

Abstract
Neuromuscular junctions (NMJs) are synapses that transmit impulses from motor neurons to skeletal muscle fibers leading to muscle contraction. Study of hereditary disorders of neuromuscular transmission, termed congenital myasthenic syndromes (CMS), has helped elucidate fundamental processes influencing development and function of the nerve-muscle synapse. Using genetic linkage, we find 18 different biallelic mutations in the gene encoding glutamine-fructose-6-phosphate transaminase 1 (GFPT1) in 13 unrelated families with an autosomal recessive CMS. Consistent with these data, downregulation of the GFPT1 ortholog gfpt1 in zebrafish embryos altered muscle fiber morphology and impaired neuromuscular junction development. GFPT1 is the key enzyme of the hexosamine pathway yielding the amino sugar UDP-N-acetylglucosamine, an essential substrate for protein glycosylation. Our findings provide further impetus to study the glycobiology of NMJ and synapses in general.
Introduction
Efficient signal transmission from the motor neuron to a skeletal muscle fiber at the neuromuscular junction (NMJ) is a prerequisite for muscle contraction.1 When an action potential reaches the presynaptic button, the neurotransmitter acetylcholine (ACh) is released into the synaptic cleft. Binding of ACh to nicotinic acetylcholine receptors (AChR) at the motor end plate of the muscle fiber induces opening of the cation selective ion channels leading to depolarization of the muscle fiber, the release of calcium ions from the sarcoplasmic reticulum, and ultimately muscle contraction. A number of different neurological conditions can result from impaired neuromuscular transmission, varying from poisoning with botulinum and snake venom toxins through the autoimmune mediated NMJ disorders such as myasthenia gravis and the Lambert-Eaton myasthenic syndrome to the hereditary congenital myasthenic syndromes (CMS; for a discussion of genetic heterogeneity of CMS, see MIM 608931).2,3 These conditions may lead to generalized paralysis and respiratory failure and, if untreated, can be life threatening. The neuromuscular junction is probably the most studied synapse, but many aspects of its formation, maturation, stabilization, and functional efficacy remain incompletely understood.4
During the last two decades, research into the molecular causes of CMS has led to the identification of mutations in several genes and has greatly influenced established or inferred concepts of NMJ pathophysiology.3 Here, we used positional cloning to determine the underlying defect in a rare autosomal recessive CMS that is characterized by a limb-girdle pattern of muscle weakness combined with the presence of tubular aggregates on muscle biopsies (myasthenia, limb-girdle, with tubular aggregates [MIM 610542]).5,6 Individuals with this condition have a recognizable pattern of weakness of shoulder and pelvic girdle muscles (Figure 1), sparing of ocular or facial muscles, neurophysiologic signs of NMJ dysfunction (decrement of the muscle action potential with repetitive nerve stimulation), and a good treatment response to acetylcholinesterase (AChE) inhibitors. Genetic evaluation of these families revealed pedigrees typical of an autosomal recessive trait, but the underlying genetic defect had not yet been identified. We localized and refined the genetic interval on chromosome 2 by homozygosity mapping, identified mutations in the gene encoding glutamine-fructose-6-phosphate transaminase 1 (GFPT1) as a so far unrecognized cause of CMS, and recapitulated human pathology in zebrafish morphants. GFPT1 catalyzes the transfer of an amino group from glutamine onto fructose-6-phosphate, yielding glucosamine-6-phosphate (GlcN-6-P) and glutamate. This transamidase reaction has been identified as the first and rate-limiting step of the hexosamine biosynthesis pathway, which is the obligatory source of essential amino sugars for the synthesis of glycoproteins, glycolipids, and proteoglycans (Figure S1, available online).7 As many key NMJ proteins are glycosylated8 our data will pave the way for new studies on the functional integrity of the neuromuscular synapse.
Figure 1.
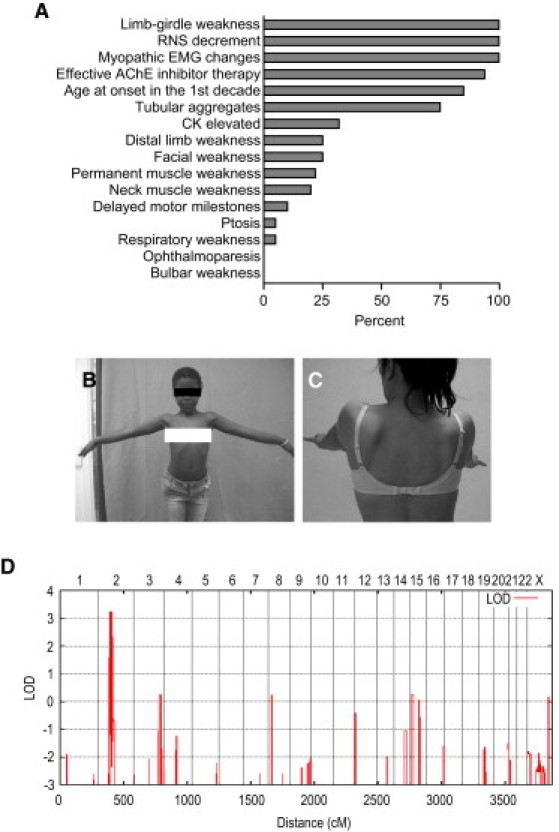
Phenotype of CMS Patients in this Study and Linkage to Chromosome 2p12-p15
(A) Relative frequency of signs and symptoms in our patient cohort (n = 23). The following abbreviations are used: RNS, repetitive nerve stimulation; EMG, electromyography; CK, creatine kinase.
(B) Patient LGM10.4 showing proximal muscle weakness and hyperlordotic posture at prolonged arm elevation.
(C) Patient LGM12.3 showing scapular winging.
(D) Genome-wide linkage analysis of family LGM3. Analysis was performed with MERLIN with the assumption of autosomal-recessive inheritance with complete penetrance and a disease allele frequency of 0.001.
Subjects and Methods
Patients
Pedigrees and countries of origin of 16 CMS families are shown in Figure S2 and a synopsis of clinical findings is shown in Figure 1. Five families, LGM3, LGM4, LGM13, LGM15, and LGM16, have been published earlier.9,10 Venous blood samples were obtained from the patients as well as from their unaffected parents and siblings. Genomic DNA was isolated with the Wizard Genomic DNA Purification Kit (Promega, Mannheim, Germany) according to the manufacturer's recommendations. All genes that are known to cause CMS when mutated (CHAT [MIM 118490], CHRNA1 [MIM 100690], CHRNB1 [MIM 100710], CHRND [MIM 100720], CHRNE [MIM 100725], COLQ [MIM 603033], DOK7 [MIM 610285], RAPSN [MIM 601592], MUSK [MIM 601296], SCN4A [MIM 603967], LAMB2 [MIM 150325], and AGRN [MIM 103320]) and two functional candidate genes (CNTN1 [MIM 600016] and AChE [MIM 100740]) were excluded by direct sequencing or by haplotype analysis.11 All studies were carried out with informed consent of the probands or their legal guardians and were approved by institutional ethics review boards in Munich and Newcastle.
