, and of moderate-to-profound severity. Representative audiograms are shown in Figure 3 and complete audiogram data of three families are presented in Figure S4. In most affected individuals, hearing impairment was more pronounced at higher frequencies (“sloping” audiogram; Figure 3A), but in some patients all frequencies were similarly affected, giving rise to a “flat” audiogram (Figure 3B). Although no longitudinal data were available, comparisons of single audiograms of different affected family members at different ages provide no evidence of progression of hearing impairment. In an affected individual of family L-1621, otoacoustic emissions (OAEs) were absent, indicating cochlear dysfunction rather than auditory neuropathy. No subjective vestibular symptoms were reported. Thus, we conclude that ILDR1 mutations cause bilateral nonprogressive moderate-to-profound sensorineural hearing impairment.
Figure 3.

Representative Audiograms of Hearing-Impaired Individuals Homozygous for ILDR1 Mutations
(A) Audiograms of selected individuals from families PKDF637, PKDF899, L-1621, L-867, and DEM4012 showing hearing impairment that is more pronounced at high frequencies.
(B) Audiograms of selected individuals from families DEM4089, DEM4098, and DEM4430 showing hearing impairment involving all frequencies similarly. Only audiograms measured from the right ear are shown. Audiograms from the left ear were similar. y, age at time of audiogram in years.
Hearing impairment appeared nonsyndromic because no associated congenital anomalies, facial dysmorphisms, or intellectual disabilities were reported by the families or detected on physical examination. Several affected individuals underwent detailed clinical and laboratory examinations, and all were normal. The most comprehensive investigations were performed in the male individual IV:6 from family PKDF899 at the age of 26 years. No anomalies were detected in blood chemistry (including urea nitrogen and creatinine), thyroid function test, and urinanalysis, and there was no ophthalmological involvement (normal funduscopy) and no cardiac arrhythmia as assessed by an electrocardiogram.
Expression of Ildr1 in the Mouse Inner Ear
ILDR1 is expressed in human prostate, testis, pancreas, kidney, heart, and liver,23 and the UniGene database (entry Mm.17807) contains an Ildr1 EST (BQ568480.1) derived from the mouse inner ear. Consistent with these observations, massively parallel signature sequencing (MPSS) of mouse transcripts has previously revealed Ildr1 expression in the inner ear and in a few other tissues.27 However, expression of Ildr1 in the various cell types of the cochlea has not been investigated. The centromeric part of the DFNB42 linkage region on chromosome 3q13.31-q22.3 shows conserved synteny with mouse chromosome 16qB3 where the orthologous Ildr1 gene is located. Because human ILDR1 and mouse Ildr1 mRNAs are highly similar with 81% sequence identity at the nucleotide level (79% sequence identity at the protein level), we investigated Ildr1 expression in the mouse cochlea as a model system. Expression of Ildr1 was first examined in P2 mouse organs by nonquantitative RT-PCR. We detected Ildr1 expression in all tissues tested, including the cochlea (Figure 4A). In this experimental setting, the most abundant transcript contained exon 6, potentially representing a transcript encoding the mouse full-length 537 amino acid isoform.
Figure 4.

Expression of Ildr1 in the Mouse Inner Ear
(A) Nonquantitative RT-PCR with exonic Ildr1 primers on cDNA derived from P2 mouse organs. gDNA, genomic DNA; H2O, control PCR with water as a template.
(B–E) In situ hybridization of Ildr1 in the mouse cochlea at P4. Antisense (AS) probe (B and D); control sense (S) probe (C and E). Arrowheads in (B) point at the organ of Corti.
(F–N) In situ hybridization of Ildr1 in the organ of Corti at high magnification at P1 (F–H), P4 (I–K), and P10 (L–N) with Ildr1 antisense (F, I, L) and sense (G, J, M) probes. Yellow dotted lines in (F), (I), and (L) indicate the positions of inner and outer hair cells. Ildr1 is expressed at low-to-intermediate levels in hair cells and at higher levels in the Pillar and Hensen cells adjacent to hair cells. In (H), (K), and (N), the hair cells are stained with a Loxhd1 antisense probe, which is used as a positive control (i.e., Loxhd1 is highly expressed in hair cells).18
(O) Diagram of Ildr1 expression in the mouse organ of Corti at P4. IHC, inner hair cell; OHC, outer hair cells.
Scale bars represent 200 μm in (B) and (C), 100 μm in (D) and (E), and 20 μm in (F)–(N).
