

Abstract
An uncharacterized multisystemic mitochondrial cytopathy was diagnosed in two infants from consanguineous Palestinian kindred living in a single village. The most significant clinical findings were tubulopathy (hyperuricemia, metabolic alkalosis), pulmonary hypertension, and progressive renal failure in infancy (HUPRA syndrome). Analysis of the consanguineous pedigree suggested that the causative mutation is in the nuclear DNA. By using genome-wide SNP homozygosity analysis, we identified a homozygous identity-by-descent region on chromosome 19 and detected the pathogenic mutation c.1169A>G (p.Asp390Gly) in SARS2, encoding the mitochondrial seryl-tRNA synthetase. The same homozygous mutation was later identified in a third infant with HUPRA syndrome. The carrier rate of this mutation among inhabitants of this Palestinian isolate was found to be 1:15. The mature enzyme catalyzes the ligation of serine to two mitochondrial tRNA isoacceptors: tRNASerAGY and tRNASerUCN. Analysis of amino acylation of the two target tRNAs, extracted from immortalized peripheral lymphocytes derived from two patients, revealed that the p.Asp390Gly mutation significantly impacts on the acylation of tRNASerAGY but probably not that of tRNASerUCN. Marked decrease in the expression of the nonacylated transcript and the complete absence of the acylated tRNASerAGY suggest that this mutation leads to significant loss of function and that the uncharged transcripts undergo degradation.
Main Text
Progressive renal failure in infancy may result from a number of etiologies including congenital anomalies of the kidneys and urinary tract, ciliopathies (whether syndromic or isolated renal disease), and congenital nephrotic syndrome. Acquired multisystemic diseases, such as vasculitides, are uncommon at this age group. Mitochondrial cytopathies are among the rare syndromes that manifest in infancy and result in multiorgan disease. The most common renal phenotype of mitochondrial cytopathy is tubulopathy (Fanconi and Bartter-like syndromes as well as renal tubular acidosis), but tubulo-interstitial disease and nephrotic syndrome resulting from focal segmental glomerulosclerosis have also been described.1,2
We investigated three infants of Palestinian descent who presented with a multisystem disease in infancy. Two infants (F1:III-1 and F1:III-6, Figure 1) were included in the mapping of the gene, which, in its mutated form, is responsible for the disease. The third child (F2:II-2) was diagnosed after the completion of the genetic study. Infants F1:III-1 and F1:III-6 belong to a consanguineous kindred of Palestinian descent from the Greater Jerusalem area (Figure 1). The nonconsanguineous parents F2:I-1 and F2:I-2 are, to the best of their knowledge, unrelated to that kindred but live in the same village. All affected individuals were born prematurely and presented during infancy with a multiorgan disease including kidney involvement, manifesting with hyperuricemia, hypochloremic metabolic alkalosis, progressive renal failure, and primary pulmonary hypertension, collectively designated HUPRA syndrome. They all experienced feeding difficulties and developmental delay. The clinical characteristics of patients with the HUPRA syndrome are summarized in Table 1.
Figure 1.
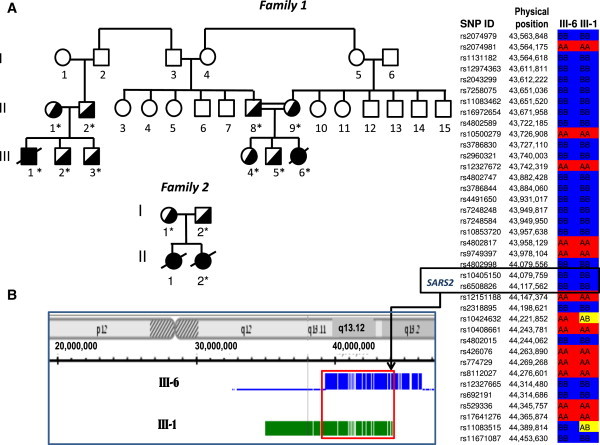
Implementation of SNP Microarray Analysis for the Identification of Mutation in SARS2
(A) Pedigrees of the families. DNA samples from individuals marked with asterisk were analyzed for the c.1169A>G mutation in SARS2.
(B) Homozygosity mapping of DNA derived from patients F1:III-1 and F1:III-6. Detection of a common homozygous region on chromosome 19 by Genotyping Console (left) and KinSNP (right) software. SARS2 is located at the very end of the shared fragment.
Table 1.
Clinical Characteristics of Patients with the HUPRA Syndrome
| Patient F1:III-1 | Patient F1:III-6 | Patient F2:II-2 | |
|---|---|---|---|
| Renal Disease | |||
| Hyperuricemia | + | + | + |
| Low FeUA | + | + | + |
| Salt wasting | + | + | + |
| Polyuria | + | ND | + |
| Elevated BUN (disproportionate to serum creatinine) | + | + | + |
| Hypochloremic metabolic alkalosis | + | + | + |
| Hypomagnesemia | + | + | + |
| High FeMg | + | + | + |
| Progressive renal failure | + | + | + |
| Extrarenal Manifestations | |||
| Prematurity | + | + | + |
| Pulmonary hypertension | + | + | + |
| Elevated serum lactate | + | + | + |
| Failure to thrive | + | + | + |
| Global developmental delay | + | + | + |
| Diabetes mellitus | + | ± | + |
| Pancytopenia | + | - | - |
Abbreviations: FeUA, fractional excretion of uric acid; FeMg, fractional excretion of magnesium; ND, not determined; plus and minus signs, stress hyperglycemia.
