

Abstract
Epilepsy is heritable, yet few causative gene mutations have been identified, and thus far no human epilepsy gene mutations have been found to produce seizures in invertebrates. Here we show that mutations in prickle genes are associated with seizures in humans, mice, and flies. We identified human epilepsy patients with heterozygous mutations in either PRICKLE1 or PRICKLE2. In overexpression assays in zebrafish, prickle mutations resulted in aberrant prickle function. A seizure phenotype was present in the Prickle1-null mutant mouse, two Prickle1 point mutant (missense and nonsense) mice, and a Prickle2-null mutant mouse. Drosophila with prickle mutations displayed seizures that were responsive to anti-epileptic medication, and homozygous mutant embryos showed neuronal defects. These results suggest that prickle mutations have caused seizures throughout evolution.
Introduction
Epilepsy is a disabling and costly disease characterized by recurrent seizures. Affecting approximately 1% of the population, it requires long-term treatment and reduces productivity.1 For most patients, however, the underlying cause of epilepsy remains unknown, impeding effective diagnosis and treatment.1 Recently, a gene mutation in human prickle homolog 1 (Drosophila) (PRICKLE1 [MIM 608500]) was associated with an ataxia syndrome accompanied by myoclonus and seizures in patients with normal brain imaging and normal intelligence,2 thus raising the possibility that mutations in prickle genes might play a critical role in epilepsy. However, the ability of prickle proteins to regulate seizures has not yet been established. First discovered in Drosophila,3 prickle proteins are highly conserved throughout evolution. Characterized by PET and LIM domains,4 the prickle proteins function in the noncanonical WNT signaling pathway, which regulates intracellular calcium release and planar cell polarity (PCP).5 At the amino acid level, Drosophila Prickle is 65% identical to human PRICKLE1 and 62% identical to PRICKLE2, and PRICKLE1 and PRICKLE2 are 59% identical to each other (Deans et al.6 and poster abstract presented at the 14th International Conference on Genome Informatics: Katoh, M., and Katoh, M. [2003]. Genome Informatics 14, 587–588). Given the strong conservation of prickle proteins throughout evolution, we hypothesized that mutations in the prickle genes would lead to seizures and epilepsy-related phenotypes in diverse species.
Subjects and Methods
Human Subjects
All human DNA samples were collected with proper informed consent and with approval from an institutional review board (IRB). Eighty-eight DNA samples from patients with myoclonus epilepsy and 176 samples from ethnically matched controls without myoclonus epilepsy were collected from either the NIH Coriell Institute for Medical Research collection or from patients phenotyped by an EEG showing generalized epileptiform discharges, a physical exam, and history that included myoclonic seizures. The history, physical exam, and EEG were performed by C.G., T.B., R.P.S., E.B.-K., A.G.B., and H.E.-S. Patients who met criteria included those with generalized tonic-clonic epilepsy, juvenile myoclonic epilepsy, and myoclonic-astatic epilepsy. Patients for whom a physical exam revealed any focal findings, patients with focal epileptiform abnormalities, and patients who experienced simple febrile seizures were excluded. All patients gave informed consent according to local IRB-approved protocols. Signature Genomic Laboratories is a CLIA-approved laboratory conducting clinical microarray-based comparative-genomic hybridization. A search of their laboratory database of cytogenetic aberrations identified one epileptic patient (patient 6, Table 1) with a deletion encompassing PRICKLE2 (MIM 608501).
Table 1.
Prickle Mutations in Patients with Myoclonus Epilepsy
| Patient # | Mutation Nucleotide | Amino Acid Change | Sex | Clinical Details |
|---|---|---|---|---|
| 1 | PRICKLE1 c.311G>A | p.Arg104Gln | both | previously described mutation in familial myoclonus-ataxia epilepsy patients2 |
| 2 | PRICKLE1 c.431G>A | p.Arg144His | male | myoclonic seizures, generalized EEG pattern, mild mental retardation |
| 3 | PRICKLE1 c. 1414T>C | p.Tyr472His | female | juvenile myoclonic epilepsy |
| 4 | PRICKLE2, c.[443G>A; 457G>A], | p.[Arg148His; Val153Ile] | female | myoclonic seizures, generalized EEG pattern, ataxia.5 brother with same mutations and similar clinical phenotype with myoclonic seizures and generalized discharges |
| 5 | PRICKLE2 c.1813G>T | p.Val605Phe | male | myoclonic seizures, generalized EEG pattern |
| 6 | PRICKLE2 deletion: chr3:62,665,527 – 64,890,116 (hg18 coordinates). | deletion of gene | male | developmental delay, epilepsy, autisic disorder |
PRICKLE1 and PRICKLE2 Resequencing
Primers and conditions for PRICKLE1 (NM_153026.2) sequencing including all exons and intron-exon boundaries were used as previously described.1 Primers for PRICKLE2 (NM_198859.3) sequencing (Table S1), including all exons and intron-exon boundaries, were used at annealing temperature of 60.5°C (F = forward primer, R = reverse primer).
Quantitative PCR
Primers were designed and ordered in accordance with the TaqMan Gene Copy Number Assay protocol from Applied Biosystems. Primers were designed within the exon boundaries of PRICKLE1 and PRICKLE2.
We made serial dilutions from 100–3.125 ng from genomic DNA of control samples to establish a standard curve for all of the primers used in the assay. We compared primer efficiencies and confirmed that primers passed all quality-control tests defined in the 2−ΔΔCt method. In accordance with ABI protocol, endogenous RNase P was used as a reference gene to be present in every reaction so novel primers could be compared to a confirmed diploid gene.
TaqMan real-time PCR was carried out with 8 ng of dried gDNA in a reaction volume of 5 μl, which was distributed as follows: 2.5 μl 2× TaqMan universal PCR Master Mix, .25 μl VIC probe RNaseP, .25 μl FAM probe custom-designed primers, and 2 μl DNase and RNase free water.
An ABI 7900HT Fast Real-Time PCR system was used with the 2−ΔΔCt method for quantitative analysis. All samples were analyzed in triplicate and compared to the RNaseP endogenous control for a reference of diploid copy number. Irregular samples were analyzed twice via the same protocol. Samples with ΔΔCt values < .7 were implicated as having possible deletions, and samples with ΔΔCt values > 1.3 were implicated as having possible duplications.