Reagents
If not stated otherwise, all chemicals were obtained from Sigma-Aldrich (Munich, Germany). Sequences of oligonucleotide primers are available upon request.
Linkage Analysis
In order to identify the disease locus, we performed homozygosity mapping12 by genome-wide genotyping of SNP for family LGM3 using the Illumina 300K chip (Illumina, San Diego, CA). Multipoint linkage analysis was performed with MERLIN13 assuming autosomal recessive inheritance, a frequency of the deleterious allele of 0.001, and complete penetrance. For targeted linkage and homozygosity analysis of chromosome 2p12-p15, DNA samples from families LGM1, LGM2, LGM5, LGM6, LGM7, LGM8, LGM10, LGM11, and LGM12 were analyzed with short tandem repeat (STR) markers. Twenty-three markers were retrieved from the National Center for Biotechnology Information UniSTS database, and four new polymorphisms were identified by using the Repeat-Masker program. PCR primers for new STRs were designed by the Primer3 program. Sense primers were labeled with FAM fluorophores (MWG Biotech, Ebersberg, Germany) for detection on an ABI 377 DNA sequencer (Applied Biosystems, Foster City, CA). Multipoint LOD scores were calculated with the GeneHunter v2.1r5 program14 in the EasyLinkage software package.15 The genomic localization of the markers was derived from the Marshfield map and the University of California Santa Cruz (UCSC) human genome assembly.
Candidate-Gene Analysis
For selection of candidate genes in the linkage region on chromosome 2p12-p15, we used the February 2009 freeze of the UCSC human genome assembly. Forty-six known genes and uncharacterized transcripts were located in the 5.92 Mb interval between STR markers D2S296 and D2S2977 (Table S1). We ranked the annotated sequences with respect to neuromuscular junction and skeletal muscle biology and disease. We used information from public databases and characterized proteins in silico by BLAST alignments to protein database entries and comparison of domain composition and organization with the SMART algorithm. We also investigated positional candidate genes for structural and functional homology with the known proteins involved in CMS and related disorders. Sixteen genes were screened for mutations (Table S1). Primer pairs for each exon, including flanking intron sequences, were designed from genomic sequence with Primer3 (for GFPT1: genomic DNA AC114772.5; mRNA, NM_002056.2 and AF334737.1 [muscle isoform16]; protein, NP_002047.2 and AAK15342.1 [muscle isoform16]). PCR products were directly sequenced with ABI PRISM 3730 DNA Analyzer and BigDye Terminator Cycle Sequencing Kit version 1.1 according to the protocols of the manufacturer (Applied Biosystems). Sequence variants were identified by visual inspection of electropherograms. Control samples were screened by restriction enzyme digestion or pyrosequencing. PCR primers for pyrosequencing were designed with Primer SNP Design version 1.01 software (Biotage, Uppsala, Sweden). Pyrosequencing was carried out as defined by the supplier on a PSQ HS96A instrument (Biotage). Samples were analyzed with the PSQ HS96A version 1.2 software (Biotage).
Plasmid Constructs
Human GFPT1 cDNA was amplified from human skeletal muscle cDNA and inserted into the EcoRI and NotI sites of the pCMV-Myc vector (Clontech, Mountain View, CA) allowing expression of human GFPT1 with an N-terminal Myc-tag. For enzyme activity assays, we cloned the GFPT1 constructs into the pEGFP-N1 plasmid (Clonetech) and simultaneously removed the EGFP open reading frame to obtain untagged GFPT1. The mutants p.Thr15Ala, p.Asp43Val, p.Arg111Cys, p.Ile121Thr, p.Asp348Tyr, p.Arg434His, and p.Met492Thr were generated by site-directed mutagenesis with mismatch primers.17 Orientation of the inserts and absence of PCR-induced mutations were verified by sequencing.
Cell Culture
HEK293, C2C12, and SW13 cells were cultured in Dulbecco's Modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum. Primary human myoblasts were isolated as previously described.18 Myoblasts from patients with GFPT1 mutations and control myoblasts were obtained from the Medical Research Council (MRC) Centre for Neuromuscular Diseases Biobank, Newcastle, UK, and the Muscle Tissue Culture Collection, Friedrich-Baur-Institut, Munich, Germany, and grown in skeletal muscle growth medium (PromoCell, Heidelberg, Germany). For differentiation of myoblasts to myotubes, the growth medium was removed at a cell density of around 80% and cells were incubated in DMEM supplemented with 2% horse serum for 7 days.
Transfection of HEK293, SW13, and C2C12 Cells
HEK293 cells were transfected with 3–6 μg of wild-type and mutated GFPT1 plasmid DNA (with and without N-terminal Myc tag) with Polyplus jetPEI transfection reagent (Biomol, Hamburg, Germany) according to the manufacturer's recommendations. Transfection of SW13 cells was carried out with FuGene6 transfection reagent (Roche Diagnostics, Mannheim, Germany) and transfection of C2C12 cells was performed with Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's recommendations. Cells were analyzed 48 hr after transfection by immunoblot or immunofluorescence staining.
Preparation of Cell and Muscle Lysates for Protein Analysis
Cultured cells were scraped off the plastic dish in PBS containing Complete Protease Inhibitor Cocktail Tablets (Roche Diagnostics) and pelleted by centrifugation (14,000 rpm, 5 min, 4°C). Subsequently, cell pellets were homogenized in lysis buffer (10 mM Tris-HCl, 1% SDS [pH 7.4] or PBS with 1% SDS). Muscle biopsy samples were homogenized in PBS containing protease inhibitors with a handheld rotor-stator homogenizer (TissueRuptor, QIAGEN, Hilden, Germany) and SDS was added to the lysis buffer to a final concentration of 1%. Homogenized cell or muscle samples were then incubated at 95–100°C for 5 min. Debris was removed by 5 min centrifugation at 14,000 rpm and supernatants were used for immunoblot analysis.