Patient F1:III-1
A 4-month-old baby boy, the first child of nonconsanguineous parents of Palestinian origin, was referred to our emergency department because of acute bronchiolitis and failure to thrive (FTT). Medical history was significant for premature birth at 34 weeks gestation, with an uneventful neonatal course. Routine blood tests revealed renal failure (serum creatinine, 0.99 mg/dl [age-appropriate normal range (N), 0.2–0.4 mg/dl]; BUN, 44 mg/dl [N, 5–18 mg/dl]; estimated creatinine clearance of 29 ml/min/1.73 m2 [N, 39–114 ml/min/1.73 m2]), anemia (Hb, 9 g/dl [N > 10.5 g/dl]), and hyperuricemia (13.8 mg/dl [N, 2.4–6.4 mg/dl]). Urinalysis demonstrated specific gravity of 1.010, mild proteinuria (protein/creatinine ratio, 1.26 mg/mg [N < 0.5 mg/mg]), and unrevealing cellular sediment. Blood pressure was mildly elevated (115/60 mmHg [95th percentile 115/60]) with borderline left ventricular hypertrophy on echocardiography. Abdominal ultrasound showed normal sized kidneys with hyperechogenic appearance. Because renal failure persisted after fluid replacement therapy and recovery from the respiratory illness, a kidney biopsy was performed. Renal histology showed immature glomeruli, consistent with his age, which were otherwise normal. Various tubulo-interstitial changes were noted including dedifferentiated, atrophic tubules with thick basement membrane or tubules that were completely denuded. Hyperplastic arteriolitis was found in the interstitium. Immunofluorescent staining of immunoglobulins and components of the complement system was negative.
During the ensuing weeks, he developed moderate polyuria (urine output 4 ml/Kg/d) with salt wasting resulting in hyponatremia (116 mEq/l [N, 133–146 mEq/l]), which required supplementation. Azotemia was always disproportionate to serum creatinine levels and hyperuricemia was associated with decreased fractional excretion of uric acid (FeUA, 5.7% [N > 7%]). Hypochloremic metabolic alkalosis persisted (32 mEq/L [N, 16–24 mEq/l]) in the face of moderately elevated blood lactate concentrations (5–7 mmole/l [N, 0.5–2.4 mmol/l]) and despite worsening renal function. Hypomagnesemia (0.9 mg/dl [N, 1.58–2.4 mg/dl]) and elevated fractional excretion of magnesium (FeMg, 12.8% [N < 5%]) were also noted. Additional characteristics of his multiorgan disease included thrombocytopenia and leukopenia with normal bone marrow examination, diabetes mellitus, hypotonia, global developmental delay, and ongoing FTT. Liver function tests were within normal limits and brain ultrasound was also normal. At 11 months of age, he developed hypoxia resulting from primary pulmonary hypertension (estimated pulmonary pressure 60 mmHg). He reached end-stage renal failure and chronic peritoneal dialysis was initiated.
Metabolic workup showed high blood and CSF lactate levels (10.2 mmol/l [N, 0.5–2.4 mmol/l] and 8.9 mmol/l [N, 0.8–2.2 mmol/l], respectively) and increased serum alanine concentration (1029 μM [N < 547 μM]). Muscle biopsy demonstrated significant variations in fiber size, consistent with type 2 fiber atrophy. Immunohistochemical staining for cytochtome C oxidase (COX) and succinate dehydrogenase (SDH) showed COX deficiency and normal SDH in a number of the muscle fibers. No ragged red fibers were identified on Gomori stain. On electron microscopy some fibers were shown to have enlarged mitochondria. Re-evaluation of the electron microscopy of the renal tissue demonstrated that a portion of the tubular epithelial cells contained markedly enlarged mitochondria with paracrystalline lesions (Figure 2), as previously reported in conjunction with mitochondrial cytopathies. His overall medical condition gradually deteriorated and he succumbed to multiorgan failure at 14 months of age. The parents subsequently had two healthy male offspring.
Figure 2.
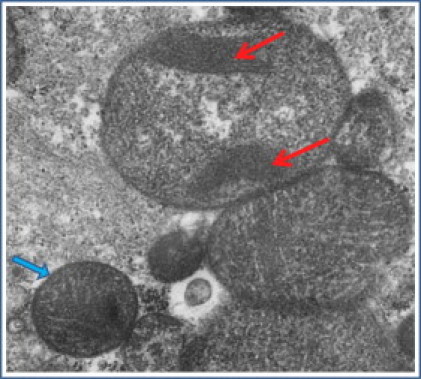
Electron Micrograph of Renal Tissue Obtained from Patient F1:III-1
Abnormal enlarged mitochondria with paracrystalline inclusions in a portion of the tubular epithelial cells (red arrows). Normal mitochondrion is marked in a blue arrow.