Zebrafish
Adults were maintained in a 14 hr light/10 hr dark cycle at 28°C. Embryos were collected from natural pairwise matings and staged by hours post-fertilization (hpf) at 28.5°C. Morphological criteria were described in Kimmel et al.7 Wild-type prickle2 was cloned from a mixture of cDNAs for 0–24 hpf zebrafish embryos with GeneAmp High Fidelity polymerase (Applied Biosystem). The Quick Change II site-directed mutagenesis kit (Stratagene) was used for generating prickle2 point mutants encoding amino acids orthologous to the human double mutations p.[Arg148His;Val153Ile] (p.[Arg144His;Val149Ile] (NM_183343.1:c.[431G>A;432A>T;445G>A]) in zebrafish) and human p.Val605Phe (p.Leu596Phe [NM_183343.1:c.(1786C>T;1788G>C] in zebrafish). To be clear about which human mutations we are evaluating in the zebrafish system, we will refer to the zebrafish reagents that encode the ortholog of the human p.[Arg148His;Val153Ile] mutations as prickle2p.Arg148His;Val153Ile. Likewise, we will refer to zebrafish reagents that encode the ortholog of the human p.Val605Phe mutation as prickle2Val605Phe. Synthetic RNAs were made with mMessage mMachine Capped RNA transcription kit (Ambion). Equivalent RNA amounts were injected into 1- to 8-cell-stage embryos (approximately 6 nl of 100 ng/μl RNA). Myc-tagged forms of the wild-type and mutant proteins all expressed stable protein of the same mobility in cell culture (data not shown). Embryos were scored by their morphology at both the 6–8 somite stage (SS) and 28 hpf. At the 6–8 SS, embryos that showed wider somites than wild-type uninjected embryos were scored as having a CE defect (data not shown). At 28 hpf, embryos with a shorter A/P axis or with eye or forebrain patterning defects were scored separately. Live embryos were photographed at 28 hpf after orienting in 3% methylcellulose. For statistical analysis of morphological defects, embryos with CE defects and eye defects were considered altered. We performed Fisher's Exact Test to compare mutants to uninjected embryos. p values smaller than 0.05 were considered statistically significant.
Intracellular Ca2+ Imaging
The ratiometric Ca2+-sensing dye Fura-2 (Molecular Probes) was injected into 1-cell zebrafish embryos. The excitation spectra are different between Ca2+-bound Fura-2 (340 nm) and Ca2+-free (380 nm) forms. By taking the ratio of the fluorescence intensity at these wavelengths, one can derive an estimate of intracellular free Ca2+. prickle2 or prickle2 mutant RNA (75 ng/μl RNA) was co-mixed with Fura-2, and 4–6 nl was injected into embryos. Embryos were oriented in a lateral position in a glass-bottomed dish on a Zeiss axiovert epifluorescence microscope. Image pairs at 340 and 380 nm excitation wavelengths (510 nm emission) were collected at 15 s intervals. Each imaging session collected 200 image pairs. The ratio image, a pixel-by-pixel match of both excitation wavelengths, is calculated by computer software (RatioTool, Inovision). The sequence of ratio images was processed, and the Ca2+ fluxes (transients) were determined with a subtractive analog as described.8 The ratio image (340 nm, Ca2+ saturated; and 380 nm, Ca2+ free) imported for publication was encoded in 8 bits and converted to pseudocolor ; a low ratio (low Ca2+) is represented by blue, and a high ratio (high Ca2+) is represented by yellow and red.
In Situ Hybridization
An RNA probe against mouse prickle2 (nucleotides 1262–1891, NM_001081146.2) was generated by in vitro transcription from a plasmid containing prickle2 cDNA, and this probe was then labeled with digoxigenin. Mice were fixed by perfusion with 4% paraformaldehyde, and the brains were left to fix in the skull overnight. After brains were dissected from the skull, they were split sagittally and digested with 10 μg/ml Proteinase K (Ambion, Austin, TX, USA) for 40 min. The half brains were hybridized overnight at 60°C in hybridization solution with 50% formamide, 50% 2× saline sodium citrate, and 6% dextran sulfate. Excess probe was digested and washed off. Each half brain was then incubated overnight with anti-digoxigenin antibody (1:2000, Roche diagnostics, Mannheim, Germany) conjugated with alkaline phosphatase. Nitroblue phosphate (BM purple substrate, Roche Diagnostics, Germany) was used for probe detection. After reactions were stopped, each half brain was washed, and images were taken with a Leica M205 FA microscope and Leica Suite V3 software.
Immunohistochemistry of Mouse Prickle2
Wild-type or mutant Prickle2 brain was harvested, fixed with 4% PFA, and sectioned with a vibratome. Sections were 100 μm thick. The sections were stained while they were floating. After staining, they were placed on a slide and mounted. The staining conditions were as follows: sections were washed with a Ca2+- and Mg2+-free phosphate-buffered saline (PBS) and permeabilized with 0.5% Triton X-100 PBS for 30 min at room temp. This was followed by incubation with a blocking solution (0.25% Triton X-100, 2% bovine serum albumin [BSA], and 1× PBS at pH 7) for 1 hr at room temperature. They were then stained with primary Prickle2 rabbit antibody at a 1:500 dilution in blocking solution for 3–4 hr; washed with 0.25% Triton X-100 PBS three times for 30 min at room temp; and incubated with anti-rabbit secondary antibody coupled to Alexa488 (Invitrogen) at a 1:500 dilution. Subsequently, the slices were again washed three times for 30 min in 0.25% Triton X-100 PBS. After these three washes, each slice was washed overnight before being mounted in 0.25% Triton X-100 PBS at 4°C. Mounting was done with Flouromount-G (Southern Biotech).
Production and Genotyping of Prickle1 and Prickle2 Mutant Mice
Prickle1 Riken Center for Developmental Biology accession number CDB0431K; GenBank accession number NM_001033217.4) mutant mice were produced as described previously.9 The Prickle2 (accession number CDB0435K; NM_001081146.2) mutant mice were generated via gene targeting in TT2 ES cells10,11 as described. We analyzed Prickle1 and Prickle2 mutant mice produced from the eighth backcross on C57BL/6. The genotyping of Prickle2 mutant mice was routinely performed by PCR with a mixture of three primers. The sizes of the PCR products were 796 bp for the wild-type allele and 609 bp for the mutant allele. The PCR primers were as follows: P1 (5′-GAC CTC ATC TAC TTT TAC CAA-3′) and P3 (5′-TAC TAC CAC CCA CTT TAT TCT-3′) for the Prickle2 wild-type allele; P2 (5′-GGC TCT TTA CTA TTG CTT TAT-3′) and P3 for the Neo gene. The PCR conditions were 95°C for 2 min, 35 cycles of 95°C for 10 s, 56°C for 10 s, and 72°C for 30 s, followed by 72°C for 10 min.9–11
Mice with the p.Phe141Ser variant (Prickle1Phe141Ser) and mice with the p.Cys251X variant (Prickle1Cys251X) mutant mice were generated from an ENU-mutagenesis screen from Ingenium Pharmaceuticals (Munich, Germany). The point mutant lines identified from the mutagenesis screen was backcrossed to C57/BL6 via a rapid congenic protocol12 (genome-wide microsatellite markers used for genotyping are available upon request). Like Prickle1-null mutants, no live homozygous Prickle1Cys251X were identified in newborn pups, but Prickle1Cys251X heterozygotes and wild-type pups were found in the expected numbers and were used for maximal electroshock seizure threshold (MEST) testing. Prickle1Phe141Ser live-born mice were found in all expected genotype combinations at birth, but there were insufficient numbers of homozygous Prickle1Phe141Ser mice for MEST testing at the time of submission.