Immunoblot Analysis
Protein concentrations in the extracts were measured with the BCA protein assay (Thermo Scientific, Waltham, MA). Protein samples were separated on NuPage Novex 4%–12% gradient mini gels (Invitrogen) according to the manufacturer's guidelines. After gel separation, proteins were transferred to a polyvinylidene fluoride membrane (GE Healthcare, Buckinghamshire, UK) for 1 hr at 350 mA. Membranes were washed in Tris buffered saline with Tween 20(TBS-T) and blocked for 1 hr at room temperature in TBS-T with 5% nonfat milk. Primary antibodies were incubated overnight at 4°C. The following antibodies were used: rabbit polyclonal anti-GFPT1 (Proteintech Group, Chicago, IL; dilution 1:1,000), mouse monoclonal anti-Myc 9E10 (Clontech, dilution 1:100), mouse monoclonal RL2 antibody to O-linked N-acetylglucosamine (Abcam, Cambridge, MA; dilution 1:1,000), mouse monoclonal anti-actin Ab-5 (BD Transduction Laboratories, San Diego, CA; dilution 1:5,000), and goat polyclonal anti-actin (Santa Cruz Biotechnology, Santa Cruz, CA; dilution 1:200). Membranes were washed three times for 10 min with TBS-T and incubated with secondary goat anti-mouse immunoglobuline G (IgG) antibody conjugated to horseradish peroxidase (HRP) (Invitrogen, dilution 1:5,000), goat anti-rabbit IgG conjugated to HRP (Invitrogen, dilution 1:5,000), or donkey anti-goat IgG conjugated to HRP (Jackson ImmunoResearch, West Grove, PA; dilution 1:10,000) for 1 hr at room temperature. Bands were detected with an enhanced chemiluminescence kit (GE Healthcare) and quantified with the ImageJ program.
125I-α-bungarotoxin Binding
The number of AChRs per motor end plate was estimated with 125I-α-bungarotoxin binding and AChE staining as described previously.19
GFPT1 Enzyme Activity Assay
The enzymatic activity of untagged wild-type and mutant GFPT1 was measured with the glutamate dehydrogenase method.20,21 HEK293 cells transfected with GFPT1 expression constructs were lysed in GFPT buffer (50 mM KH2PO4, 10 mM EDTA, 5 mM reduced L-glutathione, 12 mM D-glucose-6-phosphate Na2, and 1 mM PMSF [pH 7.6]), and 100 μl aliquots of the lysates were mixed with an equal volume of the reaction buffer (100 mM KH2PO4, 10 mM D-fructose 6-phosphate, 6.0 mM L-glutamine, 0.3 mM 3-acetylpyridine adenine dinucleotide, 50 mM KCl, and 6 U L-glutamate dehydrogenase from bovine liver [pH 7.6]) and incubated at 37°C for 45 min. The change in absorbance was monitored at 370 nm with a Spectra Max 250 microplate reader (Molecular Devices, Sunnyvale, CA). The enzymatic activity of each mutant was normalized to GFPT1 expression levels determined by immunoblot analysis of cell lysates used for enzyme activity measurements. All transfections and measurements were done in triplicates.
Zebrafish Husbandry and Observation
We used the wild-type strain ∗AB (Zebrafish International Resource Center, Eugene, OR). Zebrafish embryos were raised and staged according to standard procedures.22,23 Video recordings of embryos and larvae and light microscopy images were taken with a Leica dissection stereomicroscope equipped with a Moticam 1000 camera (Leica, Wetzlar, Germany). Touch-evoked swimming response was elicited by touching the head or the tail of the embryos with a pipette tip.
Antisense Morpholino Oligonucleotide Knockdown
Antisense morpholino oligonucleotides (MOs) were purchased from Gene Tools (Pilomath, OR). The MOs were designed with the mRNA sequence of the zebrafish GFPT1 ortholog gfpt1 and genomic sequences available from public databases (GeneID: 567861, accession number: NM_001034981) and the corresponding genomic DNA sequence obtained from the zebrafish chromosome 8 assembly. We established two MOs to target gfpt1, one translation-blocking MO binding to the region around the start codon (MO1: 5′-TCAGATACGCAAATATGCCACACAT-3′) and one splice-blocking MO directed against the splice donor site of intron 5 (MO2: 5′-TGCTGGAATGTGTTACTTGCCAGAA-3′). MO2 is expected to interfere with the splicing process and lead to a disruption of the protein open reading frame and premature translation stop. The Gene Tools standard control MO (5′-CCTCTTACCTCAGTTACAATTTATA-3′) targeting a human β-hemoglobin gene was used as a negative control for the effects of MO injection. Embryos were injected with 5–8 ng gfpt1 translation-blocking MO1, 2–10 ng of gfpt1 splice-blocking MO2 or 15–20 ng of control MO following standard procedures.22,23 Three independent MO injection experiments were performed for each MO and at least 500 injected embryos were evaluated in total for each MO.
RNA Isolation and RT-PCR
RNA from zebrafish embryos and human cultured myoblasts, myotubes, or muscle biopsies was isolated with Trizol reagent (Invitrogen) following the manufacturer's instructions. Reverse transcription was performed with 1 or 2 μg total RNA as template with the Superscript III First-Strand Synthesis System (Invitrogen). Regions of interest in the GFPT1 and gfpt1 cDNA were amplified with suitable primer sets. For segregation analysis in families, PCR products from patients' cDNA containing GFPT1 mutations were subcloned into the pGEM-T Easy vector (Promega), and individual clones were isolated and sequenced.
Immunofluorescence Stainings
For immunofluorescence analysis, cells were grown on glass coverslips and transfected as described above. Forty-eight hours after transfection, coverslips were washed in PBS, fixed in 3.7% formaldehyde in 1× CSK buffer (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, and 10 mM PIPES [pH 6.8]) for 10 min at room temperature, and permeabilized with 0.1% Triton X-100 in 1× CSK buffer for 15 min. After three washes in PBS, nonspecific binding sites were blocked with PBS containing 5% horse serum for 1 hr, followed by overnight incubation at 4°C with a mouse monoclonal anti-Myc 9E10 antibody (Clontech, dilution 1:100) in PBS with 5% horse serum. After three washes in PBS, cells were incubated with the secondary antibody, goat anti-mouse IgG conjugated to Alexa Fluor 488 (Invitrogen, dilution 1:500), for 1 hr at room temperature. Nuclei were visualized with bisbenzimide H 33258 (40 μg/ml). Digital images were captured with a Zeiss Axiovert 200 M fluorescence microscope and a Zeiss AxioCam HR photo camera (Zeiss, Oberkochen, Germany). Whole-mount immunofluorescence staining and imaging of zebrafish embryos was done as previously described.24 In brief, NMJ were visualized with Alexa Fluor 594 conjugated α-bungarotoxin (Invitrogen, 1 μg/ml) and a mouse monoclonal anti-SV2 (synaptic vesicle protein 2) antibody (Developmental Studies Hybridoma Bank, Iowa City, IA; dilution 1:200). Slow-twitch muscle fibers were visualized with a mouse monoclonal anti-F59 (slow muscle myosin heavy chain, slow MyHC) antibody (Developmental Studies Hybridoma Bank, dilution 1:50).
Statistical Analysis
The data show the mean ± SD. Statistical significance was determined with a two-tailed Student's t test. Significance was set at p < 0.05 (∗), p < 0.01 (∗∗), or p < 0.001 (∗∗∗).