Patient F1:III-6
A 7-month-old baby girl was admitted to the hospital because of recurrent vomiting and FTT. She was the third child of healthy consanguineous parents of Palestinian descent. She was born prematurely at 27 weeks of gestation (birth weight 1080 g) and had an uneventful neonatal course, except for mild metabolic alkalosis (serum bicarbonate 26–29.5 mEq/L, venous pCO2 43–45 mmHg). Upon admission she weighed 3350 g (<3rd percentile). Vital signs were normal. Physical examination was unremarkable except for severe cachexia. Laboratory tests were significant for leukopenia (WBC, 2100/mm3) and anemia (Hb, 4.8 g/dl), salt wasting resulting in hyponatremia (124 mEq/L), renal failure (BUN, 87 mg/dl; creatinine, 1.14 mg/dl; estimated creatinine clearance of 22 ml/min/1.73 m2), and metabolic alkalosis (pH 7.48; bicarbonate, 30 mEq/L; pCO2, 39 mmHg) with elevated blood lactate (5.54 mmol/L). She was also noted to have marked hyperuricemia (26.8 mg/dl) associated with decreased FeUA (<5.0%) and increased urinary magnesium leak leading to hypomagnesemia (1.2 mg/dl).
During her hospital stay she developed systemic and pulmonary hypertension with biventricular hypertrophy, on echocardiography, and estimated pulmonary pressure of 67/25 that partially responded to sildenafil. Additionally, she suffered from global developmental delay. Her renal function had gradually deteriorated and she experienced occasional stress hyperglycemia (324 mg/dl). She died at 10 months of age because of respiratory insufficiency.
Patient F2:II-2
A female baby who was born at 27 weeks gestation to apparently nonconsanguineous parents of Palestinian origin had an uneventful neonatal course. One year earlier, her mother gave birth to a baby girl, also at 27 weeks gestation, who suffered from pulmonary hypertension and died of respiratory failure at the age of 39 days. She also had moderate renal failure. At 4 months of age, our patient was referred to another hospital because of poor feeding and failure to thrive, and she developed a sepsis-like episode with hemodynamic shock and severe metabolic acidosis requiring mechanical ventilation and vasopressor support. Blood and CSF cultures were negative. After recovering from the acute episode, she was found to have several medical problems. (1) Severe pulmonary hypertension and right ventricular hypertrophy, detected by echocardiography, which partially responded to sildenafil. (2) Renal failure (estimated creatinine clearance, 31 ml/min/1.73 m2) associated with normal sized hyperechogenic kidneys. (3) Electrolyte imbalance: hyperuricemia (14.1 mg/dl) with low FeUA, urinary magnesium leak resulting in hypomagnesemia (0.97 mg/dl), salt wasting leading to hyponatremia (122 mEq/l), metabolic alkalosis (bicarbonate 29.4 mEq/L) concurrent with high blood lactate (8 mmol/l). (4) Mild developmental delay associated with normal brain ultrasound. (5) Failure to thrive and vomiting. Metabolic workup showed high urinary lactate and Krebs cycle intermediates. Muscle biopsy was morphologically normal. Immunohistochemical staining demonstrated focal COX deficiency in 5% of the fibers. Enzymatic analysis of respiratory chain complexes revealed mildly reduced activity of all complexes except for complex II. These results are expressed as percentage of the mean control values: citrate synthase, 95%; NADH-ferricyanide reductase, 48%; NADH-cytochrome C reductase, 40%; succinate dehydrogenase, 115%; succinate-cytochrome C oxidoreductase, 96%; and cytochrome C oxidase, 57%.3 At 1 year of age she was suffering from failure to thrive despite feeding enriched formula provided via percutaneous gastrostomy tube, worsening renal failure, slow psychomotor development, severe pulmonary hypertension, and diabetes mellitus. One month later she died of refractory pulmonary hypertension.
Analysis of the pedigree suggested an autosomal-recessive disorder, associated with compromised energy production, and led to the assumption that this is a defect in nuclear DNA rather than in mitochondrial DNA (mtDNA).
The enzymatic activity of the mitochondrial respiratory chain complexes in mitochondria isolated from patients' muscle specimens were determined by standard spectrophotometric methods. The activities of complex I, III, and IV, normalized to control reference values, were reduced and only complex II activity was relatively preserved. Because complex II is the only complex that is encoded solely by the nuclear genome, this pattern suggested a defect in the synthesis of the mtDNA-encoded proteins. However, the ratio of mtDNA to nuclear DNA in patient F1:III-1 muscle was found to be within the normal range (70% of the age-matched control mean), which excludes mtDNA depletion.
Nuclear-encoded genes are central to a number of processes pertinent to the synthesis of mtDNA-encoded proteins (mitochondrial transcription, translation, and replication machinery) as well as assembly and stability of the mitochondrial respiratory complexes. The consanguinity in family 1 implied that the gene bearing a pathogenic mutation would be located within a homozygous identity-by-descent region. We undertook a genome-wide SNP homozygosity analysis of these two patients, aiming to identify a candidate gene.