Pentylenetetrazol Evoked Seizures and Electrocorticography (ECoG) Recordings
Three 3.2 mm stainless steel screws (Stoelting) were implanted above the right frontal lobe, left parietal lobe, and right cerebellum. The first two electrodes were to serve as epidural recording electrodes, and the latter were to serve as a reference or ground electrode. The coordinates relative to the bregma were as follows. Frontal: anterioposterior +1.5 mm, lateral −1.5 mm. Parietal: anteroposterior −3.0 mm, lateral +1.5 mm. Cerebellar reference: anteroposterior −6.0 mm. At least 3 days were allowed for mice to recover prior to testing. Baseline ECoG activity was recorded for 15 min prior to intraperitoneal injection of pentylenetetrazol (PTZ; 50 mg/kg). Recordings were continued for 75 min after injection. ECoG activity was captured with a TDT MEDUSA preamplifier, and the base station and was recorded at a sampling rate of 2034.5 Hz with TDT OpenX software with high- and low-pass filters at 1 Hz and 3000 Hz, respectively. ECoG recordings were analyzed with MATLAB R2007b software. The threshold for spike scoring was an excess of five times the standard deviation of the mean preinjection baseline amplitude. The number of spikes was statistically analyzed with a Student's t test. Experimenters were blinded to the mice genotypes during all steps, including PTZ testing and data analyses. All animal experiments complied with the NIH Guide for the Care and Use of Laboratory Animals.
Testing MEST
The electrical stimulus required to reach maximal electroconvulsive seizure was determined as described previously13 in Prickle2+/−, Prickle2−/−, Prickle1+/− , Prickle1+/Cys251X, Prickle1+/Phe141Ser, and wild-type mice. The electroshock was given for 0.2 s at 60 Hz with a maximal voltage of 500 V via the Rodent Shocker-type 221 (Harvard Apparatus, Holliston, MA) with ear electrodes moistened with saline. Generalized seizure with hind-limb extension was assessed. Fischer's exact test was used for statistical analysis. All mice were 10–14 weeks old. Wild-type mice (n = 43) were C57/B6 littermates of prickle mutant mice tested at 7 mA and pooled into one large group. Seizure rates between different sets of wild-type mice were similar (data not shown). The following numbers of mice were used for testing: The following numbers of mice were tested: Prickle1+/− (n = 23), Prickle1Cys251X (n = 5), Prickle1Phe141Ser (n = 7), Prickle2+/− (n = 14), and Prickle2 −/− (n = 7). Seizure rates were also recorded for wild-type, Prickle1+/−, Prickle2+/−, and Prickle2−/− at 8 mA and 9 mA. There was no statistical difference between genotypes at 8 mA and 9 mA, although overall seizure rates increased (not shown).
Drosophila Lines and Genetics
The pksple1/pksple1 flies were obtained from the Bloomington Drosophila Stock Center housed at Indiana University. pksple1arose spontaneously.3 Laboratory yw67 or Oregon-R (kindly provided by Chun-Fang Wu) males were crossed with pksple1homozygous mutant female flies to produce the pksple1/+ heterozygous flies; these crosses produced flies whose background was either 50% yw67 or 50% Oregon-R. Both the control flies and pksple1 heterozygotes were subjected to identical culture conditions and standard Drosophila cornmeal food media.
Immunocytochemistry of Fly Brains and Embryos
Crawling third-instar larvae from the laboratory strain yw67 were bisected, and the interior halves were inverted in PBS. After fixation in 4% formaldehyde in PBS for 30 min, the carcasses were washed five times for 10 min in PBST (PBS and 0.1% Triton X-100), blocked with PBSTB (PBST and 1% BSA) for 40 min, and incubated with a 1:2000 dilution of rabbit polyclonal antibody raised against Drosophila prickle (provided by Jeff Axelrod) overnight at 4° centigrade. The carcasses were washed five times for 10 min in PBST, blocked with PBSTB for 40 min, and incubated with a fluorphore-conjugated secondary antibody (Alexa Fluor 488 goat anti-rabbit IgG (H+L); Invitrogen catalog number A-11008) for 90 min at room temperature. The carcasses were washed five times for 10 min with PBST, and the brains were dissected from the carcasses and mounted in Vectashield containing DAPI (Vector Laboratories). Images were obtained with a Leica TCS SPE confocal microscope.
Peripheral nervous system neurons were visualized in 14- to 16-hr-old yw67 or pksple1 homozygous mutants via a 22C10 primary antibody (1:200 dilution of antibody concentrate, Developmental Studies Hybridoma Bank, University of Iowa, Department of Biology) followed by an Alexa Fluor 488 goat anti-mouse IgG (H+L) (Invitrogen catalog number A-11001) secondary antibody. Embryo fixation and antibody incubations were performed as previously described,14 and images were obtained via fluorescence microscopy.
Modified Bang-Sensitivity Behavioral Assay
The “bang-sensitivity” behavioral assay measuring recovery from epileptic seizures was performed as previously described with slight variation15–17. Twenty flies (ten females and ten males) aged up to 7 days after eclosion were collected under C02 and transferred to an empty glass vial demarcated with centimeter markers up to 7 cm. Vials with either control or mutant flies were mechanically stimulated by insertion into a Fisher Scientific vortex mixer set to “10” (highest setting). The flies were first prevortexed for 20 s to ensure that all flies were able to survive the assay and climb after vortexing (control and mutant flies were age matched and vortexed simultaneously). Two minutes later, flies were simultaneously vortexed for 20 s and then placed on a flat lab bench. Flies were digitally videotaped with a Panasonic SDR-S7 camera. Digital video was then reviewed frame by frame at 5 s intervals up to 25 s. As previously defined, flies were scored as “recovered” when they climbed to the top of the vial (7 cm). In addition to the climbing assay, we scored flies for recovery “off bottom” if they were able to recover from seizure activity, begin coordinated locomotive behavior, and leave the bottom of the vial. All scoring was statistically analyzed with Chi-Square analysis.