Results
We had access to 11 so far unreported and 2 previously reported families with limb-girdle CMS with tubular aggregates of various ethnic origins (Figure S2). Direct sequencing or haplotype analysis excluded all known genes and loci previously known to be involved in CMS.3 Consanguinity in a Libyan multiplex family with five affected children (family LGM3) presented the possibility of locating the disease locus by homozygosity mapping.12 Genome-wide genotyping with approximately 300,000 SNP markers revealed a single hit on chromosome 2p12-p15 with a maximum LOD score of 3.24 (Figure 1), indicating linkage to this locus. Localization of so far unknown CMS mutations on chromosome 2p was further supported by the results of STR marker analysis in additional families suitable for homozygosity mapping or linkage analysis (LGM1, LGM2, LGM5, LGM6, LGM7, LGM8, LGM10, LGM11, and LGM12; Figure S2). Although these pedigrees were too small to give significant LOD scores, reconstruction of haplotypes was in full agreement with linkage to this locus. We systematically exploited the genotyping data to detect potential recombination events. Under the assumption of genetic homogeneity, historic recombination events suggested from regions of homozygosity in families LGM10 and LGM11 narrowed the critical genetic interval to a region of interest of 5.92 Mb (Figure S3).
We evaluated and ranked the 46 genes in this region (Table S1) on the basis of expression pattern and function, and sequenced the entire coding region and exon-intron boundaries of 16 genes in the index cases from the three families that produced the highest LOD scores (LGM1, LGM3, and LGM10). We identified different homozygous missense mutations in the gene encoding GFPT1 in these three families (Figure 2) and no disease-related changes in any of the other sequenced genes. We obtained further evidence for a causative role of GFPT1 mutations when extending GFPT1 mutation screening to the remaining 13 families, yielding biallelic mutations in all pedigrees except for families LGM4, LGM15, and LGM16 (Figure S4). We identified a total of 18 different GFPT1 mutations consisting of 13 missense mutations, 3 frameshift mutations, 1 nonsense mutation and 1 variant in the 3′-UTR (Figure 2, Table 1). When DNA from family members was available, we observed that the disease cosegregated with recessive inheritance of the GFPT1 mutations. The parents carried mutations in the heterozygous state, and unaffected siblings carried either one heterozygous mutation or were homozygous for the wild-type alleles. When DNA from family members was not available (families LGM9 and LGM13), subcloning and sequencing of amplicons from cDNA derived from muscle biopsies demonstrated that the mutations were on separate alleles. We observed that some GFPT1 missense mutations caused rather mild amino acid changes or affected residues that are not strictly conserved in orthologs (Table 1 and Figure S5). Nevertheless, analysis of extended numbers of healthy control individuals, adjusted to the type of mutation (at least 103 for each truncating and 221 for each missense mutation; 635 for the 3′-UTR variant), did not yield any of the identified genotypes (Table 1).
Figure 2.
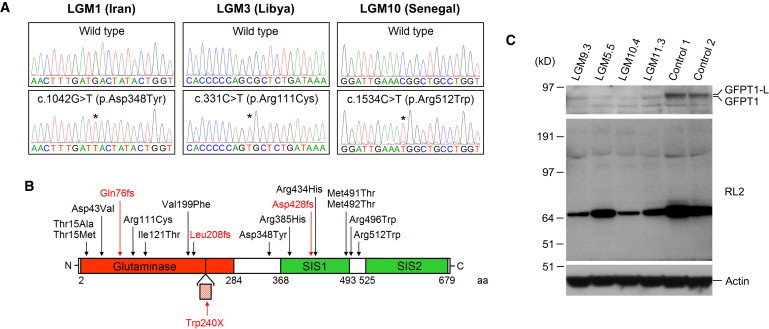
Identification of GFPT1 Mutations
(A) GFPT1 mutations detected in families used for candidate-gene sequencing. Chromatogram pairs of an unaffected individual and an affected patient are shown. The asterisks indicate the position of the mutations. Base and amino acid changes with the corresponding position in the nucleotide and protein sequence are given above chromatograms displaying GFPT1 mutations. The numbering of nucleotides and amino acids follows NM_002056.2 and NP_002047.2, which represent the short, ubiquitous isoform of GFPT1. Nucleotide numbering uses the A of the ATG translation initiation start site as nucleotide +1.
(B) Domain structure of GFPT1 and position of mutations. GFPT1 contains a glutaminase and two sugar isomerase (SIS) domains. The site of insertion of 18 additional amino acids (hashed box) encoded by a muscle specific exon16 is indicated. Missense mutations are in black, truncating mutations are in red. The 3′-UTR mutation is not shown. The numbering of nucleotides and amino acids is as in (A) except for the c.719G>A (p.Trp240X) mutation in the muscle specific exon, for which we used numbering of the longer GFPT1-L sequence.16 aa is an abbreviation for amino acid.
(C) Immunoblot of GFPT1 and O-GlcNAcylation in muscle biopsies of GFPT1 patients. Muscle lysates of patients LGM9.3, LGM5.5, LGM10.4, and LGM11.3 and two healthy control individuals were immunoblotted with an anti-GFPT1 antibody (top), the RL2 antibody (detects single N-acetylglucosamine at serine or threonine residues, middle) and an anti-actin antibody (as loading control, bottom). GFPT1 band intensities were normalized to actin bands in the same lane. Total GFPT1 levels (GFPT1-L and the shorter ubiquitous isoform) in patients' muscles were reduced to 29%, 8%, 9%, and 26% of the level in control muscle 1. The levels of O-linked N-acetylglucosamine on proteins were also markedly decreased in patients' muscles. The membrane was first decorated with the anti-GFPT1 antibody and later reprobed with the RL2 and anti-actin antibodies.
Table 1.