Genomic DNA was extracted from blood samples of patients, their parents, and control individuals. All experiments involving DNA and cell cultures from patients, as well as DNA from parents and healthy controls, were performed after obtaining informed consent and were approved by the Shaare Zedek Medical Center Ethics Committee (number 78/09) and by the National Committee for Genetic Studies of the Israeli Ministry of Health (number 019-2010). Homozygosity mapping of DNA from patients F1:III-1 and F1:III-6 was performed on GeneChip Human Mapping 250K Nsp Array from Affymetrix. The experiments were performed according to protocols provided by the manufacturer. Quality control and data analysis (genotyping) were performed with the Genotyping Console. The genotypes were then analyzed with the KinSNP software. The children shared two homozygous fragments, each one of whom was more than 2 Mbp in length: 5 Mbp on chromosome 19 (from 39.3 Mbp [rs12611086] to 44.4 Mbp [rs11083515]; total 220 SNPs) and 2.1 Mbp on chromosome 4 (from 67.9 Mbp [rs11131698] to 70.0 Mbp [rs7677996]; total 147 SNPs). These two homozygous loci contained altogether approximately 150 genes.
Molecular diagnostic algorithms for the selection of candidate genes causing mitochondrial disorders have been formulated recently.4–7 Taking into account these considerations, we selected the following candidate genes, all located on chromosome 19, given that their products are imported into the mitochondria and associated with energy supply: COX6B1 (MIM 124089), COX7A1 (MIM 123995), MRPS12 (MIM 603021), and SARS2 (MIM 612804). Direct DNA sequencing of all the exons and the adjacent exon-intron boundaries was carried out and yielded only one mutation. The single homozygous mutation, c.1169A>G, resulting in p.Asp390Gly, was in exon 13 of SARS2, the gene encoding mitochondrial seryl-tRNA synthetase (NCBI Reference Sequence: NM_017827.3). The causative nature of this mutation was substantiated by the detection of the same missense mutation in a homozygous fashion in patient F2:II-2, who has an identical phenotype. The parents of patients F1:III-1, F1:III-6, and F2:II-2 were tested and, as expected, were found to be carriers of this mutation (Figure 3). Their four unaffected siblings were heterozygous for the mutation. Collectively, this mutation segregates with the disease in these families. The PolyPhen predictor defines this mutation as probably damaging. The mutation was absent in 212 chromosomes derived from healthy unrelated individuals of Palestinian descent. Targeted screening of this mutation was performed on anonymous DNA samples of individuals from the same village. This revealed a very high carrier rate of 1:15 (7 out of 103 samples tested).
Figure 3.
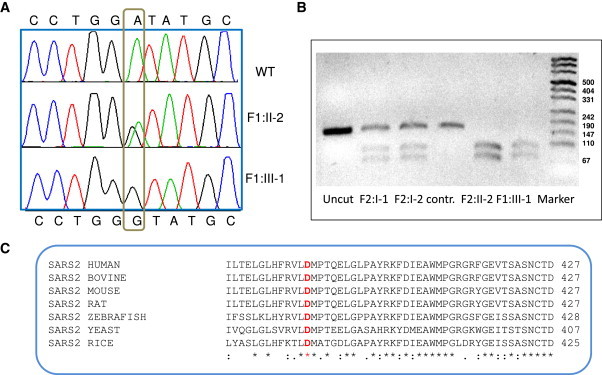
Mutation Analysis of DNA Samples from Patients, Carriers, and Normal Individual and Confirmation of the Conservation among Species of the Mutated Amino Acid in SARS2
(A) DNA sequence identifying the c.1169A>G mutation in SARS2.
(B) Restriction enzyme analysis with BsaJI: this mutation introduces a restriction site for this enzyme, which results in the digestion of the 166 bp oligonucleotide into 94 bp and 72 bp fragments.
(C) Multiple sequence alignment of SARS2 in various species with ClustalW program. In red: the amino acid corresponding to the mutation in human DNA c.1169A>G (p.Asp390Gly).