Valproic Acid Treatment of Drosophila
Either no valproic acid or 10 mM valproic acid (Sigma; final concentration) was added to standard cornmeal-based fly food, and 1- to 7- day-old flies were allowed to consume the agar for 84 hr, after which the bang-sensitivity assay was performed immediately. This dosage of valproic acid was lethal in homozygous pksple1 flies, and only heterozygotes were used for this testing.
Statistical Methods
For statistical evaluation of association between missense variants in prickle genes and myoclonus epilepsy, we used Fisher's two-tailed exact test. Missense variation or no missense variation are the possible categorical outcomes for each chromosome. Affected and normal (ethnically matched controls) are the two groups. For the affected group, 5/176 chromosomes were missense, and for the control group, 0/352 chromosomes were missense.
For statistical evaluation of neurodevelopmental anomalies in pksple1/pksple1 versus yw67 control flies, we used Fisher's two-tailed exact test and neurodevelopmental anomaly versus no neurodevelopmental anomaly as the possible categorical outcomes for these two groups of flies. For pksple1/ pksple1, 11/230 presented with neurodevelopmental anomalies; for yw67 controls, 0/399 were anomalous.
MEST and the bang-sensitivity assay were evaluated by Chi-Square analysis. PTZ testing was evaluated with the Student's t test. The zebrafish development assay was analyzed with the Fisher's exact test. n values for all experiments are included in the figure legends.
Results
Heterozygous Mutations in PRICKLE1 or PRICKLE2 in Epilepsy Patients
Our original report of a human PRICKLE1 mutation described a homozygous mutation in PRICKLE1 in families with myoclonus epilepsy-ataxia syndrome.2 However, the role of aberrant PRICKLE1 and the closely related PRICKLE2 in the general population of patients with myoclonus epilepsy has not been examined. To test whether mutations in prickle genes are associated with epilepsy, we sequenced PRICKLE1 and PRICKLE2 in 88 unrelated patients with myoclonus epilepsy. We found two patients with missense mutations in PRICKLE1 and two patients with missense mutations in PRICKLE2 (Table 1, patients 2–5).18 In addition, we identified a patient with epilepsy and an interstitial chromosomal deletion encompassing PRICKLE2. (Table 1, patient 6). All four patients with missense changes were heterozygous for the variants and had neither deletions nor duplications in PRICKLE1 or PRICKLE2 exons. We did not identify the variants observed in our patients in any of the 2100 Centre d'Etudes du Polymorphisme Humain Human Genome Diversity (CEPH-HGD) control chromosomes we screened, none of the variants are reported in the 1000 genomes database or in dbSNP, and we found no novel variants or deletions in the PRICKLE1 and PRICKLE2 genes in 352 ethnically matched control chromosomes (two-tailed Fisher's exact test, p value for association = 0.004; see statistical methods). (An additional patient with a PRICKLE2 deletion and one with a PRICKLE1 deletion were also identified by Signature Genomics [not shown]. They are known to have neurological phenotypes, but epilepsy was not reported at the time of testing, and additional information could not be gathered). No patients with PRICKLE1 or PRICKLE2 exon-encompassing deletions have been reported in the SickKids Foundation Database of Genomic Variants the Children's Hospital of Philadelphia, the Database of Genomic Variants Archive (DGVa) at the European Bioinformatics Institute, or the Database of Genomic Structural Variation (dbVar) at the National Center for Biotechnology Information. These results suggest that the heterozygous variants we observed in the prickle genes are associated with myoclonus epilepsy.
Mutant Prickle2 Shows Decreased Activity in Zebrafish Overexpression Assays
Misregulation of WNT/PCP has been shown to disrupt convergent-extension (C-E) movements during gastrulation in vertebrates19.We have previously demonstrated that overexpression of mutant PRICKLE1 identified in epilepsy patients has a decreased ability to produce C-E defects in developing zebrafish embryos.2 To investigate whether mutant PRICKLE2 variants identified in epilepsy patients are functionally different from the wild-type in vivo, we injected RNA encoding either the wild-type or mutant prickle2 RNAs (for which mutations in the zebrafish were orthologous to those found in epilepsy patients 4 and 5; Table 1) into zebrafish embryos. Overexpression of wild-type Prickle2 alters C-E movements in gastrulation, resulting both in a reduced anterior-posterior length in comparison to that of uninjected embryos (Figures 1A and 1B) and in forebrain and eye defects (Figures 1C and 1D). Embryos expressing prickle2 mutant RNA showed a significantly different phenotype than did those expressing wild-type prickle2. The p.[Arg148His;Val153Ile] prickle2 ortholog showed increased activity compared to wild-type activity, and the p.Val605Phe prickle2 ortholog showed decreased activity compared to wild-type activity (Table 2). Thus, in an overexpression assay, the identified human PRICKLE2 mutations show altered activity in zebrafish.
Figure 1.
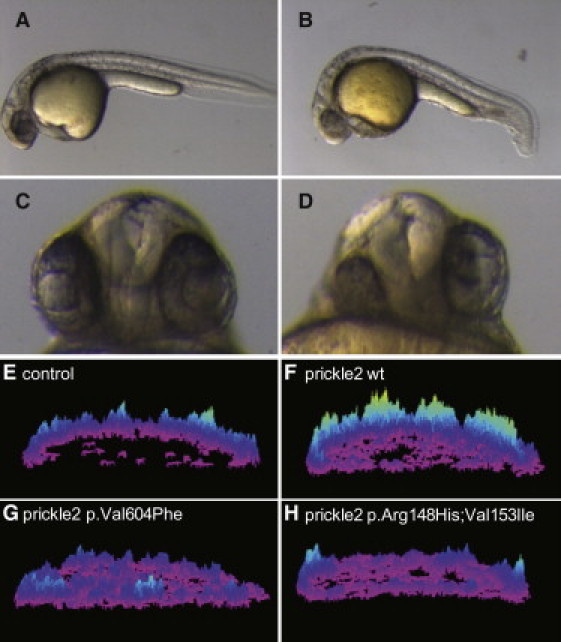
Zebrafish Prickle Mutations Orthologous to Those Identified in Human Epilepsy Patients Show Altered Activity in CE and Calcium Release
(A–D) Morphological phenotypes at 28 hpf. Compared to wild-type uninjected embryos (A), prickle2-injected embryos (B) display a shorter A-P axis and a kinky tail. The lateral view is shown, and anterior is to the left. Moreover, prickle2-injected embryos (D) show defects in eye and forebrain patterning at 28 hpf. (C) Uninjected embryo. Ventral view. The numbers of embryos with defects in each RNA-injected group are shown in Table 2.