GFPT1 Mutations Found in CMS Families
| Exon | Nucleotide Change | Effect on the Protein | Families with This Mutation | Ethnic Origin |
Number of Control Samples |
Granthama | SIFTb | ||
|---|---|---|---|---|---|---|---|---|---|
| German | Further Ethnically Matched | Total | |||||||
| 2 | c.43A>G | p.Thr15Ala | LGM13 | Italy | 341 | 102 Italian | 443 | 58 | 0.10 |
| 2 | c.44C>T | p.Thr15Met | LGM8 | UK | 324 | 82 British | 406 | 81 | 0.01 |
| 3 | c.128A>T | p.Asp43Val | LGM6 | Germany | 223 | 223 | 152 | 0.00 | |
| 4 | c.222_223insA | p.Gln76fs | LGM14 | Sweden | 117 | 117 | |||
| 4 | c.331C>T | p.Arg111Cys | LGM3, LGM14 | Libya, Sweden | 173 | 67 North African | 240 | 180 | 0.03 |
| 5 | c.362T>C | p.Ile121Thr | LGM6 | Germany | 432 | 432 | 89 | 0.01 | |
| 7 | c.595G>T | p.Val199Phe | LGM9 | Germany | 425 | 425 | 50 | 0.01 | |
| 8 | c.621_622 del | p.Leu208fs | LGM13 | Italy | 45 | 58 Italian | 103 | ||
| 8A | c.719G>A | p.Trp240X | LGM2 | Turkey | 92 | 115 Turkish | 207 | ||
| 11 | c.1042G>T | p.Asp348Tyr | LGM1 | Iran | 376 | 66 Iranian | 442 | 160 | 0.05 |
| 13 | c.1154G>A | p.Arg385His | LGM7 | UK | 340 | 97 British | 437 | 29 | 0.00 |
| 14 | c.1278_1281 dup | p.Asp428fs | LGM12 | Spain | 57 | 83 Spanish | 140 | ||
| 14 | c.1301G>A | p.Arg434His | LGM7 | UK | 331 | 95 British | 426 | 29 | 0.02 |
| 15 | c.1472T>C | p.Met491Thr | LGM11 | Spain | 452 | 86 Spanish | 538 | 81 | 0.45 |
| 15 | c.1475T>C | p.Met492Thr | LGM5 | Spain | 327 | 84 Spanish | 411 | 81 | 0.01 |
| 15 | c.1486C>T | p.Arg496Trp | LGM8 | UK | 136 | 85 British | 221 | 101 | 0.00 |
| 15 | c.1534C>T | p.Arg512Trp | LGM10 | Senegal | 146 | 84 African American | 230 | 101 | 0.03 |
| 19 | c.∗22C>A | LGM5, LGM9, LGM12 | Spain, Germany | 543 | 92 Spanish | 635 | |||
The numbering of nucleotides and amino acids follows NM_002056.2 and NP_002047.2 except for the c.719G>A (p.Trp240X) mutation in the muscle specific exon, for which we used numbering of the longer GFPT1-L sequence, AF334737.1 and AAK15342.1.16
The Grantham scale categorizes codon replacements into classes of chemical dissimilarity between the encoded amino acids (0–50: conservative; 51–100: moderately conservative; 101–150: moderately radical; ≥151: radical).40,41
Sorting Intolerant from Tolerant (SIFT) predicts the functional importance of amino acid substitutions based on the alignment of orthologous sequences (0.0-0.05: intolerant; 0.051-0.1: potentially intolerant; 0.101-0.2: borderline; 0.201-1.0: tolerant).42
We noted that the crystal structures of the Escherichia coli ortholog for GFPT1 (GlmS) and of the isomerase domain of human GFPT1 had been solved (Protein Data Bank accession numbers 2J6H and 2ZJ3).25,26 With this data as the basis of a structural model for human GFPT1, the c.1154G>A (p.Arg385His) mutation would be predicted to impair GFPT1 function because the arginine residue at position 385 in human GFPT1 corresponds to E. coli GFPT1 arginine 312, which contributes to the GFPT1 dimer interface. Other GFPT1 missense mutations do not affect amino acid residues required for oligomerisation or involved in the catalytic mechanism. Consistent with this observation, heterologous protein expression of the GFPT1 mutants p.Thr15Ala, p.Asp43Val and p.Asp348Tyr had only small effects on enzymatic activity and of the mutants p.Arg111Cys and p.Arg434His had no effect at all (Figure S6). We also did not observe any major effect of the GFPT1 mutations on subcellular localization of the protein in transfected cells (Figure S6). By contrast, we found substantially reduced GFPT1 levels in muscle biopsies (Figure 2) and cultured primary myoblasts and myotubes obtained from GFPT1 patients (Figure S6). Reduced amounts of GFPT1 were also seen in patients with the 3′-UTR mutation c.∗22C>A (LGM5.5, LGM9.3).
We also set up experiments to follow potential indirect or direct functional consequences of GFPT1 mutations. Radiometric assays showed a strongly reduced number of AchR available for 125I-α-bungarotoxin binding at motor end-plates of GFPT1 patient LGM8.3 (to around 25% of normal). The end product of the hexosamine pathway, uridine diphospho-N-acetylglucosamine (UDP-GlcNAc), is involved in multiple glycosylation processes,7 including the dynamic modification of serine or threonine residues of intracellular proteins (O-GlcNacylation)27 (Figure S1). We therefore immunoblotted muscle samples of GFPT1 patients and controls with the RL2 antibody, which selectively detects O-linked N-acetylglucosamine (O-GlcNAc) residues on numerous proteins.28 As expected, the antibody recognized several bands whose intensities were markedly decreased in muscles from GFPT1 patients (Figure 2).
In a final series of experiments, we used zebrafish as an animal model. GFPT1 is conserved in zebrafish, with high expression levels during early somite development (data not shown). We knocked down the expression of the zebrafish GFPT1 ortholog gfpt1 by injection of antisense MOs, a translation-blocking MO targeting the region surrounding the start codon (MO1) and a splice-blocking MO targeting the splice donor site of intron 5 (MO2). Both MOs induced a clear reduction of gfpt1 levels at 48 hr postfertilization (hpf) (Figure S7). Knockdown of neuromuscular junction proteins usually affects motility and swimming behavior of injected embryos. Indeed, MO-injected embryos displayed altered tail morphology (curled tails and shortened tails, Figure 3), and swimming and touch-evoked escape response at 48 hpf were severely impaired (Movie S1). Histologically, we observed abnormal muscle morphology and delayed NMJ development. Somites had lost their regular chevron shape with rounding of myosepta to a C shape instead of the V shape in control embryos. Staining for slow-twitch muscle fibers revealed morphological abnormalities ranging from wavy fibers to severely damaged fibers that were detached from the vertical myoseptum (Figure 3). NMJ development progressed normally up to 24 hpf, but from that time point onward, NMJ development was delayed. Motor neuron axons had completely or partially lost their ability to project branches into laterally located muscle fibers and to form synapses with them (Figure 3). Both MOs resulted in strikingly similar phenotypes, whereas embryos injected with a standard control MO were indistinguishable from noninjected embryos (data not shown).
Figure 3.
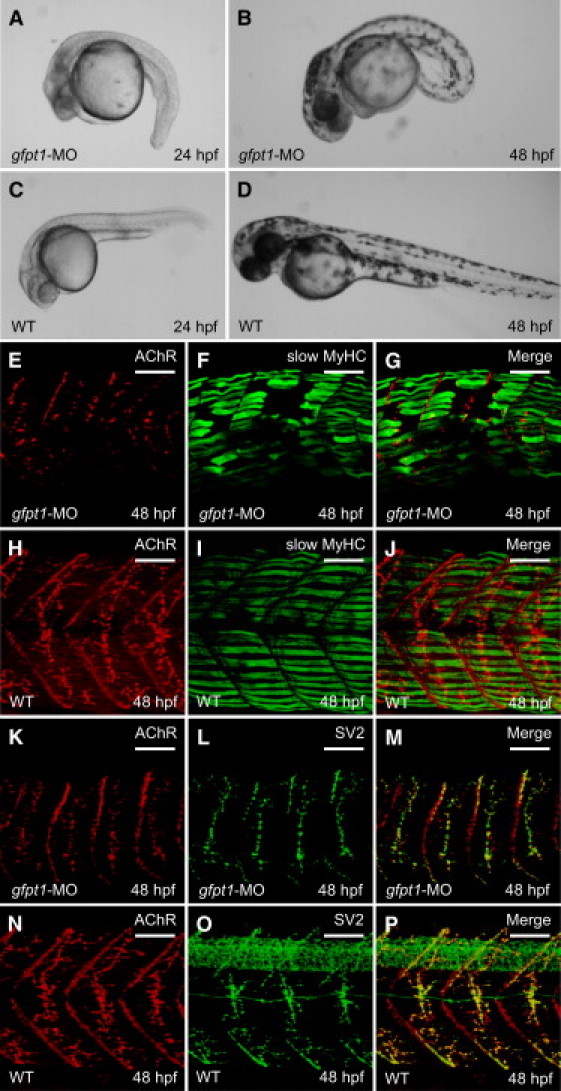
Gpft1 Knockdown in Zebrafish Embryos
(A–D) Live embryos at 24 and 48 hpf injected with the translation-blocking gfpt1 antisense morpholino oligo MO1 (A and B) and noninjected controls (C and D). Note increased tail curvature in gfpt1 morphants.
