

Abstract
The importance of intracellular folate metabolism is illustrated by the severity of symptoms and complications caused by inborn disorders of folate metabolism or by folate deficiency. We examined three children of healthy, distantly related parents presenting with megaloblastic anemia and cerebral folate deficiency causing neurologic disease with atypical childhood absence epilepsy. Genome-wide homozygosity mapping revealed a candidate region on chromosome 5 including the dihydrofolate reductase (DHFR) locus. DHFR sequencing revealed a homozygous DHFR mutation, c.458A>T (p.Asp153Val), in all siblings. The patients' folate profile in red blood cells (RBC), plasma, and cerebrospinal fluid (CSF), analyzed by liquid chromatography tandem mass spectrometry, was compatible with DHFR deficiency. DHFR activity and fluorescein-labeled methotrexate (FMTX) binding were severely reduced in EBV-immortalized lymphoblastoid cells of all patients. Heterozygous cells displayed intermediate DHFR activity and FMTX binding. RT-PCR of DHFR mRNA revealed no differences between wild-type and DHFR mutation-carrying cells, whereas protein expression was reduced in cells with the DHFR mutation. Treatment with folinic acid resulted in the resolution of hematological abnormalities, normalization of CSF folate levels, and improvement of neurological symptoms. In conclusion, the homozygous DHFR mutation p.Asp153Val causes DHFR deficiency and leads to a complex hematological and neurological disease that can be successfully treated with folinic acid. DHFR is necessary for maintaining sufficient CSF and RBC folate levels, even in the presence of adequate nutritional folate supply and normal plasma folate.
Main Text
Various inherited and acquired disorders affecting folate uptake, transport, and metabolism have been described. Nutritional folate deficiency is one of the most common micronutrient deficiencies worldwide. Depending on the severity of folate deficiency, it is associated with megaloblastic anemia, neurologic and mental disorders, cardiovascular disease, embryonic defects (in particular neural tube defects), and, possibly, malignancies.1 Furthermore, the importance of intracellular folate metabolism for normal psychomotor and cognitive development is illustrated by the severity of symptoms in disorders of folate metabolism, such as methylenetetrahydrofolate reductase deficiency (MTHFR [MIM 236250]) or glutamate formiminotransferase deficiency (FTCD [MIM 229100]).2
Dihydrofolate reductase (DHFR [MIM 126060]) catalyzes the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF) and, at a lower rate, of folic acid (FA) to DHF. DHFR plays a key role in maintaining intracellular folate homeostasis and is an important target for cytostatic drugs. To date, three cases with initially suspected inherited DHFR deficiency have been reported.3,4 Later analyses revealed normal DHFR activity in one patient and the presence of transcobalamin II deficiency (TCN2 [MIM 275350]) as the underlying disorder in another of the reported patients.5,6 Follow-up for the third patient was not reported. A distinct genetic defect associated with DHFR deficiency has not been described.
We examined three children of healthy, distantly related parents of European descent (Figure 1A). Patient 1 (VII-1 in Figure 1) presented at age 11 without any clinical symptoms or findings. Peripheral-blood analysis revealed macrocytosis without anemia (Table 1). Electroencephalogram (EEG) at that time showed relatively slow 8–9/s waves, particularly in the parieto-occipital region, and intermittently repetitive generalized high-amplitude delta waves but no epilepsy-specific potentials.
Figure 1.
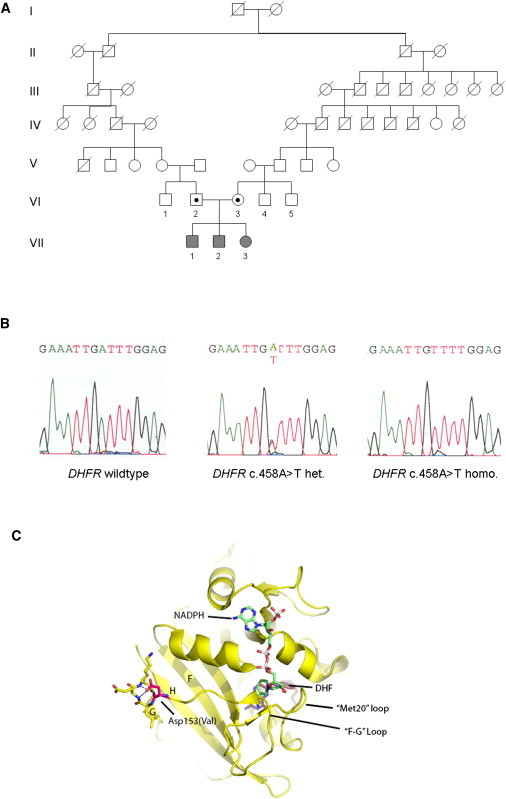
Identification of the DHFR c.458A>T Mutation in a Family and Tertiary Structure of Human DHFR
(A) Pedigree of the family. Genetic analysis was performed in the affected siblings and their parents.