(E–H) Surface plots of calcium release activity were generated from images of live zebrafish embryos. The height and color of the peaks indicates the number of calcium fluxes observed over the course of the experiment; the embryos are oriented in a lateral position. (E) A control injected embryo. (F) An embryo overexpressing wild-type prickle2 RNA. (G) An embryo overexpressing prickle2Val605Phe-mutant-encoding RNA. (H) An embryo overexpressing prickle2 Arg148His; Val153Ile-mutant-encoding RNA.
Table 2.
Summary of Overexpression Phenotypes in Zebrafish Embryos
| CE Defects | CE+ Eye Defects | Normal | p Value, Compared to prickle2 Wild-Type | |
|---|---|---|---|---|
| Uninjected (n = 58) | 0 | 0 | 100% | <0.01 |
| prickle2 wild-type (n = 139) | 79% | 34.5% | 20.1% | |
| prickle2Arg148His;Val153Ile (n = 205) | 88.3% | 28.3% | 11.7% | 0.045 |
|
prickle2Val605Phe (n = 148) |
66.2% | 34.5% | 33.8% | 0.012 |
prickle2 Mutations Result in Reduced Calcium Activation in Zebrafish
We next sought to evaluate the in vivo physiological consequences of identified human PRICKLE2 mutations. Overexpression of zebrafish prickle1 in the developing embryo is sufficient to activate calcium release frequency.5 We therefore evaluated the impact of wild-type and mutant prickle2 on calcium release dynamics in vivo. Embryos injected with the calcium sensor Fura-2 and expressing wild-type or mutant prickle RNAs were imaged continuously at 15 s intervals during early epiboly stages (Figures 1E–1H). We used a subtractive algorithm to evaluate calcium release dynamics,5 and changes are represented as a color-scale surface plot showing the number of events relative to the location in the embryo for the duration of the time course. In uninjected embryos, we observed a characteristic frequency of endogenous calcium release (Figure 1E, Movie S1). Expression of wild-type prickle2 RNA in zebrafish embryos was sufficient to activate calcium release (Figure 1F, Movie S2). Embryos expressing either prickle2Val605Phe (Figure 1G and Movie S3) or prickle2Arg148His;Val153Ile (Figure 1H and Movie S4) were less active in stimulating calcium release than those expressing wild-type prickle2 RNA. These results suggest that the missense PRICKLE2 mutations identified in epilepsy patients alter the in vivo physiological activity of Prickle2 in zebrafish.
Decreased Seizure Threshold in Mouse Models Deficient for the Prickle Genes
To directly test whether heterozygous mutations in prickle genes might cause epilepsy, we evaluated seizure threshold in the following mutant mouse lines: (1) Prickle1+/−, (2) Prickle1 Cys251X, which has a mutation encoding a nonsense amino acid change that truncates the Prickle1 protein shortly after the PET and LIM domains, (3) Prickle1 Phe141Ser heterozygous mutant mice, which have a mutation encoding a Phe141Ser amino acid change that alters an amino acid in the PET/LIM domain that is conserved throughout evolution, and (4) in mice heterozygous for null mutations in Prickle2 . Seizure threshold was tested via the maximal electroconvulsive seizure threshold (MEST) test, which scores the occurrence of tonic hind-limb seizures at a specific amperage of applied electrical shock. Prickle1+/−, Prickle1+/Cys251X, Prickle1+/Phe141Ser, and Prickle2 heterozygous (Figure 2A) mice all displayed a decreased seizure threshold compared to that of wild-type littermates. These results demonstrate that heterozygous null or point mutations in the prickle genes are sufficient to lower seizure threshold. Moreover, the seizure predisposition in the Prickle1+/Phe141Ser mice suggests that disruption of the highly conserved PET/LIM domain is sufficient to lower seizure threshold.
Figure 2.
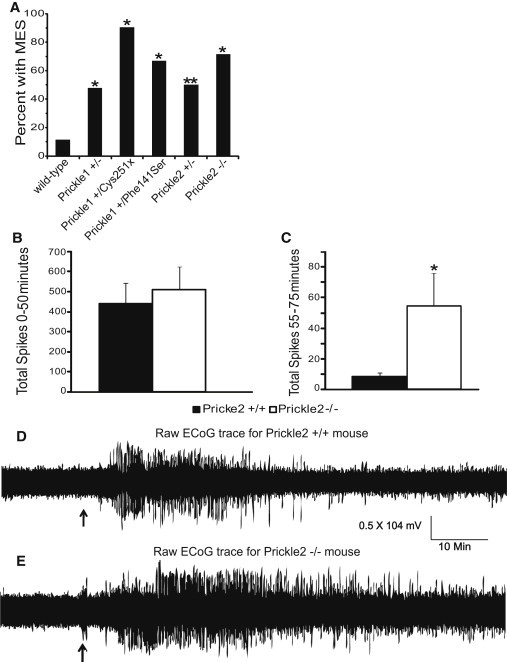
Mouse Models Deficient for the Prickle Genes Show an Increased Seizure Phenotype
(A) Maximal electroshock seizure threshold (MEST) in wild-type mice versus mice deficient for the prickle genes. Prickle1+/−, Prickle1+/Cys251X, Prickle1+/Phe141Ser, Prickle2+/−, and Prickle2−/− mice show a significantly lower MEST than wild-type mice at 7 mA (∗p < 0.001, ∗∗p < 0.005; wild-type n = 43, Prickle1+/− n = 23, Prickle1+/Cys251X n = 10, Prickle1+/Phe141Ser n = 12, Prickle2+/− n = 14, and Prickle2−/− n = 7).
(B and C) Total number of electrographic spikes recorded from electrocorticography (ECoG) in response to pentylenetetrazol (PTZ) injection for the first 50 min after injection (B) and the last 25 min of recording after injection (C). During the first 50 min after injection, there is no difference in the total number of spikes between wild-type and Prickle2−/− mice. However, there is a significant difference seen during the last 25 min of recording: Prickle2−/− mice had significantly more seizures than their wild-type counterparts. ∗p = 0.039 as determined by the Student's t test; wild-type n = 7, Prickle2−/− n = 6. Error bars represent SEM.
(D and E) Representative ECoG traces for wild-type and Prickle2−/− mice upon stimulation with PTZ. PTZ was injected (black arrow) after 15 min of baseline recording. ECoG was recorded for 75 min after injection. A wild-type raw trace recording is shown in (D). A Prickle2−/− raw trace recording is shown in (E). In comparison to wild-type mice, Prickle2−/− mice continue to seize for prolonged periods after injection.