To determine the spatio-temporal expression pattern of Ildr1 in the mouse inner ear, we performed mRNA in situ hybridization experiments on P1, P4, and P10 mice. Ildr1 mRNA was detected in the cochlea and the vestibule at P4 (Figures 4B–4E and Figure S5, respectively). Expression in the organ of Corti was low at P1 (Figures 4F–4H) and increased gradually at P4 and P10 (Figures 4I–4N). Expression was detected at low-to-intermediate levels in hair cells and at higher levels in a subset of supporting cells (Figures 4F, 4I, and 4L). Supporting cells expressing high levels of Ildr1 at P4 are presumably the Pillar cells and a subset of Hensen cells (Figures 4I and 4O). A schematic overview of the Ildr1 expression pattern in the mouse organ of Corti at P4 is given in Figure 4O.
Expression of ildr1 in Zebrafish
In the zebrafish genome (build Zv9), a single ILDR1 ortholog is annotated. Danio rerio ildr1 shares 40% amino acid sequence identity with human ILDR1. A BLAST search28 of the nonredundant (NR) protein database with the human full-length 546 amino acid ILDR1 sequence identified ildr1 and a second protein, the product of zgc:64227, as putative ILDR1 orthologs in zebrafish. ildr1 and its paralog encoded by zgc:64227 share 47% sequence identity at the amino acid level and are presumably derived from a single ancestral gene after the genome duplication that has occurred in the fish lineage (Figure S6). Although zgc:64227 has previously been shown to be expressed in the zebrafish otic placode at the 14–19 somite stage (corresponding to 16–19 hr postfertilization [hpf]) and in the lateral line from the 20–25 somite stage (19–22 hpf) on (ZFIN homepage), the tissue distribution of ildr1 has not been determined. Therefore, we performed ildr1 whole-mount in situ hybridization in wild-type zebrafish embryos and larvae between early segmentation stages (11 hpf) and day 6 of development. No distinct signal was obtained with the control sense probe (data not shown). We did not detect ildr1 expression at 11 hpf. At 15 hpf, however, a weak signal was present in the developing endoderm (data not shown), and a broader expression pattern can be seen from 24 hpf onward (Figure 5). At this stage, ildr1 is expressed in several derivatives of the preplacodal ectoderm including the adenohypophyseal placode, the olfactory epithelium and, notably, the otic vesicle and the primordium of the posterior lateral line, which later gives rise to the neuromast-containing lateral line organ (Figures 5A and 5B). In addition, transcripts can be detected in the developing gastrointestinal duct (Figures 5A and 5B). ildr1 expression persists at least up to day 6 (144 hpf) in the endoderm, the nose, the neuromasts (which are the sensory organs of the lateral line), and in the ear where expression is high at 48 hpf (Figures 5C and 5D). Notably, the otic vesicle from approximately 20 hpf onward, the ear, and the lateral line primordium contain hair cells.8
Figure 5.

Expression of ildr1 in Zebrafish
Whole-mount in situ hybridization with probes for ildr1 transcripts in wild-type zebrafish embryos and larvae at ages indicated in the bottom left corners. (A, C, D) Lateral views with anterior to the left. (B) Cross section at the level of the posterior lateral line primordium. The inset in (C) depicts a magnification of the ildr1-positive anterior pituitary gland of the 48 hpf larvae. Arrowheads in (D) point to neuromasts, the sensory organs of the lateral line system. ap, anterior pituitary; e, ear; en, endoderm; n, nose; nc, notochord; oc, oral cavity; oe, olfactory epithelium; ov, otic vesicle; pllp, posterior lateral line primordium.