(B) Sequence analysis of DHFR exon 5 detected a homozygous base change, c.458A>T, in all siblings and a heterozygous mutation in both parents. Amplifications were performed with the HotStar-Taq DNA Polymerase Kit (QIAGEN) with 35 cycles in a GeneAmp PCR System 9700 (Applied Biosystems). Sequencing reactions were performed with BigDye Terminator Cycle Sequencing Ready Reaction (Applied Biosystems) and analyzed on an ABI 3100 DNA Genetic Analyzer (Applied Biosystems).
(C) Crystal structure of human DHFR (yellow ribbon model with highlighted secondary structure, Protein Data Bank entry 2W3M) bound to NADPH (color-coded stick model with green carbons, white phosphors, red oxygens, and blue nitrogens) and DHF (ruby stick model). The mutation site Asp153Val is highlighted as a magenta stick model. Asp153 stabilizes one side of the “F-G” loop, indicating that the mutation alters DHFR activity by influencing the fold and dynamics of the catalytically important “F-G” and “Met20” loops. The figure was prepared with the program PyMOL (Schrödinger).
Table 1.
Hematological Parameters, Total RBC Folate, and RBC Folate Metabolites and CSF 5-MTHF before and after 5-FTHF Substitution
|
RBC Folates |
CSF |
||||||
|---|---|---|---|---|---|---|---|
| Hb (g/l) | MCV (fl) | Total (nmol/l) | 5-MTHF (nmol/l) | NonmethylTHF (nmol/l) | FA+DHF (nmol/l) | 5-MTHF (nmol/l) | |
| DHFR p.D153V hetero. | |||||||
| Mother | 149 | 89 | 773 | 760 | 12.7 | 0 | |
| Father | 165 | 92 | 342 | 328 | 13.6 | 0 | |
| DHFR p.D153V homo.: untreated | |||||||
| Pt. 1 | 135 | 108 | 66 | 55 | 11.3 | 0 | 13.6 |
| DHFR p.D153V homo.: treated with FA | |||||||
| Pt. 2 | 132 | 92 | 204a | <5 | |||
| Pt. 3 | 144 | 99 | 152a | <5 | |||
| DHFR p.D153V homo.: treated with 5-FTHF | |||||||
| Pt. 1 | 139 | 90 | 241 | 165 | 31.0 | 46 | n.e. |
| Pt. 2 | 153 | 80 | 641 | 153 | 398 | 92 | 61 |
| Pt. 3 | 135 | 89 | 702 | 374 | 226 | 102 | 59 |
| Reference range | 111–166 | 76–90 | 264–1153b | 264–1086 | 0–67 | 0 | 41–117 |
5-FTHF, 5-formyltetrahydrofolate. NonmethylTHF (nonmethyltetrahydrofolate) contains 5,10-methenylTHF + 5-formylTHF + 10-formylTHF + 5,10-methyleneTHF + THF. n.e., not examined.
Analyzed by chemiluminescence immunoassay (CLIA, Roche Diagnostics; assay-dependent normal values: 397–1589).
Assay-dependent normal values for LC-MS/MS.