The eukaryotic mitochondria presumably evolved from the fusion of an oxidative bacterium with a glycolytic proto-eukaryotic cell. Its genetic machinery includes several nonuniversal codons and the translation system uses 22 tRNAs encoded by mtDNA. Eighteen tRNAs correspond to one amino acid each. Two additional amino acid residues (leucine and serine) acylate two tRNA isoacceptors each. A set of 20 aminoacyl-tRNA synthetases (ARSs), each one corresponding to a single amino acid, is encoded in the nucleus and the product is imported into the mitochondria. Only three ARSs (not including SARS) are found both in the cytosol and the mitochondria. Aminoacyl-tRNA synthetases provide the specific attachment of amino acids to the 3′-ends of their cognate tRNA. They are subdivided into two groups based on their protein structural characteristics. SARS2 is a homodimeric enzyme whose catalytic domain is composed of the antiparallel β sheet architecture with three short conserved motifs, belonging to the class II ARS. The mutated aspartic acid residue is positioned at the C-terminal globular domain and located in the second of eight β-strands that assemble the active site.8
Mutations in mitochondrial ARSs resulting in decreased aminoacylation by the corresponding amino acid are expected to adversely affect mitochondrial translation systems and lead to derangements in the synthesis of mitochondrial proteins and consequently in energy supply. Thus far, mutations in three mitochondrial ARS-encoding genes have been found to be pathogenic: DARS2 (MIM 610956), resulting in leukoencephalopathy with brain stem and spinal cord involvement associated with elevated lactate concentrations (LBSL; MIM 611105);9 RARS2 (MIM 611524) leading to infantile encephalopathy, predominantly affecting the cerebellum (MIM 611523);10 and YARS2 (MIM 610957) manifesting as myopathy, lactic acidosis, and sideroblastic anemia (MLASA; MIM 613561).11
The major determinant of specific recognition by SARS in prokaryotic, eukaryotic, and archaeal systems is the characteristic long variable arm of the serine isoacceptors. Of note, SARS2, which represents the mitochondrial seryl-tRNA synthetase, provides serine aminoacylation to two mitochondrial tRNAs. In both of them this elongated variable arm is absent. In addition, tRNASerAGY lacks the entire D domain12 whereas the tRNASerUCN is missing a single nucleotide in the connector between the acceptor and the D-arm and has an unusual cloverleaf configuration with an extended anticodon stem13,14 (Figure 4). Moreover, these two tRNAs do not share sequence or structural similarities.8 It is therefore assumed that the recognition elements that define the specificity of SARS2 to two isoacceptors are different.15
Figure 4.
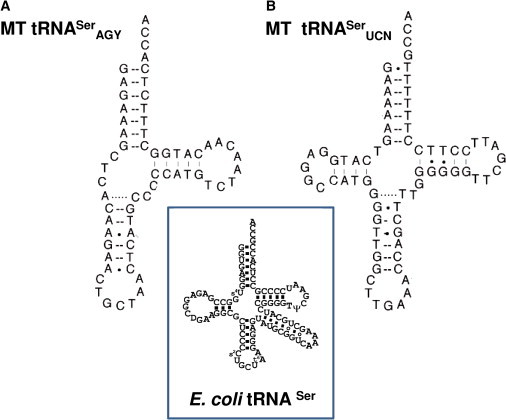
Comparison of the Secondary Structures of E. coli tRNASer with Two Human Mitochondrial tRNAsSer
Human tRNASerAGY (A) and tRNASerUCN (B) are shown.
The impact of structural dissimilarities between the two mitochondrial serine isoacceptors on the binding and acylation mechanisms of SARS2 was confirmed, in part, by the aminoacylation assay of the protein product of SARS2 containing specific mutations performed by Chimnaronk et al.8 The authors demonstrated that some mutants lead to a decrease in activity with respect to one of the isoacceptors without having any effect on the second one. These experiments confirm the dual-mode recognition of two substrate tRNAs. Unfortunately, this analysis does not include mutations in the area of the SARS2 corresponding to the A2 β-strand, which is relevant to our cases. In order to evaluate the pathogenic effect of the c.1169A>G SARS2 mutation on tRNAsSer acylation, we used EBV-immortalized peripheral lymphocyte cultures derived from patients. Total RNA was isolated under acidic conditions in order to preserve aminoacylation and was resolved on an acidic gel as described by Varshney16 and modified by Chernyakov.17 Aminoacylated RNAs were separated by electrophoresis on a 6.5% polyacrylamide gel containing 8 M urea. The aminoacylated tRNA migrates slower than its corresponding deacylated species. After separation, RNA was transferred to a membrane. Specific tRNAs were visualized with oligonucleotide DNA probes for MT tRNASerAGY and tRNASerUCN labeled with 32P at the 5′ end via standard northern blotting techniques (Figure 5). The signals were quantified, normalized to the signal of MT-tRNALeu (loading control), and expressed as a ratio compared to tRNASerAGY or tRNASerUCN signals in normal lymphocyte culture. Normalization to probes of genomic tRNASer and genomic tRNALeu yielded similar results (data not shown). The amount of total tRNASerAGY in patient's cells was reduced to 10%–20% of the control. The residual pool of this tRNA was nonacylated. In contrast, the total amount of tRNASerUCN was nearly identical to the control. We conclude that the c.1169A>G mutation in SARS2 significantly impairs the ability of the enzyme to acylate tRNASerAGY. These data support the concept that uncharged tRNA undergoes degradation and that mutations in ARS decrease the amount of not only the charged product but also of the uncharged substrate.10 Experimental data show that failure of aminoacylation leads to degradation and decrease in the steady-state levels of mutated tRNAs.18,19 If we extrapolate these observations to tRNASerUCN, we can conclude that the level of its acylation was not significantly decreased. However, because separation of the charged from the uncharged form of tRNASerUCN could not be achieved because of similar mobility of these two species, the possibility that in the patient's sample this isoacceptor remained mostly in the uncharged form cannot be entirely excluded.