Although we observed no live-born Prickle1 homozygous knockout mice9 or Prickle1Cys251X/Cys251X mice (see Subjects and Methods), we found viable Prickle2−/− mice at expected Mendelian ratios (Figures S1 and S2. Prickle1Phe141Ser/Phe141Ser mice were born at apparently normal Mendelian ratios and appear normal, but there were insufficient numbers at the time of submission to test for seizures). As we previously showed for Prickle1,2 Prickle2 is expressed in epilepsy-associated brain regions, including the hippocampus and cerebral cortex (Figure S3), and expression was lost in Prickle2-null mice (Figures S2C and S2D). Prickle2−/− mice displayed higher seizure rates than Prickle2+/− mice (Figure 2A). We also observed increased epileptiform discharges in Prickle2−/− mice compared to controls after treatment with the seizure-inducing GABA agonist pentylenetetrazole (PTZ) (Figures 2B–2E). These data suggest that Prickle2 dosage is directly related to seizure threshold.
Drosophila with Homozygous prickle Mutations Display Seizures
To test whether a prickle mutation would lead to seizures in Drosophila, we obtained homozygous pricklespiny-leg-1 (pksple1) mutant flies; this mutation was described more than 60 years ago as a recessive loss-of-function allele showing typical planar cell polarity abnormalities, including anomalies in the body epidermis and legs (and including duplications of leg tarsal joints).4,20 Despite diffuse localization of Prickle in the developing fly brain (Figure 3), neurological phenotypes have not been described for any mutant line deficient for the prickle genes. To formally test whether prickle mutations in flies lead to an epilepsy-like disorder, we subjected pksple1 homozygous mutants to a variation of the classic “bang sensitivity” assay.16,21 In this model for seizure sensitivity, a glass vial containing flies is vortexed, inducing seizures in flies genetically predisposed to this disorder (such flies demonstrate dramatic twitching behavior similar to seizures observed in mice and humans [Figure 4A], and electrographic recordings have substantiated these findings).16,21 The seizures prevent or delay flies from climbing the vial wall, which is the normal behavior of wild-type flies. Homozygous pksple1mutant flies displayed a severely delayed recovery resulting in a prolonged climbing response compared to that of yw67control flies (Figures 4B and 4C; see also Movie S5), even though the homozygous pksple1 flies climb in a manner similar to control flies both before and minutes after bang testing (Movies S6 and S7, respectively).
Figure 3.
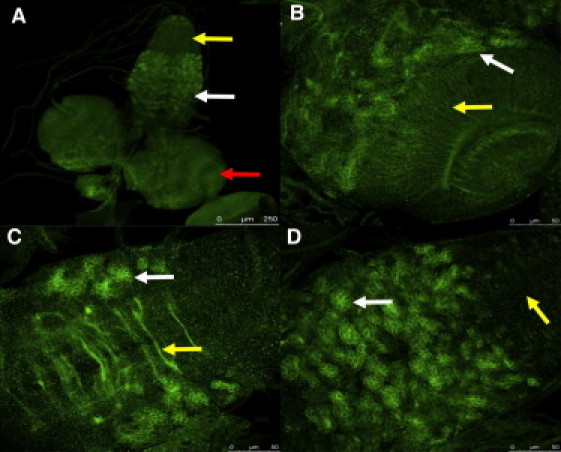
Prickle Is Expressed Widely in the Drosophila CNS
A Prickle-specific polyclonal antibody was used for visualization of Prickle in the crawling third-instar larval CNS. Most neurons of the larval CNS persist through metamorphosis and join groups of newly born adult-specific neurons to form the adult CNS.
(A) Prickle is localized in the optic lobes (one lobe is indicated by the red arrow), as well as central brain structures between the lobes, and to part of the ventral segmental ganglia (white arrow). High-level staining is not seen in ventral segmental ganglia that correspond to the abdominal segments (yellow arrow).
(B) A higher magnification of the optic lobe region of the brain shows Prickle staining in clusters of bundled neurons (white arrow) as well as neuronal projections spanning the optic lobe (yellow arrow).
(C) A higher magnification of the ventral segmental ganglia shows Prickle staining in the commisures that cross the ganglia (yellow arrow) as well as in clusters of neurons similar to those seen in the optic lobe regions of the brain (white arrow).
(D) A higher magnification of the surface of the ventral segmental ganglia shows Prickle staining in clusters of neurons (white arrow); note that the ventral segmental ganglia corresponding to the abdominal segments is largely devoid of high Prickle staining (yellow arrow).
Figure 4.
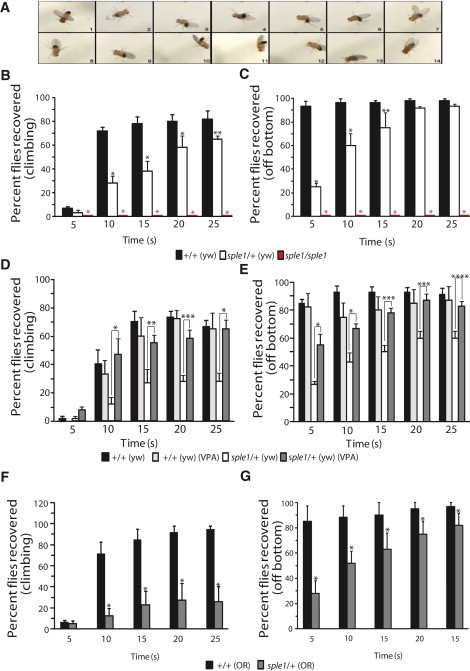
Drosophila that are Homozygous or Heterozygous for the prickle mutation pksple1Are Predisposed to Seizures, and Seizures are Diminished by Treatment with Valproic Acid
(A) Seizure in a pksple1 fly. Photos of typical homozygous pksple1-seizing fly were captured every 0.5 s after the bang assay. All control flies have already climbed up the side of the vial for the time period presented. For a live side-by-side comparison of control and pksple1 flies, see Movies S5–S9.
(B) pksple1 homozygous flies and pksple1 heterozygous flies in the yw67 (yw) background [pksple1/+ (yw)] have significantly impaired seizure recovery when they are compared to same-aged control yw67 [+/+(yw)] flies. ∗p < 0.001, ∗∗p = 0.039.
(C) pksple1 homozygous and heterozygous flies have significantly impaired recovery off the bottom in comparison to same-aged control +/+(yw) flies. ∗p < 0.001, ∗∗p = 0.001.