Patient 2 (VII-2 in Figure 1) presented at age 5 with hemoglobin level at 56 g/l, mean corpuscular volume (MCV) at 117 fl, 3.3% reticulocytes, and serum lactate dehydrogenase activity at 2066 U/l without any congenital physical abnormality. Bone marrow examination revealed typical features of megaloblastic anemia. Serum folate, cobalamin, transcobalamin I and II binding capacity, and homocysteine, as well as urinary excretion of methylmalonic, orotic, and formiminoglutamic acid, were normal. Treatment with hydroxycobalamin and FA (5 mg per day) normalized hemoglobin levels and MCV. At age 8, increasing learning difficulties developed and the patient presented with short episodes of involuntary blinking and winking, partially associated with impaired consciousness. EEG revealed atypical childhood absence epilepsy with eyelid myoclonia. A bone marrow smear still displayed mild megaloblastic changes despite an almost normal peripheral-blood analysis (Table 1).
Patient 3 (VII-3 in Figure 1) had a complicated febrile seizure at age 2. Blood analysis was normal apart from marked macrocytosis with MCV 109 fl. Physical examination did not reveal any pathological findings. Subsequent psychomotor and cognitive development was normal. At age 5, the girl presented with frequent episodes of impaired vision, blinking, and squinting associated with impaired consciousness and repetitive eyeball movements during sleep. EEG showed severe pathologic changes compatible with an atypical childhood absence epilepsy with eyelid myoclonia. Blood examination still showed isolated macrocytosis, whereas a bone marrow smear displayed megaloblastic changes (Table 1). The girl was treated with FA (5 mg per day) after consideration of the apparent effectiveness of FA in her brother, who was neurologically asymptomatic at that period.
After the occurrence of neurologic symptoms in patient 2 despite FA supplementation, 5-methyltetrahydrofolate (5-MTHF) and neurotransmitter concentrations in the cerebrospinal fluid (CSF) were determined for further assessment of the metabolic changes of the disease. 5-MTHF analysis was performed precisely as previously described.7 It revealed very low 5-MTHF in patient 1. In patients 2 and 3, CSF 5-MTHF was undetectable despite preceding FA treatment (Table 1). Total red blood cell (RBC) folate was low in all patients. Initially, treatment with folinic acid (5-formyltetrahydrofolate; 5-FTHF) at 1 mg/kg per day resulted in CSF 5-MTHF normalization in patients 2 and 3; patient 1 was not reexamined. Total RBC folate increased in all patients (Table 1), associated with normalization of MCV and bone marrow morphology. Subsequently, patient 1 received 5-FTHF very irregularly. Three years later, he presented with focal epilepsy as his first clinical impairment. In patient 2, 5-FTHF treatment led to improved school performance, but persistent epilepsy. Patient 3 became transiently independent of anticonvulsive treatment and had no additional neurological symptoms. Later, irregular 5-FTHF treatment was associated with the recurrence of epileptic symptoms. The clinical and biochemical features of the patients were suggestive for an inherited disorder of folate transport or metabolism. By genomic sequencing, we excluded FOLR1 mutations known to cause congenital cerebral folate deficiency (CFD [MIM 613068]).8
Consistent with a recessive inheritance model, we performed homozygosity mapping by using genome-wide SNP analysis performed on the Affymetrix GeneChip platform. This and all subsequent examinations were performed in accordance with the ethical standards of the ethics committee of the University of Ulm. Written informed consent was obtained from both parents. Genome-wide SNP analysis revealed a short overlap on the long arm of chromosome 2 and a second common region on the long arm of chromosome 5 (nucleotide 79613515–nucleotide 82033415, Human Genome Assembly, NCBI Human Genome Browser, build 36 [hg18]). This region, spanning 2.42 Mb, contains 28 known genes, including DHFR, the gene encoding for dihydrofolate reductase. Another gene locus known to be associated with folate transport or metabolism was not found within the overlapping homozygous regions. Genomic sequencing of DHFR exons and neighboring intronic nucleotides was performed after genomic DNA PCR reactions (for primer sequences, see Table S1 available online). Sequence analysis revealed a homozygous DHFR mutation, c.458A>T, in exon 5 (RefSeq NM_000791.3) leading to a p.Asp153Val substitution in all siblings, thus affecting an amino acid that, according to the HomoloGene database, is highly conserved (Figure 1B). The mutation has been deposited to the NCBI dbSNP. Both parents (VI-2 and VI-3 in Figure 1) are heterozygous for the mutation. The mutation was not detected in 120 control samples.