(E–P) Whole-mount immunostainings of zebrafish embryos. Slow-twitch muscle fiber structure in 48 hpf gfpt1-MO1 injected morphants (E–G) and noninjected controls (H–J). Embryos were stained for AChR (α−bungarotoxin) and slow-twitch muscle fibers (anti-slow muscle myosin heavy chain [slow MyHC], F59 antibody). Gfpt1 morphants show severe muscle fiber defects with detachment of fibers from the vertical myoseptum. Somites have lost their chevron shape. (K–P) Neuromuscular junctions in 48 hpf gfpt1 morphants (K–M) and noninjected controls (N–P). Embryos were stained for AChR (α−bungarotoxin) and presynaptic nerve endings (anti-SV2 antibody). In controls, motor axons have branched out into all parts of the somite where they form contacts with AChR clusters. In gfpt1-MO1 injected embryos AChR clusters are predominantly located along the medial surface of the somite, and motor axons form fewer branches than in controls. The scale bars represent 50 μm.
Discussion
In this study, we provide two lines of genetic evidence that GFPT1, the key enzyme of the hexosamine pathway yielding essential amino sugars for carbohydrate modifications, is required for critical events in neuromuscular transmission. Specifically, we localized and refined the genetic interval for a so far genetically undetermined autosomal recessive CMS on chromosome 2 and identified GFPT1 mutations as the molecular defect. This finding was recapitulated by a neuromuscular morphant phenotype observed in zebrafish embryos lacking GFPT1. We found 18 different homozygous and compound heterozygous GFPT1 mutations in 13 unrelated families, and GFPT1 mutations occurred in CMS cohorts of various ethnic origins. Although failure to detect mutations in three additional cases is probably related to so far unexplored mutation mechanisms or involvement of additional unknown genes, our data indicate that GFPT1 mutations represent the major cause for limb-girdle CMS with good treatment response to AChE inhibitors or tubular aggregates in muscle biopsies. With respect to genetic diagnosis and counselling, limb-girdle CMS patients with effective response to AChE inhibitors or tubular aggregates in muscle biopsies should undergo GFPT1 sequencing, whereas all other limb-girdle CMS patients should be screened for DOK7 mutations first and—if negative—for GFPT1 mutations.
In metazoa, there are two GFPT genes, GFPT1 and GFPT2, encoding isozymes with different tissue distributions.29 GFPTs are homodimeric, cytoplasmic enzymes that transfer an amino group from glutamine onto fructose-6-phosphate, yielding glucosamine-6-phosphate (GlcN-6-P) and glutamate. This reaction is the first and rate-limiting step of the hexosamine biosynthesis pathway (Figure S1), which is the obligatory source of essential building blocks for the glycosylation of proteins and lipids.7 GFPT1 is the only or predominant isoform in kidney, pancreas and liver. Because glycosylation is essential for cell survival, it might be expected that GFPT1 mutations create hypomorphic alleles. Indeed none of our patients carries two null mutations in the constitutive exons of the GFPT1 gene. The homozygous p.Trp240X mutation (family LGM2) is predicted to lead to decreased GFPT1 levels only in heart and skeletal muscle as it occurs in an alternative 18 amino acid exon, exclusively incorporated in the predominant GFPT1 species in striated muscle.16 The expected residual function of GFPT1 mutants is also consistent with our finding that some GFPT1 missense mutations result in amino acids with similar physicochemical properties or affect residues that are not strictly conserved in orthologs (Figure S5).
With the notable exception of p.Arg385His, which potentially affects GFPT1 dimerization, in silico structural modeling and heterologous expression studies did not reveal definite pathophysiological effects of GFPT1 mutants. However, protein expression data in patients' muscle cells and biopsy specimens suggest that GFPT1 mutations may lead to substantially reduced GFPT1 levels. The mechanism by which the mutants inhibit protein expression remains to be determined but may involve reduced levels of GFPT1 mRNA, increased GFPT1 turn-over, or interference with protein translation. Notably, the 3′-UTR mutation c.∗22C>A (patients LGM5.5, LGM9.3) was also associated with reduced amounts of GFPT1. For this particular mutation, the combination of an observed decrease in protein levels, its presence in three unrelated families, its absence in a large number of control chromosomes, and compound heterozygosity with a truncating mutation in one family strongly suggests that c.∗22C>A is a disease-causing variant. Nevertheless, the pathogenic mechanism at the molecular level of c.∗22C>A is not yet clear. Analysis of the GFPT1 skeletal muscle mRNA by RT-PCR in two patients excluded effects on splicing and showed similar levels of wild-type and mutated position c.∗22 mRNA expression (Figure S6). It is possible that this mutation, like many 3′-UTR mutations in other genes, affects translational efficiency.30
Other CMS related genes known to date encode structural components of the neuromuscular junction, whereas GFPT1 is believed to play a more general role in the cell. The product of the transamidase reaction catalyzed by GFPT1, GlcN-6-P, is used in the hexosamine pathway to synthesize UDP-GlcNAc that serves as common precursor for all amino sugars used for the synthesis of glycoproteins, glycolipids, and proteoglycans (Figure S1).7 Many key NMJ proteins are glycosylated,8 including MuSK, AChR subunits, agrin, dystroglycan, and integrins. Interestingly, a missense mutation removing one of the N-glycosylation sites of the ɛ subunit of the AChR causes CMS.31 Moreover, treatment with tunicamycin (an inhibitor of protein glycosylation) and in vitro mutations of AChR subunits that prevent glycosylation severely reduce cell surface expression of AChR, either through a decrease of metabolic stability or through a failure in efficient assembly of the pentameric AChR.32 UDP-GlcNAc is also extensively involved in intracellular signaling in a wide range of species. N-acetylglucosamine is reversibly attached to serine or threonine residues of cytoplasmic and nuclear proteins, similar to phosphorylation (O-GlcNAcylation).27 Reduced endplate AChR numbers and decreased protein O-GlcNAcylation in patients' muscles observed in our study may provide a small glimpse at the molecular and cellular consequences of GFPT1 mutations. Lectin stainings, digestions with glycosidases, antibodies to carbohydrate-linked proteins, or mass spectrometry-based techniques are likely to demonstrate specific changes in protein glycosylation. Identification of such changes may also help to clarify why, although universally required, the functions of the NMJ is especially vulnerable to alterations of the hexosamine pathway.