Discussion
We have identified mutations of ILDR1 underlying autosomal-recessive nonsyndromic hearing impairment at the DFNB42 locus. Seven of ten distinct ILDR1 mutations are nonsense, frameshift, or splice site mutations predicted to introduce premature stop codons that may lead to NMD and/or protein truncation. Thus, complete loss of ILDR1 function appears to underlie hearing impairment in most if not all cases. The DFNB42 locus was originally mapped in a single Pakistani family.11 Our identification of ILDR1 mutations in nine Pakistani and two Iranian families raises the possibility that mutations of ILDR1 may be among the more prevalent causes of ARHI, a genetically highly heterogeneous condition. On the other hand, the large allelic heterogeneity at the DFNB42 locus in families from Pakistan alone is similar to allelic heterogeneity for other genes associated with hearing impairment that have been extensively studied in this population.21,22
ILDR1 belongs to an evolutionarily conserved family of Ig domain-containing proteins of unknown function. Mouse Ildr2 (also known as Lisch-like) has been reported as a type 2 diabetes susceptibility gene in DBA mice.29 Another homolog, LSR, encodes the lipolysis-stimulated lipoprotein receptor that binds free fatty acids and contributes to the control of plasma triglyceride and cholesterol levels.30,31 Notably, the zebrafish ildr2 ortholog is highly expressed in the otic vesicle at 24 hpf and the ear at 48 hpf.29 We note that human ILDR2 maps to the DFNA7 locus on 1q24.132 and is therefore an interesting candidate gene for autosomal-dominant deafness. Although the function of ILDR1 remains elusive,23 its expression in the mouse cochlea and vestibule as well as in the zebrafish ear and in lateral line neuromasts supports an essential role in hearing in vertebrates. A necessary function of ILDR1 only for hearing is supported by the lack of obvious additional clinical signs outside the auditory system in affected individuals despite the wide tissue distribution of ILDR1. This is not an unusual observation; there are mutant alleles of several ubiquitously expressed genes associated with nonsyndromic deafness such as TPRN (MIM 613354), MARVELD2 (also known as TRIC, MIM 610572), HGF (MIM 142409), and ACTG1 (MIM 102560).17,33–36
Many genes that are mutated in DFNB hearing impairment are highly expressed in cochlear hair cells and predominant expression in supporting cells is an exception.37 Our in situ hybridization experiments in the mouse inner ear show that Ildr1 is weakly expressed in hair cells and at higher levels in a subset of supporting cells. Whether DFNB42-linked hearing impairment is due to disruption of ILDR1 function in hair cells, supporting cells, or both remains to be addressed in future studies.
In conclusion, mutations of ILDR1 constitute a previously unreported cause of DFNB hearing impairment. ILDR1 is a gene of unknown function but with a consistent expression pattern in auditory and vestibular tissues in mouse and zebrafish. Future studies will address the spectrum and prevalence of ILDR1 mutations in ARHI families of different ethnic and geographical origins, the localization of the ILDR1 protein in the mammalian cochlea, the tissue distribution and functions of the different ILDR1 isoforms and possible ligands, interaction partners, and downstream effectors of this putative transmembrane receptor.
Acknowledgments
We wish to thank the family members for their invaluable participation and cooperation. We thank Dennis Drayna and Changsoo Kang for critically reading the manuscript. This work was funded by the Deutsche Forschungsgemeinschaft (DFG; BO2985/3-1; to G.B.), the European Commission FP6 Integrated Project EUROHEAR, Grant LSHG-CT-20054-512063 (to B.W. and C.K.), NIH RO1 DC002842 (to R.J.H.S), NHMRC Career Development Award (to M.B.), NHMRC Overseas Biomedical Fellowship (to M.S.H.), Doris Duke Fellowship (to A.E.S), NIH DC007704 and Skaggs Institute for Chemical Biology (to U.M.), NIH intramural funds from NIDCD DC000039-14 (to T.B.F.), Higher Education Commission (HEC), Government of Pakistan (to W.A.), NIH-NIDCD grant DC03594 (to S.M.L.), and the ICGEB, Trieste, Italy (to Sheikh R.). J.A. thanks Dost Muhammad Baloch and Ahmed Farooq Bazai, VC, BUITEMS, for faculty funds. Genotyping services were partially provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the NIH to The Johns Hopkins University, Contract Number N01-HG-65403 (to S.M.L.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes browser, http://browser.1000genomes.org/index.html