DHFR possesses an α/β fold with a central 8-stranded sheet, flanked by four α helices (Figure 1C). DHF and NADPH cofactor are bound in a deep cleft (active site cleft) that partitions the structure into two subdomains. Two prominent loops, denoted “Met20” and “F-G loop,” emerge from one subdomain and form a lid that buries and positions NADPH's nicotinamid moiety in the cleft for hydride transfer. A variety of studies indicate that the dynamics of conformational states of these active site loops are critical for efficient catalysis.9 Asp153 is situated at the C-terminal end of the “F-G” loop, which connects β sheets F and G. It forms a hydrogen bond with the backbone nitrogen of Gly155, thereby stabilizing a helical turn that is involved in anchoring sheet G and loops “F-G” as well as “G-H” to the remainder of the structure. Val153 will lack this hydrogen bond as well as the negative charge. The mutation likely affects fold, conformational stability, and dynamics of the “F-G” loop and its closely associated Met20 loop, resulting in an enzyme with reduced catalytic efficiency.
Assuming DHFR deficiency, we analyzed folate metabolites in RBCs and plasma by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as previously described.10 This assay distinguishes 5-MTHF, FA (sum of FA and DHF, which converts to FA during assay), nonmethylTHF, and unsubstituted THF. Total RBC folate was obtained by addition of the separate folate fractions or by a chemiluminescence immunoassay. Before treatment, patient 1 had low RBC folates despite normal plasma folates (Table 1). During 5-FTHF treatment, RBC 5-MTHF remained low in all patients. In addition, nonmethylTHF and FA were present. Appreciable concentrations of nonmethyl folates in RBCs are normally observed only in individuals with the methylenetetrahydrofolate reductase (MTHFR [MIM 607093]) 677 TT but not the MTHFR 677 CC genotype.11 The latter genotype was found in all siblings. Therefore, the occurrence of non-methylTHF, which includes 5-FTHF, is probably caused by 5-FTHF treatment and not by DHFR deficiency. During 5-FTHF treatment, FA+DHF was remarkably high in RBCs, whereas THF remained low, which supports impaired DHF-to-THF conversion. DHF is the likely source for the measured FA+DHF, because no FA was used at the time and spontaneous formation of FA in vivo is believed to be virtually absent. Given that THF is the sole substrate for polyglutamate synthase and that polyglutamation is required for cellular storage of folates, the insufficient supply of THF from DHF may explain low total RBC folate levels.
Plasma and CSF homocysteine (Hcy) and methionine were normal. Apparently, in this situation, with normal nutritive folate supply and normal plasma folate, Hcy regulation by S-adenosylmethionine via MTHFR and cystathionine beta-synthase (CBS [MIM 618833] pathways is sufficient to compensate for dysfunctional DHFR. This is in contrast to the Hcy increase observed in patients treated with methotrexate (MTX), which represents a high-affinity substrate for DHFR. However, because MTX is inhibiting not only DHFR but also other folate-related transport proteins and enzymes (including MTHFR, dihydropteridine reductase, etc.), its effect on Hcy is certainly of a more complex nature than being due to only DHFR inhibition. Neopterin, biopterin, and neurotransmitter levels in the CSF and S-adenosyl-methionine and S-adenosyl-homocysteine concentrations in the plasma and in RBC were all within the normal range. Leukocyte global DNA methylation, determined by LC-MS/MS12 and expressed as the methyl-cytosine percentage of total cytosine, was also normal and not influenced by 5-FTHF substitution (data not shown).
To examine DHFR function directly, we studied the formation of THF from DHF in EBV-immortalized lymphoblastoid cells and fibroblasts from patients and controls. Cells were incubated with NADPH and DHF, then stabilized with mercaptoethanol and frozen. Cells were lysated by freeze-thaw cycles. After centrifugation, the supernatant was acidified, an internal standard ([13C5]-5-methenylTHF) was added, and proteins were removed. THF concentration was determined by LC-MS/MS.10 DHFR activity in lymphoblastoid cells was severely reduced to less than 10% of control levels in all patient cells, whereas cells of the heterozygous mother (VI-3 in Figure 1) exhibited DHFR activity close to the lower limit of the control range (Figure 2A). A confirmation of these results in fibroblasts was not possible because under the assay conditions that were used, DHFR activity in fibroblasts was very low, even in wild-type cells (data not shown). Nevertheless, in the context of the genetic and biochemical data, the results for DHFR activity obtained in lymphoblastoid cells have to be regarded as representative for the disorder.