(D) The addition of valproic acid to fly food significantly diminishes seizures in pksple1 heterozygotes [pksple1/+ (yw)] and returns climbing ability to that of control +/+(yw) flies. ∗p < 0.001, ∗∗p = 0.002, ∗∗∗p = 0.001.
(E) The addition of valproic acid to fly food significantly recovers the ability of the flies to right themselves off the bottom of the vial and climb. ∗p = 0.002, ∗∗p = 0.010, ∗∗∗p = 0.001, ∗∗∗∗p = 0.005.
(F and G) pksple1 heterozygous flies are bang sensitive in multiple backgrounds. (F) pksple1 heterozygous flies in the Oregon-R (OR) background [sple1/+(OR)] have significantly impaired seizure recovery in comparison to same-aged wild-type Oregon-R [+/+ (OR)] flies. ∗p = 0.007. (G) sple1/+(OR) heterozygous flies have significantly impaired recovery off bottom in comparison to same-aged wild-type +/+(OR) flies. ∗p = 0.007. For all experiments, wild-type n = 60 and pksple1 n = 60. Error bars represent standard error of the mean; the data were statistically analyzed with the Chi-square test.
Heterozygous prickle Mutations Cause Seizures in Drosophila
To test whether the heterozygous prickle mutation was sufficient to decrease seizure threshold, we tested pksple1 heterozygotes (which we produced by crossing pksple1 homozygous mutants with yw67 control flies) for bang sensitivity. We found that pksple1 heterozygotes displayed none of the morphological abnormalities found in the homozygotes, such as tarsal duplications or altered bristle polarity (data not shown), yet the pksple1 heterozygotes were significantly more bang sensitive than control flies of the same age (Figure 4B and Movie S8). In addition to using the classic bang-sensitivity parameters, we also scored the time it took for the flies to leave the bottom of the vial. Again, pksple1 heterozygotes were significantly impaired compared to controls (Figure 4C) and displayed an agitated recovery period in which they visibly exhibited excessive excitability in the form of grooming themselves more than usual and flipping over on their backs and then righting themselves (Figure 4A and Movie S9).
To test whether anti-epileptic drugs reverse the bang sensitivity, we used valproic acid (VPA), an anti-epileptic medication used for treatment of myoclonus epilepsy in humans (including humans with PRICKLE1 mutations2) and shown to decrease seizure activity in other bang-sensitive flies.22 VPA strongly decreased bang sensitivity in pksple1 heterozygotes, whose climbing behavior resembled that of the control fly group (Figures 4D and 4E).
In order to test whether the prickle epilepsy phenotype could be observed in two different genetic backgrounds (which would reduce the likelihood that a particular genetic background had a role in the phenotype), we crossed the pksple1 homozygotes to a second laboratory strain, Oregon-R. Both the Oregon-R and yw67 flies are standard “workhorse” laboratory strains that have been utilized for many years. The Oregon-R strain is derived from wild flies that were collected in 1925 or earlier by DE Lancefield at Roseburg, Oregon.23 The yw67 flies have a less clear origin and contain two mutations (in the yellow and white genes) useful in molecular genetic manipulations, such as P-element-mediated transformation. These mutations affect body and eye color, respectively. yw67 is one of the most common laboratory strains utilized by Drosophila researchers worldwide because of its ease of culture and usefulness in transformation. The w67 mutation was generated by Lefevre, probably in a Canton S background because this was the wild-type strain he often used for his mutagenic studies on the white gene.24 Nonetheless, because of their different genotypic backgrounds, yw67 and Oregon-R have been cultured separately for many years and thus can be appropriately considered to be separate fly strains25,26. We compared pksple1/+ (Oregon-R) heterozygous flies to the wild-type Oregon-R flies. As in the yw67 background, pksple1 heterozygous flies in the Oregon-R background showed increased seizures in a comparison with controls (Figures 4F and 4G).
Homozygous pksple1 Mutant Embryos Display Neuronal Defects
In order to determine whether a developmentally controlled neuronal defect might be causative for the epilepsy phenotype in flies, we stained pksple1 homozygous mutant embryos with the 22C10 antibody, widely used to stain neurons of the peripheral nervous system (PNS). Because we had observed a decreased viability of pksple1 homozygous mutant flies (data not shown), we specifically looked for gross defects in the architecture of the developing nervous system (Figure 5). We focused on obvious defects represented by altered patterns of 22C10 staining that were present in fully intact embryos showing no damage from the antibody staining and mounting procedure. Thus, we identified 11 out of 230 prickle mutant embryos that showed such defects, whereas 0 out of 399 control embryos showed such defects (two-tailed Fisher's exact test, p value = 0.0001). One striking feature of the mutant embryos was a generalized disorganization of the PNS: aberrant migration of neuronal processes resulted in improper connections. For example, Figure 5B shows a mutant embryo whereby two neuronal processes associated with contiguous chordotonal organs have improperly fused.
Figure 5.
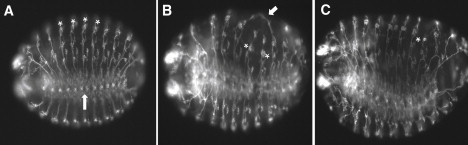
Neuronal Defects Are Observed in prickle Mutant Embryos
Images (20×) of control 14–16 hr yw67 embryos (A) are compared to two 14–16 hr pksple1homozygous mutant embryos (B and C) that have been stained with the 22C10 antibody so that PNS neurons are visualized. Note the location of chordotonal organs (asterisks in A) as well as the location of the ventral nerve chord (upward arrow in A) in the control embryo. For the mutant embryo in (B), note the aberrant neuronal processes that have joined (arrow) as well as the abnormal location of two chordotonal organs (asterisks). For the mutant embryo in (C), note the overall disorganized PNS staining pattern, as well as the improperly positioned neurons associated with the chordotonal organ in abdominal segment 6 (asterisk). Anterior is to the left, and ventral is down but slightly rotated toward the viewer.
Discussion
Most generalized epilepsy-causing mutations have been identified in genes that encode ion channels.1,27–31 The work presented here is atypical in that it affects a non-ion-channel protein. The original descriptions of non-ion-channel-generalized epilepsy-associated mutations showed autosomal-recessive inheritance,32–34 with the exception of EF-HAND DOMAIN (C-TERMINAL)-CONTAINING PROTEIN 1 (EFHC1, [MIM 608815]) mutants, which are autosomal dominant.35,36 However, both humans and mice heterozygous for mutations in the myoclonus epilepsy gene CYSTATIN B (CSTB, [MIM 601145]) have been reported to display EEG and clinical abnormalities37–39 (also see Table S2). Similarly, in the case of PRICKLE1, both heterozygous and homozygous genotypes can be associated with myoclonus epilepsy in humans. Finally, the finding that both heterozygous and homozygous mutations in both vertebrates and invertebrates can lead to epilepsy suggests that the dosage of the various prickle proteins is critical in carrying out their normal functions.