Figure 5.
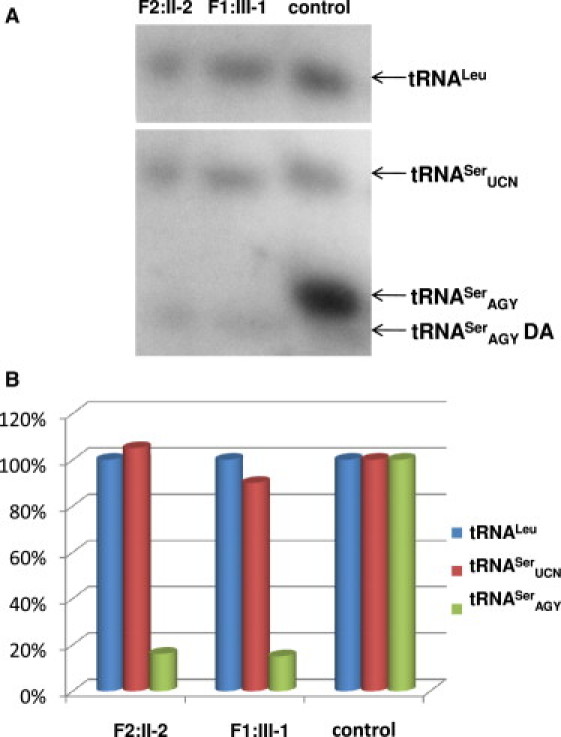
Quantitative Analysis of tRNASerAGY and tRNASerUCN Amino Acylation in EBV-Transformed Lymphocyte Cultures
(A) Northern blot: equal amounts (2 μg) of total RNA purified from normal and patient-derived lymphocyte lines were separated at 4°C on an acidic 6.5% polyacrylamide-7 M urea gel. Hybridization with 32P-labeled probes for tRNASerAGY and tRNASerUCN. The blots were then stripped and rehybridized with probes for MT-tRNALeu. One representative northern hybridization, out of three experiments, is presented. DA denotes deacylated tRNA.
(B) Histogram presentation of the data from (A). The intensity of hybridization signals was normalized by the signal of loading control MT-tRNALeu and is presented as percentage of the signal of the corresponding probe in the control.
We speculate that other pathogenic mutations in SARS2 may result in inhibition of acylation of tRNASerUCN causing a different phenotype. For instance, sensorineural deafness has been shown to be caused by mutations in the mitochondrial gene encoding tRNASerUCN.20 However, the consequences of MT mutations are hardly predictable. Indeed, mutations in the mitochondrial gene encoding tRNASerAGY, which theoretically prevent its ability to be acylated by serine, similar to the effect of the p.Asp390Gly mutation in SARS2, were reported to result in a completely different phenotype consisting primarily of a neurological disease affecting the central nervous system (basal ganglia and cerebral atrophy) and the proximal muscles.21
The most striking features of the phenotype resulting from a homozygous missense mutation in SARS2 were prematurity, progressive renal disease, and primary pulmonary hypertension. Feeding problems, resulting in failure to thrive and developmental delay, were also noted without obvious morphological alterations in the brain after brain ultrasound. The most common renal phenotype attributed to mitochondrial cytopathy is the Fanconi syndrome. Our patients presented with a tubulopathy but without perturbations in proximal tubular functions, which often manifest as glycosuria, phosphaturia, metabolic acidosis, or generalized aminoaciduria. Reduced energy production may account for impaired tubular function especially in the thick ascending limb of the loop of Henle, which is known to have a high energy requirement, primarily needed to support the Na-K-ATPase cotransporter activity, and is therefore susceptible to anoxic damage. Our patients showed features of the thick ascending limb dysfunction that resemble those observed in patients treated with excessive doses of loop diuretics, known to inhibit the luminal Na-K-2Cl cotransporter. They had salt wasting resulting in hyponatremia, urinary magnesium leak leading to hypomagnesemia, and decreased concentration capacity associated with polyuria and significantly elevated blood urea levels disproportionate to the creatinine concentrations. Marked metabolic alkalosis, despite decreasing renal function, may have resulted from the combination of hypochloremia and relative volume depletion, a phenomenon known as contraction alkalosis. The latter may also be responsible for the hyperuricemia with decreased urinary fractional excretion of uric acid. The accepted mechanism underlying hyperuricemia within the context of pulmonary hypertension is increased production of uric acid in ischemic/hypoxic cardiac tissue.22 This would result in normal, if not increased, fractional excretion of uric acid. The markedly decreased FeUA observed in our patients supports a primary renal tubular defect as the main cause of hyperuricemia. The clinical signs of tubulopathy were supported by the tubular abnormalities observed under electron microscopy. The combination of urinary concentration defect, salt wasting, hyperuricemia, and renal insufficiency was previously described in two familial cases with a mitochondrial point mutation.2 They also shared similar histological findings. It is quite intriguing that morphological alterations were not detected by brain ultrasound despite the fact that the brain is a tissue with high energy requirement and that SARS2 mRNA is highly expressed in this organ.