Figure 2.
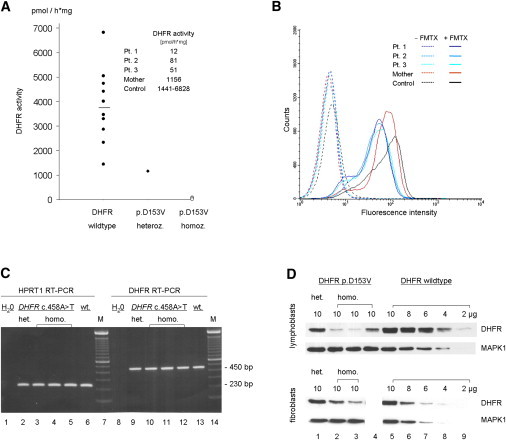
Functional Characteristics of the DHFR Mutant
(A) DHFR activity in lymphoblastoid cells of control individuals (DHFR wild-type), the patients' mother (heterozygous for DHFR p.Asp153Val = p.D153V), and the three siblings (homozygous for DHFR p.D153V) as measured by the formation of THF out of DHF per hour and mg protein. A DHFR Kit (Sigma) was used for the assay (for details, see text).
(B) Fluorescence-activated cell sorting (FACS) analysis of lymphoblastoid cells after incubation with 10 μM fluorescein-labeled methotrexate (FMTX; Invitrogen). Cells were additionally stained with 7-Aminoactinomycin D (7-AAD; 2,5 μg/ml) to sort vital cells for FMTX-FACS analysis.
(C) DHFR RT-PCR after isolation of mRNA from lymphoblastoid cells of the heterozygous mother (lanes 2 and 9), the three homozygous siblings (lanes 3–5 and 10–12; patients 1–3), and a control individual (DHFR wild-type) (lanes 6 and 13) revealed a single ∼450 bp cDNA fragment (agarose gel, 1.5%). cDNA was synthesized with the use of random hexamers and Superscript II RNase HT (Invitrogen) at 42°C for 50 min. DHFR PCR was performed in 35 cycles at 55°C (for primer sequences, see Table S1).
(D) DHFR protein expression in DHFR p.D153V heterozygous (lane 1, mother) and homozygous (lane 2, patient 2; lane 3, patient 3; lane 4, patient 1) lymphoblastoid cells (upper panel) and fibroblasts as compared to control cells (stepwise dilution of total blotted protein: lanes 5–9). In the lower panel, lane 4 is waste because fibroblasts from patient 1 were not available. After separation on a 15% SDS gel, proteins were blotted on an Immobilon-P membrane (Millipore). Primary antibodies (DHFR [E-18], MAPK1 [D-2], Santa Cruz Biotechnology) were diluted to a concentration of 1:200 and 1:10,000; secondary antibodies (anti-goat IgG-HRP, Santa Cruz Biotechnology; goat anti-mouse IgG [H+L]-HRP, BioRad), 1:2000 and 1:10,000, respectively. Blots were developed with the use of the Super Script West Pico Chemoluminescent Substrate Kit (Thermo Scientific).
To assess binding of the substrate to the mutated DHFR, we performed a fluorescence-based assay,13 showing that binding of fluorescein-labeled methotrexate (FMTX) to DHFR is severely reduced in lymphoblastoid cells of all three patients and slightly decreased in cells of the heterozygous mother (Figure 2B).
To assess whether the reduced DHFR activity was caused by an altered mRNA and protein expression, we performed RT-PCR and immunoblot analyses (Figures 2C and 2D). In contrast to the DHFR mRNA expression, which did not differ between patients and controls (Figure 2C), protein expression was reduced in mutated cells as compared to wild-type cells. Immunoblotting revealed reduced intact DHFR in heterozygous (∼70%) as well as in homozygous (∼20%–50%) lymphoblastoid cells (Figure 2D). Protein reduction was less severe in mutated fibroblasts (heterozygous ∼90%, homozygous 70%–80%). Probably, the mutation is responsible for the reduced protein expression by changing protein folding, binding to chaperones, or influencing the stability of other protein complexes involving DHFR.