In order to obtain further proof that a reduction of GFPT1 levels leads to NMJ or muscle dysfunction, we utilized zebrafish as an animal model. Although overall the muscle structure of zebrafish differs from mammalian muscles,33 NMJ formation occurs in a similar stepwise sequence, and zebrafish mutants and morphants have previously proved a good model system for studies of myasthenic syndromes and NMJ function.24,34–36 Morpholino-mediated knockdown of gfpt1 in zebrafish embryos induced a neuromuscular phenotype and resulted in altered muscle histology and delayed NMJ maturation. These data provide another level of genetic evidence and independently confirms that GFPT1 is required for normal NMJ formation.
Although the precise pathogenic mechanisms involved remain to be determined, our work highlights amino sugar metabolism as a pathophysiological mechanism that can underlie NMJ disorders and paves the way for treatment options. Because the vertebrate NMJ is a classic model synapse becasue of its comparatively large size and easy accessibility,37 further investigation of the role of GFPT1 for NMJ function is likely to also contribute to our understanding of synapse development and organization in general. Although there are no clear-cut associations in our patients, we note that GFPT1 and the hexosamine pathways have been implicated in signaling pathways that may become deregulated in diseases of the immune system, diabetes mellitus, cancer, cardiovascular disease, and neurodegenerative diseases.38,39 Our findings give additional impetus for investigating the role of GFPT1 in human health and disease.
Acknowledgments
We wish to thank the patients and their families for participating in this study, C. Jepson, M. Reza, and M. Ritso for technical assistance; A. Vincent for reporting results from end-plate studies; and D. Burns and the staff of the Institute of Human Genetics Zebrafish Facility for expert fish care. We are grateful to the Research Institute for Rare Diseases within the Instituto de Salud Carlos III, Madrid, for providing control DNA samples; the Medical Research Council (MRC) Centre for Neuromuscular Diseases Biobank, Newcastle; and the Muscle Tissue Culture Collection, Friedrich-Baur-Institut, Munich, for providing primary human myoblasts. All three institutions are partners of the EuroBioBank Network. The present study was supported by a grant from the Association Française contre les Myopathies (AFM) to H.L. and J.S.M., by Science City Newcastle, and by the MRC as part of the MRC Centre for Neuromuscular Diseases. J.S. is supported by a grant from the Gebert Rüf Foundation, A.A. by a grant from the Deutsche Forschungsgemeinschaft (DFG, Ab 130/2-1) and D.B., J.C., and S.M. by grants from the MRC, the Myasthenia Gravis Association, and the Muscular Dystrophy Campaign. J.S. is a Heisenberg fellow of the DFG. J.S.M. receives a research fellowship from the Faculty of Medical Sciences, Newcastle University. V.G. is a research fellow of the Alexander von Humboldt Foundation. N.M. received a fellowship from the Instituto de Salud Carlos III and Fundación para la Investigación del Hospital Universitario La Fe (CM06/00154). J.J.V. and N.M. are members of CIBER de Enfermedades Neurodegenerativas (CIBERNED), Valencia, Spain. Newcastle University is the coordinating partner of TREAT-NMD (EC, 6th FP, proposal #036825; www.treat-nmd.eu).
Contributor Information
Jan Senderek, Email: jan.senderek@cell.biol.ethz.ch.
Hanns Lochmüller, Email: hanns.lochmuller@ncl.ac.uk.
Supplemental Data
Noninjected embryos swim away quickly when touched with a fine pipette tip, whereas gfpt1 deficient embryos hardly move when touched.
Web Resources
The URLs for data presented herein are as follows:
BLAST alignments, http://blast.ncbi.nlm.nih.gov
ClustalW algorithm, www.ebi.ac.uk/clustalw/
Image J program, http://rsb.info.nih.gov/ij
Marshfield map, http://research.marshfieldclinic.org/genetics/GeneticResearch/compMaps.asp
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3 program, http://frodo.wi.mit.edu/primer3
RCSB Protein Data Bank, http://www.pdb.org/
Repeat-Masker program, http://www.repeatmasker.org
SMART algorithm, http://smart.embl-heidelberg.de
UniSTS database, www.ncbi.nlm.nih.gov/sites/entrez?db=unists
University of California Santa Cruz (UCSC) human genome database, build hg19, February 2009, http://genome.ucsc.edu
Zebrafish genome assembly, http://www.sanger.ac.uk/Projects/D_rerio
References
- 1.Kandel E.R., Schwartz J.H., Jessell T.M. McGraw-Hill; New York: 2000. Principles of Neural Science. [Google Scholar]
- 2.Newsom-Davis J. The emerging diversity of neuromuscular junction disorders. Acta Myol. 2007;26:5–10. [PMC free article] [PubMed] [Google Scholar]
- 3.Engel A.G., Shen X.M., Selcen D., Sine S.M. What have we learned from the congenital myasthenic syndromes. J. Mol. Neurosci. 2010;40:143–153. doi: 10.1007/s12031-009-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu H., Xiong W.C., Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McQuillen M.P. Familial limb-girdle myasthenia. Brain. 1966;89:121–132. doi: 10.1093/brain/89.1.121. [DOI] [PubMed] [Google Scholar]
- 6.Johns T.R., Dreifuss F.E., Crowley W.J., Fakadej A.V. Familial non-progressive myasthenic myopathy. Neurology. 1966;16:307. [Google Scholar]
- 7.Haltiwanger R.S., Lowe J.B. Role of glycosylation in development. Annu. Rev. Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 8.Martin P.T. Glycobiology of the synapse. Glycobiology. 2002;12:1R–7R. doi: 10.1093/glycob/12.1.1r. [DOI] [PubMed] [Google Scholar]
- 9.Sieb J.P., Tolksdorf K., Dengler R., Jerusalem F. An autosomal-recessive congenital myasthenic syndrome with tubular aggregates in a Libyan family. Neuromuscul. Disord. 1996;6:115–119. doi: 10.1016/0960-8966(95)00034-8. [DOI] [PubMed] [Google Scholar]
- 10.Rodolico C., Toscano A., Autunno M., Messina S., Nicolosi C., Aguennouz M., Laurà M., Girlanda P., Messina C., Vita G. Limb-girdle myasthenia: clinical, electrophysiological and morphological features in familial and autoimmune cases. Neuromuscul. Disord. 2002;12:964–969. doi: 10.1016/s0960-8966(02)00137-2. [DOI] [PubMed] [Google Scholar]