When considering the various possible functions of SARS2, it should be noted that according to all the bioinformatic annotations, it could participate in the biosynthesis of selenocysteinyl-tRNA in the mitochondria (see UniProt, UCSC Browser, GeneCards). We assume that this process is unlikely to occur, because the human mitochondrial genome does not encode selenoproteins and, furthermore, UGA used by selenocysteinyl-tRNA codes for tryptophan in the human mitochondrial genome.23 We therefore disregarded this possibility in our experiments.
Quantitative analysis of the northern blot revealed that the total amount of tRNASerAGY was significantly reduced to 10%–20% of the control, whereas the acylated form was undetectable. The subunits of all three MT-encoded complexes of the respiratory chain contain the AGY codons, but in varying proportions. 11 out of 13 MT protein-encoding genes possess these codons and can therefore be truncated during expression. Absence of the charged tRNASerAGY may alter the balance between the nuclear-encoded and MT-encoded subunits of these complexes. Given the residual activity of the mutated SARS2, we speculate that it maintains most tissues above their necessary threshold but fails to do so in tissues with high energy requirement. This phenomenon has been described in patients with neuronal mitochondrial cytopathy resulting from mutations in DARS2, in whom the overall mitochondrial function in patients' fibroblasts and lymphoblasts were seemingly normal in the face of severe neuronal disease.9 Accordingly, mutations in SARS2 resulting in a significant decrease of the respiratory chain activity may be critical for the thick ascending limb, which has a high density of mitochondria needed to provide the excessive energy level required for the maintenance of the various transporters. This concept, in addition to explaining the tissue specificity of the disease, may also account for the disparity in the activity of the respiratory complexes in the muscle biopsies of our patients. Further studies are warranted in order to confirm this hypothesis.
We describe three infants diagnosed with an uncharacterized multisystem fatal mitochondrial cytopathy, which we named HUPRA syndrome (hyperuricemia, pulmonary hypertension, renal failure in infancy, and alkalosis). The genetic basis underlying this recessively inherited clinical entity is a homozygous founder mutation in SARS2 that is highly prevalent among the inhabitants of a Palestinian village. This information will enable the implementation of targeted screening and the provision of premarital and prenatal diagnosis to couples at risk. In fact, we have recently analyzed a chorionic-villus sample derived from F2:I-1 and found that she carries an unaffected fetus. The combination of signs of thick ascending limb dysfunction (primarily hyperuricemia and alkalosis) with progressive renal failure in infancy should raise the possibility of mitochondrial cytopathy.
Acknowledgments
The authors are grateful to Rachel Beeri for assistance with tissue cultures, to Zvi Neeman for the electron micrograph of the kidney biopsy, and to Nina Entelis for insightful advice.
Web Resources
The URLs for data presented herein are as follows:
Basic Local Alignment Search Tool, http://blast.ncbi.nlm.nih.gov/Blast.cgi/
ClustalW multiple sequence alignment, http://www.ebi.ac.uk/Tools/clustalw/
GeneCards, http://www.genecards.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
Primer-blast tool for finding specific primers, http://www.ncbi.nlm.nih.gov/tools/primer-blast/
UCSC Browser, http://genome.ucsc.edu/
UniProt, http://www.uniprot.org/
References
- 1.Hall A.M., Unwin R.J., Hanna M.G., Duchen M.R. Renal function and mitochondrial cytopathy (MC): More questions than answers? Q. J. Med. 2008;101:755–766. doi: 10.1093/qjmed/hcn060. [DOI] [PubMed] [Google Scholar]
- 2.Tzen C.Y., Tsai J.D., Wu T.Y., Chen B.F., Chen M.L., Lin S.P., Chen S.C. Tubulointerstitial nephritis associated with a novel mitochondrial point mutation. Kidney Int. 2001;59:846–854. doi: 10.1046/j.1523-1755.2001.059003846.x. [DOI] [PubMed] [Google Scholar]
- 3.Saada A., Shaag A., Elpeleg O. mtDNA depletion myopathy: Elucidation of the tissue specificity in the mitochondrial thymidine kinase (TK2) deficiency. Mol. Genet. Metab. 2003;79:1–5. doi: 10.1016/s1096-7192(03)00063-5. [DOI] [PubMed] [Google Scholar]
- 4.Wong L.J., Scaglia F., Graham B.H., Craigen W.J. Current molecular diagnostic algorithm for mitochondrial disorders. Mol. Genet. Metab. 2010;100:111–117. doi: 10.1016/j.ymgme.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 5.Finsterer J., Harbo H.F., Baets J., Van Broeckhoven C., Di Donato S., Fontaine B., De Jonghe P., Lossos A., Lynch T., Mariotti C. European Federation of Neurological Sciences. EFNS guidelines on the molecular diagnosis of mitochondrial disorders. Eur. J. Neurol. 2009;16:1255–1264. doi: 10.1111/j.1468-1331.2009.02811.x. [DOI] [PubMed] [Google Scholar]