However, both the DHFR activity and FMTX binding show an equally severe DHFR dysfunction, whereas the variable reduction of protein expression is only moderate (lymphoblasts) or mild (fibroblasts). Therefore, the interindividual protein variation probably reflects differences that are not intrinsic to DHFR but also depend on extrinsic factors that target DHFR directly or target its binding to stabilizing intracellular factors and, hence, contribute to the endogenous variability of cellular protein processing. With regard to the equally severe reduction of DHFR activity, the variation of the moderate protein reduction as a single factor cannot explain the phenotypic variability seen in patients with DHFR deficiency.
The causes underlying the different phenotype expression in individual patients remain obscure. Additional DHFR alterations that could possibly modify the effect of the DHFR p.Asp153Val mutation were excluded (data not shown). MTHFR activity in homozygous and heterozygous lymphoblastoid cells carrying the DHFR mutation, as determined by LC-MS/MS, did not differ from that in wild-type cells (data not shown).
Biopterin and folate metabolism appear to overlap at several points, and previous in vitro studies demonstrated that human dihydropteridine reductase (DHPR [MIM 612676]) may function in THF metabolism.14,15 We therefore analyzed DHPR activity in dried blood samples16 from the three siblings and found that it did not differ from that of controls (data not shown). However, DHPR activity in the peripheral blood may not adequately reflect its activity in the brain.17
To summarize, we have shown that the homozygous DHFR mutation p.Asp153Val causes DHFR deficiency and leads to a complex neurological and hematological disease in the absence of any malformation, including neural tube defects. Apparently, intact intracellular folate metabolite recycling through DHFR is of central importance for maintaining sufficient CSF and RBC folate levels, even in the presence of adequate nutritional folate supply and normal plasma folate. CSF folate levels were comparable to those of patients with congenital or acquired cerebral folate deficiency who present in infancy or early childhood with severe neurological symptoms.8,18 It seems possible that in CFD patients, not only the low CSF folate but also other changes associated with folate receptor dysfunction contribute to the early disease manifestation and the more severe phenotype.
In conclusion, we have described patients with inherited DHFR deficiency due to homozygous DHFR mutation. This condition merits consideration in young patients who present with unexplained macrocytic anemia, moderately severe developmental disorders, and epilepsy.
Acknowledgments
This work was supported in part by the Swiss National Science Foundation grant no. 3100A0-1199852/1 (to N.B.) and by DFG Sonderforschungsbereich 684 (to KPH). The authors thank Marlies Just from the Institute for Virology, University Hospital Ulm, for her continuous support by establishing EBV-immortalized lymphoblast cell lines and Marina Jans for excellent technical assistance.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Entrez Gene, http://www.ncbi.nlm.nih.gov/gene
NCBI dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
NCBI Nucleotide database, http://www.ncbi.nlm.nih.gov/nuccore
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Accession Numbers
The NCBI dbSNP accession number for the sequence reported in this paper is rs121913223.