GenBank, http://www.ncbi.nlm.nih.gov/genbank
Hereditary Hearing Loss home page, http://hereditaryhearingloss.org
MutationTaster, http://www.mutationtaster.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Polyphen, http://genetics.bwh.harvard.edu/pph
Sorting Intolerant from Tolerant (SIFT), http://sift.jcvi.org
TMHMM (prediction of transmembrane helices in proteins), http://www.cbs.dtu.dk/services/TMHMM
UCSC Genome Bioinformatics, http://www.genome.ucsc.edu
UniGene, http://www.ncbi.nlm.nih.gov/unigene
ZFIN, http://zfin.org/cgi-bin/webdriver?MIval=aa-ZDB_home.apg
References
- 1.Fritzsch B., Beisel K.W., Bermingham N.A. Developmental evolutionary biology of the vertebrate ear: Conserving mechanoelectric transduction and developmental pathways in diverging morphologies. Neuroreport. 2000;11:R35–R44. doi: 10.1097/00001756-200011270-00013. [DOI] [PubMed] [Google Scholar]
- 2.Dror A.A., Avraham K.B. Hearing impairment: A panoply of genes and functions. Neuron. 2010;68:293–308. doi: 10.1016/j.neuron.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Gillespie P.G., Muller U. Mechanotransduction by hair cells: Models, molecules, and mechanisms. Cell. 2009;139:33–44. doi: 10.1016/j.cell.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frolenkov G.I., Belyantseva I.A., Friedman T.B., Griffith A.J. Genetic insights into the morphogenesis of inner ear hair cells. Nat. Rev. Genet. 2004;5:489–498. doi: 10.1038/nrg1377. [DOI] [PubMed] [Google Scholar]
- 5.Petersen M.B., Willems P.J. Non-syndromic, autosomal-recessive deafness. Clin. Genet. 2006;69:371–392. doi: 10.1111/j.1399-0004.2006.00613.x. [DOI] [PubMed] [Google Scholar]
- 6.Brown S.D., Hardisty-Hughes R.E., Mburu P. Quiet as a mouse: Dissecting the molecular and genetic basis of hearing. Nat. Rev. Genet. 2008;9:277–290. doi: 10.1038/nrg2309. [DOI] [PubMed] [Google Scholar]
- 7.Leibovici M., Safieddine S., Petit C. Mouse models for human hereditary deafness. Curr. Top. Dev. Biol. 2008;84:385–429. doi: 10.1016/S0070-2153(08)00608-X. [DOI] [PubMed] [Google Scholar]
- 8.Nicolson T. The genetics of hearing and balance in zebrafish. Annu. Rev. Genet. 2005;39:9–22. doi: 10.1146/annurev.genet.39.073003.105049. [DOI] [PubMed] [Google Scholar]
- 9.Nicolson T., Rusch A., Friedrich R.W., Granato M., Ruppersberg J.P., Nusslein-Volhard C. Genetic analysis of vertebrate sensory hair cell mechanosensation: The zebrafish circler mutants. Neuron. 1998;20:271–283. doi: 10.1016/s0896-6273(00)80455-9. [DOI] [PubMed] [Google Scholar]
- 10.Vrijens K., Van Laer L., Van Camp G. Human hereditary hearing impairment: Mouse models can help to solve the puzzle. Hum. Genet. 2008;124:325–348. doi: 10.1007/s00439-008-0556-y. [DOI] [PubMed] [Google Scholar]
- 11.Aslam M., Wajid M., Chahrour M.H., Ansar M., Haque S., Pham T.L., Santos R.P., Yan K., Ahmad W., Leal S.M. A novel autosomal recessive nonsyndromic hearing impairment locus (DFNB42) maps to chromosome 3q13.31-q22.3. Am. J. Med. Genet. A. 2005;133A:18–22. doi: 10.1002/ajmg.a.30508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: Graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 13.O'Connell J.R., Weeks D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 15.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 16.Ruschendorf F., Nurnberg P. ALOHOMORA: A tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 17.Li Y., Pohl E., Boulouiz R., Schraders M., Nurnberg G., Charif M., Admiraal R.J., von Ameln S., Baessmann I., Kandil M. Mutations in TPRN cause a progressive form of autosomal-recessive nonsyndromic hearing loss. Am. J. Hum. Genet. 2010;86:479–484. doi: 10.1016/j.ajhg.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grillet N., Schwander M., Hildebrand M.S., Sczaniecka A., Kolatkar A., Velasco J., Webster J.A., Kahrizi K., Najmabadi H., Kimberling W.J. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am. J. Hum. Genet. 2009;85:328–337. doi: 10.1016/j.ajhg.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwander M., Sczaniecka A., Grillet N., Bailey J.S., Avenarius M., Najmabadi H., Steffy B.M., Federe G.C., Lagler E.A., Banan R. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J. Neurosci. 2007;27:2163–2175. doi: 10.1523/JNEUROSCI.4975-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammerschmidt M., Pelegri F., Mullins M.C., Kane D.A., van Eeden F.J., Granato M., Brand M., Furutani-Seiki M., Haffter P., Heisenberg C.P. dino and mercedes, two genes regulating dorsal development in the zebrafish embryo. Development. 1996;123:95–102. doi: 10.1242/dev.123.1.95. [DOI] [PubMed] [Google Scholar]