- 11.von der Hagen M., Schallner J., Kaindl A.M., Koehler K., Mitzscherling P., Abicht A., Grieben U., Korinthenberg R., Kress W., von Moers A. Facing the genetic heterogeneity in neuromuscular disorders: linkage analysis as an economic diagnostic approach towards the molecular diagnosis. Neuromuscul. Disord. 2006;16:4–13. doi: 10.1016/j.nmd.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Lander E.S., Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 13.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 14.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 15.Lindner T.H., Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–407. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- 16.DeHaven J.E., Robinson K.A., Nelson B.A., Buse M.G. A novel variant of glutamine: fructose-6-phosphate amidotransferase-1 (GFAT1) mRNA is selectively expressed in striated muscle. Diabetes. 2001;50:2419–2424. doi: 10.2337/diabetes.50.11.2419. [DOI] [PubMed] [Google Scholar]
- 17.Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 18.Lochmüller H., Johns T., Shoubridge E.A. Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp. Cell Res. 1999;248:186–193. doi: 10.1006/excr.1999.4407. [DOI] [PubMed] [Google Scholar]
- 19.Vincent A., Cull-Candy S.G., Newsom-Davis J., Trautmann A., Molenaar P.C., Polak R.L. Congenital myasthenia: end-plate acetylcholine receptors and electrophysiology in five cases. Muscle Nerve. 1981;4:306–318. doi: 10.1002/mus.880040407. [DOI] [PubMed] [Google Scholar]
- 20.Eguchi S., Oshiro N., Miyamoto T., Yoshino K., Okamoto S., Ono T., Kikkawa U., Yonezawa K. AMP-activated protein kinase phosphorylates glutamine : fructose-6-phosphate amidotransferase 1 at Ser243 to modulate its enzymatic activity. Genes Cells. 2009;14:179–189. doi: 10.1111/j.1365-2443.2008.01260.x. [DOI] [PubMed] [Google Scholar]
- 21.Ye F., Maegawa H., Morino K., Kashiwagi A., Kikkawa R., Xie M., Shen Z. A simple and sensitive method for glutamine:fructose-6-phosphate amidotransferase assay. J. Biochem. Biophys. Methods. 2004;59:201–208. doi: 10.1016/j.jbbm.2003.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 23.Westerfield M. University of Oregon Press; Eugene: 2000. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio) [Google Scholar]
- 24.Müller J.S., Jepson C.D., Laval S.H., Bushby K., Straub V., Lochmüller H. Dok-7 promotes slow muscle integrity as well as neuromuscular junction formation in a zebrafish model of congenital myasthenic syndromes. Hum. Mol. Genet. 2010;19:1726–1740. doi: 10.1093/hmg/ddq049. [DOI] [PubMed] [Google Scholar]
- 25.Mouilleron S., Badet-Denisot M.A., Golinelli-Pimpaneau B. Glutamine binding opens the ammonia channel and activates glucosamine-6P synthase. J. Biol. Chem. 2006;281:4404–4412. doi: 10.1074/jbc.M511689200. [DOI] [PubMed] [Google Scholar]
- 26.Nakaishi Y., Bando M., Shimizu H., Watanabe K., Goto F., Tsuge H., Kondo K., Komatsu M. Structural analysis of human glutamine:fructose-6-phosphate amidotransferase, a key regulator in type 2 diabetes. FEBS Lett. 2009;583:163–167. doi: 10.1016/j.febslet.2008.11.041. [DOI] [PubMed] [Google Scholar]
- 27.Wells L., Vosseller K., Hart G.W. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science. 2001;291:2376–2378. doi: 10.1126/science.1058714. [DOI] [PubMed] [Google Scholar]
- 28.Holt G.D., Snow C.M., Senior A., Haltiwanger R.S., Gerace L., Hart G.W. Nuclear pore complex glycoproteins contain cytoplasmically disposed O-linked N-acetylglucosamine. J. Cell Biol. 1987;104:1157–1164. doi: 10.1083/jcb.104.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oki T., Yamazaki K., Kuromitsu J., Okada M., Tanaka I. cDNA cloning and mapping of a novel subtype of glutamine:fructose-6-phosphate amidotransferase (GFAT2) in human and mouse. Genomics. 1999;57:227–234. doi: 10.1006/geno.1999.5785. [DOI] [PubMed] [Google Scholar]
- 30.Chen J.M., Férec C., Cooper D.N. A systematic analysis of disease-associated variants in the 3′ regulatory regions of human protein-coding genes II: the importance of mRNA secondary structure in assessing the functionality of 3′ UTR variants. Hum. Genet. 2006;120:301–333. doi: 10.1007/s00439-006-0218-x. [DOI] [PubMed] [Google Scholar]
- 31.Ohno K., Wang H.L., Milone M., Bren N., Brengman J.M., Nakano S., Quiram P., Pruitt J.N., Sine S.M., Engel A.G. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron. 1996;17:157–170. doi: 10.1016/s0896-6273(00)80289-5. [DOI] [PubMed] [Google Scholar]
- 32.Prives J., Bar-Sagi D. Effect of tunicamycin, an inhibitor of protein glycosylation, on the biological properties of acetylcholine receptor in cultured muscle cells. J. Biol. Chem. 1983;258:1775–1780. [PubMed] [Google Scholar]
- 33.Devoto S.H., Melançon E., Eisen J.S., Westerfield M. Identification of separate slow and fast muscle precursor cells in vivo, prior to somite formation. Development. 1996;122:3371–3380. doi: 10.1242/dev.122.11.3371. [DOI] [PubMed] [Google Scholar]
- 34.Ono F., Higashijima S., Shcherbatko A., Fetcho J.R., Brehm P. Paralytic zebrafish lacking acetylcholine receptors fail to localize rapsyn clusters to the synapse. J. Neurosci. 2001;21:5439–5448. doi: 10.1523/JNEUROSCI.21-15-05439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ono F., Shcherbatko A., Higashijima S., Mandel G., Brehm P. The Zebrafish motility mutant twitch once reveals new roles for rapsyn in synaptic function. J. Neurosci. 2002;22:6491–6498. doi: 10.1523/JNEUROSCI.22-15-06491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jing L., Gordon L.R., Shtibin E., Granato M. Temporal and spatial requirements of unplugged/MuSK function during zebrafish neuromuscular development. PLoS ONE. 2010;5:e8843. doi: 10.1371/journal.pone.0008843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanes J.R., Lichtman J.W. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- 38.Hart G.W., Housley M.P., Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 39.Love D.C., Krause M.W., Hanover J.A. O-GlcNAc cycling: emerging roles in development and epigenetics. Semin. Cell Dev. Biol. 2010;21:646–654. doi: 10.1016/j.semcdb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 41.Li W.H., Wu C.I., Luo C.C. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J. Mol. Evol. 1984;21:58–71. doi: 10.1007/BF02100628. [DOI] [PubMed] [Google Scholar]
- 42.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Noninjected embryos swim away quickly when touched with a fine pipette tip, whereas gfpt1 deficient embryos hardly move when touched.