- 6.Robinson B.H. Lactic acidemia and mitochondrial disease. Mol. Genet. Metab. 2006;89:3–13. doi: 10.1016/j.ymgme.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 7.Zhu X., Peng X., Guan M.X., Yan Q. Pathogenic mutations of nuclear genes associated with mitochondrial disorders. Acta Biochim. Biophys. Sin. (Shanghai) 2009;41:179–187. doi: 10.1093/abbs/gmn021. [DOI] [PubMed] [Google Scholar]
- 8.Chimnaronk S., Gravers Jeppesen M., Suzuki T., Nyborg J., Watanabe K. Dual-mode recognition of noncanonical tRNAs(Ser) by seryl-tRNA synthetase in mammalian mitochondria. EMBO J. 2005;24:3369–3379. doi: 10.1038/sj.emboj.7600811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheper G.C., van der Klok T., van Andel R.J., van Berkel C.G., Sissler M., Smet J., Muravina T.I., Serkov S.V., Uziel G., Bugiani M. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 10.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riley L.G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., Lim S.C., Thorburn D., Ryan M.T., Giegé R. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am. J. Hum. Genet. 2010;87:52–59. doi: 10.1016/j.ajhg.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinberg S., Gautheret D., Cedergren R. Fitting the structurally diverse animal mitochondrial tRNAs(Ser) to common three-dimensional constraints. J. Mol. Biol. 1994;236:982–989. doi: 10.1016/0022-2836(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 13.Yokogawa T., Shimada N., Takeuchi N., Benkowski L., Suzuki T., Omori A., Ueda T., Nishikawa K., Spremulli L.L., Watanabe K. Characterization and tRNA recognition of mammalian mitochondrial seryl-tRNA synthetase. J. Biol. Chem. 2000;275:19913–19920. doi: 10.1074/jbc.M908473199. [DOI] [PubMed] [Google Scholar]
- 14.Helm M., Brulé H., Friede D., Giegé R., Pütz D., Florentz C. Search for characteristic structural features of mammalian mitochondrial tRNAs. RNA. 2000;6:1356–1379. doi: 10.1017/s1355838200001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimada N., Suzuki T., Watanabe K. Dual mode recognition of two isoacceptor tRNAs by mammalian mitochondrial seryl-tRNA synthetase. J. Biol. Chem. 2001;276:46770–46778. doi: 10.1074/jbc.M105150200. [DOI] [PubMed] [Google Scholar]
- 16.Varshney U., Lee C.P., RajBhandary U.L. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J. Biol. Chem. 1991;266:24712–24718. [PubMed] [Google Scholar]
- 17.Chernyakov I., Baker M.A., Grayhack E.J., Phizicky E.M. Identification and analysis of tRNAs that are degraded in Saccharomyces cerevisiae due to lack of modifications. Methods Enzymol. 2008;449:221–237. doi: 10.1016/S0076-6879(08)02411-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibbons W.J., Jr., Yan Q., Li R., Li X., Guan M.X. Genomic organization, expression, and subcellular localization of mouse mitochondrial seryl-tRNA synthetase. Biochem. Biophys. Res. Commun. 2004;317:774–778. doi: 10.1016/j.bbrc.2004.03.113. [DOI] [PubMed] [Google Scholar]
- 19.Bacman S.R., Atencio D.P., Moraes C.T. Decreased mitochondrial tRNALys steady-state levels and aminoacylation are associated with the pathogenic G8313A mitochondrial DNA mutation. Biochem. J. 2003;374:131–136. doi: 10.1042/BJ20030222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hutchin T.P., Navarro-Coy N.C., Van Camp G., Tiranti V., Zeviani M., Schuelke M., Jaksch M., Newton V., Mueller R.F. Multiple origins of the mtDNA 7472insC mutation associated with hearing loss and neurological dysfunction. Eur. J. Hum. Genet. 2001;9:385–387. doi: 10.1038/sj.ejhg.5200640. [DOI] [PubMed] [Google Scholar]
- 21.Wong L.J.C., Yim D., Bai R.K., Kwon H., Vacek M.M., Zane J., Hoppel C.L., Kerr D.S. A novel mutation in the mitochondrial tRNA(Ser(AGY)) gene associated with mitochondrial myopathy, encephalopathy, and complex I deficiency. J. Med. Genet. 2006;43:e46. doi: 10.1136/jmg.2005.040626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bendayan D., Shitrit D., Ygla M., Huerta M., Fink G., Kramer M.R. Hyperuricemia as a prognostic factor in pulmonary arterial hypertension. Respir. Med. 2003;97:130–133. doi: 10.1053/rmed.2003.1440. [DOI] [PubMed] [Google Scholar]
- 23.Papp L.V., Wang J., Kennedy D., Boucher D., Zhang Y., Gladyshev V.N., Singh R.N., Khanna K.K. Functional characterization of alternatively spliced human SECISBP2 transcript variants. Nucleic Acids Res. 2008;36:7192–7206. doi: 10.1093/nar/gkn829. [DOI] [PMC free article] [PubMed] [Google Scholar]