References
- 1.Stanger O. Physiology of folic acid in health and disease. Curr. Drug Metab. 2002;3:211–223. doi: 10.2174/1389200024605163. [DOI] [PubMed] [Google Scholar]
- 2.Whitehead V.M. Acquired and inherited disorders of cobalamin and folate in children. Br. J. Haematol. 2006;134:125–136. doi: 10.1111/j.1365-2141.2006.06133.x. [DOI] [PubMed] [Google Scholar]
- 3.Tauro G.P., Danks D.M., Rowe P.B., Van der Weyden M.B., Schwarz M.A., Collins V.L., Neal B.W. Dihydrofolate reductase deficiency causing megaloblastic anemia in two families. N. Engl. J. Med. 1976;294:466–470. doi: 10.1056/NEJM197602262940903. [DOI] [PubMed] [Google Scholar]
- 4.Walters T.R. Congenital megaloblastic anemia responsive to N5-formyl tetrafolic acid administration. J. Pediatr. 1967;70:686–687. [Google Scholar]
- 5.Erbe R.W. Inborn errors of folate metabolism (second of two parts) N. Engl. J. Med. 1975;293:807–812. doi: 10.1056/NEJM197510162931606. [DOI] [PubMed] [Google Scholar]
- 6.Hoffbrand A.V., Tripp E., Jackson B.F., Luck W.E., Frater-Schroder M. Hereditary abnormal transcobalamin II previously diagnosed as congenital dihydrofolate reductase deficiency. N. Engl. J. Med. 1984;310:789–790. doi: 10.1056/nejm198403223101217. [DOI] [PubMed] [Google Scholar]
- 7.Blau N., Opladen T. Folates. In: Blau N., Duran M., Gibson K.M., editors. Laboratory Guide to the Methods in Biochemical Genetics. Springer-Verlag; Berlin, Heidelberg: 2008. pp. 717–724. [Google Scholar]
- 8.Cario H., Bode H., Debatin K.-M., Opladen T., Schwarz K. Congenital null mutations of the FOLR1 gene-a progressive neurologic disease and its treatment. Neurology. 2009;73:2127–2129. doi: 10.1212/WNL.0b013e3181c679df. [DOI] [PubMed] [Google Scholar]
- 9.Schnell J.R., Dyson H.J., Wright P.E. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 10.Smith D.E., Kok R.M., Teerlink T., Jakobs C., Smulders Y.M. Quantitative determination of erythrocyte folate vitamer distribution by liquid chromatography-tandem mass spectrometry. Clin. Chem. Lab. Med. 2006;44:450–459. doi: 10.1515/CCLM.2006.085. [DOI] [PubMed] [Google Scholar]
- 11.Smulders Y.M., Smith D.E., Kok R.M., Teerlink T., Gellekink H., Vaes W.H., Stehouwer C.D., Jakobs C. Red blood cell folate vitamer distribution in healthy subjects is determined by the methylenetetrahydrofolate reductase C677T polymorphism and by the total folate status. J. Nutr. Biochem. 2007;18:693–699. doi: 10.1016/j.jnutbio.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 12.Kok R.M., Smith D.E., Barto R., Spijkerman A.M., Teerlink T., Gellekink H.J., Jakobs C., Smulders Y.M. Global DNA methylation measured by liquid chromatography-tandem mass spectrometry: analytical technique, reference values and determinants in healthy subjects. Clin. Chem. Lab. Med. 2007;45:903–911. doi: 10.1515/CCLM.2007.137. [DOI] [PubMed] [Google Scholar]
- 13.Santiago Y., Chan E., Liu P.Q., Orlando S., Zhang L., Urnov F.D., Holmes M.C., Guschin D., Waite A., Miller J.C. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc. Natl. Acad. Sci. USA. 2008;105:5809–5814. doi: 10.1073/pnas.0800940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollock R.J., Kaufman S. Dihydropteridine reductase may function in tetrahydrofolate metabolism. J. Neurochem. 1978;31:115–123. doi: 10.1111/j.1471-4159.1978.tb12439.x. [DOI] [PubMed] [Google Scholar]
- 15.Smith I., Howells D.W., Hyland K. Pteridines and mono-amines: relevance to neurological damage. Postgrad. Med. J. 1986;62:113–123. doi: 10.1136/pgmj.62.724.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blau, N., and Thöny, B. (2008). Pterins and related enzymes. In N. Blau MD, K.M. Gibson, ed. (Berlin, Heidelberg: Laboratory Guide to the Methods in Biochemical Genetics. Springer-Verlag), pp. 665–702.
- 17.Kaufman S. Some metabolic relationships between biopterin and folate: implications for the “methyl trap hypothesis”. Neurochem. Res. 1991;16:1031–1036. doi: 10.1007/BF00965847. [DOI] [PubMed] [Google Scholar]
- 18.Ramaekers V.T., Rothenberg S.P., Sequeira J.M., Opladen T., Blau N., Quadros E.V., Selhub J. Autoantibodies to folate receptors in the cerebral folate deficiency syndrome. N. Engl. J. Med. 2005;352:1985–1991. doi: 10.1056/NEJMoa043160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.