- 21.Santos R.L., Wajid M., Pham T.L., Hussan J., Ali G., Ahmad W., Leal S.M. Low prevalence of Connexin 26 (GJB2) variants in Pakistani families with autosomal recessive non-syndromic hearing impairment. Clin. Genet. 2005;67:61–68. doi: 10.1111/j.1399-0004.2005.00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anwar S., Riazuddin S., Ahmed Z.M., Tasneem S., Jaleel A.u., Khan S.Y., Griffith A.J., Friedman T.B., Riazuddin S. SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred's syndrome in Pakistanis. J. Hum. Genet. 2009;54:266–270. doi: 10.1038/jhg.2009.21. [DOI] [PubMed] [Google Scholar]
- 23.Hauge H., Patzke S., Delabie J., Aasheim H.C. Characterization of a novel immunoglobulin-like domain containing receptor. Biochem. Biophys. Res. Commun. 2004;323:970–978. doi: 10.1016/j.bbrc.2004.08.188. [DOI] [PubMed] [Google Scholar]
- 24.Strausberg R.L., Feingold E.A., Grouse L.H., Derge J.G., Klausner R.D., Collins F.S., Wagner L., Shenmen C.M., Schuler G.D., Altschul S.F. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. USA. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 26.Brodeur J., Larkin H., Boucher R., Thériault C., St-Louis S.C., Gagnon H., Lavoie C. Calnuc binds to LRP9 and affects its endosomal sorting. Traffic. 2009;10:1098–1114. doi: 10.1111/j.1600-0854.2009.00933.x. [DOI] [PubMed] [Google Scholar]
- 27.Peters L.M., Belyantseva I.A., Lagziel A., Battey J.F., Friedman T.B., Morell R.J. Signatures from tissue-specific MPSS libraries identify transcripts preferentially expressed in the mouse inner ear. Genomics. 2007;89:197–206. doi: 10.1016/j.ygeno.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGinnis S., Madden T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32:W20–W25. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dokmanovic-Chouinard M., Chung W.K., Chevre J.C., Watson E., Yonan J., Wiegand B., Bromberg Y., Wakae N., Wright C.V., Overton J. Positional cloning of “Lisch-Like”, a candidate modifier of susceptibility to type 2 diabetes in mice. PLoS Genet. 2008;4:e1000137. doi: 10.1371/journal.pgen.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narvekar P., Berriel Diaz M., Krones-Herzig A., Hardeland U., Strzoda D., Stohr S., Frohme M., Herzig S. Liver-specific loss of lipolysis-stimulated lipoprotein receptor triggers systemic hyperlipidemia in mice. Diabetes. 2009;58:1040–1049. doi: 10.2337/db08-1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yen F.T., Roitel O., Bonnard L., Notet V., Pratte D., Stenger C., Magueur E., Bihain B.E. Lipolysis stimulated lipoprotein receptor: A novel molecular link between hyperlipidemia, weight gain, and atherosclerosis in mice. J. Biol. Chem. 2008;283:25650–25659. doi: 10.1074/jbc.M801027200. [DOI] [PubMed] [Google Scholar]
- 32.Fagerheim T., Nilssen O., Raeymaekers P., Brox V., Moum T., Elverland H.H., Teig E., Omland H.H., Fostad G.K., Tranebjaerg L. Identification of a new locus for autosomal dominant non-syndromic hearing impairment (DFNA7) in a large Norwegian family. Hum. Mol. Genet. 1996;5:1187–1191. doi: 10.1093/hmg/5.8.1187. [DOI] [PubMed] [Google Scholar]
- 33.Rehman A.U., Morell R.J., Belyantseva I.A., Khan S.Y., Boger E.T., Shahzad M., Ahmed Z.M., Riazuddin S., Khan S.N., Riazuddin S., Friedman T.B. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am. J. Hum. Genet. 2010;86:378–388. doi: 10.1016/j.ajhg.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riazuddin S., Ahmed Z.M., Fanning A.S., Lagziel A., Kitajiri S., Ramzan K., Khan S.N., Chattaraj P., Friedman P.L., Anderson J.M. Tricellulin is a tight-junction protein necessary for hearing. Am. J. Hum. Genet. 2006;79:1040–1051. doi: 10.1086/510022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schultz J.M., Khan S.N., Ahmed Z.M., Riazuddin S., Waryah A.M., Chhatre D., Starost M.F., Ploplis B., Buckley S., Velasquez D. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. Am. J. Hum. Genet. 2009;85:25–39. doi: 10.1016/j.ajhg.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu M., Yang T., Wei S., DeWan A.T., Morell R.J., Elfenbein J.L., Fisher R.A., Leal S.M., Smith R.J., Friderici K.H. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26) Am. J. Hum. Genet. 2003;73:1082–1091. doi: 10.1086/379286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilcox E.R., Burton Q.L., Naz S., Riazuddin S., Smith T.N., Ploplis B., Belyantseva I., Ben-Yosef T., Liburd N.A., Morell R.J. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell. 2001;104:165–172. doi: 10.1016/s0092-8674(01)00200-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.