The precise mechanism for how seizures are produced remains a mystery. This work implicates prickle-dependent signaling in seizure prevention. It is not yet certain how abnormal prickle proteins cause epilepsy, but we envision two possible mechanisms. First, during vertebrate embryogenesis, prickle proteins and other planar cell polarity (PCP) core components have been shown to control the formation of multiple structures through the regulation of cell polarity,6,8,40–52 suggesting that during neurogenesis, PCP might contribute to the development of a fully functional neural network, by directing neural migration (or the migration of neuronal processes such as axons and dendrites) via a polarized plane, by polarizing a neuron that has properly migrated to its final position (either through shuttling cellular components to particular locations along a directional axis or by positioning the location of neuronal structures such as axons and dendrites), or a combination of both. This possibility is supported by the findings that prickle1b knockdown disrupts motor neuron migration in zebrafish,51 Prickle knockdown disrupts neurite extension in vitro,53 and social interaction is abnormal in mice with mutations in the PCP gene Dvl154. Our results demonstrating migration defects of neurons or neuronal processes in pksple1 homozygous mutant fly embryos are consistent with these developmentally based mechanisms and suggest that a defect specifically affecting neuronal positioning could be the root cause of epilepsy in prickle mutants.
Second, the altered calcium signaling in prickle mutants combined with decreased seizure activity in the presence of valproic acid (an anti-epileptic medication that has several effects on calcium signaling, including interference with inositol and alteration of calcium currents2,55,56) suggests that prickle-mediated calcium signaling might underlie prickle function in the nervous system and that prickle-mediated epilepsy is related to dysregulated calcium currents. In both cases, the cellular pathology of defective neurons may be attributable to the disruption of polarized trafficking of membrane and functional proteins, including channels that are required for proper regulation of neural activities and neuron networking, highlighting the essential role of PCP core components in neurogenesis and in the regulation of brain function.57
An epilepsy phenotype that is found in disparate species and crosses the invertebrate-vertebrate transition strongly suggests that the prickle proteins are part of a highly conserved evolutionary pathway for regulating seizures. Drosophila provide a powerful genetic tool for dissecting the key elements of this seizure pathway in the form of suppressor and enhancer screens. Furthermore, given the ease with which compounds can be added to fly food, and the ability to rapidly screen for seizure activity, we foresee that the results presented here will promote the efficient translation of prickle-related epilepsy into novel treatments.
Acknowledgments
This work was supported by National Institutes of Health grants R01 NS064159 and 3R01NS064159-02S1 (to A.G.B.), R01 GM059823 (to J.D.A.), RO1 CA112369 (to D.C.S.), and P50 AG05136 (to T.M.), by 1R21NS058309-01A1 (to J.W.), by a VA Merit Review Award (to J.W.), and by a grant from the Howard Hughes Medical Institute (to M.S.). We thank Jeff Murray, Jeff Neul, Val Sheffield, Bev Davidson, Zoha Kibar, Heather Mefford, and Sam Berkovic for their thoughtful reviews. We thank Jeff Murray for access to the CEPH-HGD samples and Taqman assay. L.G.S. and J.A.R. are employed by Signature Genomics Laboratories.
Contributor Information
Naoto Ueno, Email: nueno@nibb.ac.jp.
Alexander G. Bassuk, Email: alexander-bassuk@uiowa.edu.
Supplemental Data
Time-lapse video (the embryo is oriented in a lateral view) consisting of pseudocolored ratio images derived from image pairs (340 and 380 nm excitation wavelengths) collected in 15 s intervals over a 50 min period. Low Ca2+ is represented by blue, and high Ca2+ is represented by yellow and red.
Time-lapse video (the embryo is oriented in a lateral view) consisting of pseudocolored ratio images derived from image pairs (340 and 380 nm excitation wavelengths) collected in 15 s intervals over a 50 min period. Low Ca2+ is represented by blue, and high Ca2+ is represented by yellow and red.
Time-lapse video (the embryo is oriented in a lateral view) consisting of pseudocolored ratio images derived from image pairs (340 and 380 nm excitation wavelengths) collected in 15 s intervals over a 50 min period. Low Ca2+ is represented by blue, and high Ca2+ is represented by yellow and red.
Time-lapse video (the embryo is oriented in a lateral view) consisting of pseudocolored ratio images derived from image pairs (340 and 380 nm excitation wavelengths) collected in 15 s intervals over a 50 min period. Low Ca2+ is represented by blue, and high Ca2+ is represented by yellow and red.
pksple1 homozygous flies (left) display a severe bang-sensitive phenotype; most flies remain at the bottom of the vial throughout the testing period. Wild-type flies are shown on the right.
pksple1 homozygous flies (left) climb normally as compared to wild-type flies (right) prior to bang testing.
pksple1 homozygous flies (left) climb normally as compared to wild-type flies (right) after bang testing.
pksple1 heterozygous (right) flies display delayed climbing and increased seizures as compared to wild-type flies (left).
pksple1 heterozygous (right) flies display delayed climbing off the bottom and increased seizures as compared to wild-type flies (left). Note that several pksple1 heterozygous flies at the bottom of the vial are on their backs and shaking.
Web Resources
The URLs for data presented herein are as follows:
Copy Number Variation, http://cnv.chop.edu
Database for Genomic Variants. http://projects.tcag.ca/variation/
Database of Genomic Variants Archive, http://www.ebi.ac.uk/dgva/page.php
NCBI Database of Genomic Structural Variation, http://www.ncbi.nlm.nih.gov/dbvar/
NCBI Single Nucleotide Polymorphism, http://www.ncbi.nlm.nih.gov/projects/SNP/
Mutant Mice generated in Center for developmental Biology, http://www.cdb.riken.jp/arg/mutant%20mice%20list.html
1000 Genomes, A Deep Catalog of Human Genetic Variation, http://browser.1000genomes.org/index.html
Accession Numbers
The Riken Center for Developmental Biology accession number for the Prickle2 mutant mice is CDB0435K
References
- 1.Frankel W.N. Genetics of complex neurological disease: challenges and opportunities for modeling epilepsy in mice and rats. Trends Genet. 2009;25:361–367. doi: 10.1016/j.tig.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bassuk A.G., Wallace R.H., Buhr A., Buller A.R., Afawi Z., Shimojo M., Miyata S., Chen S., Gonzalez-Alegre P., Griesbach H.L. A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am. J. Hum. Genet. 2008;83:572–581. doi: 10.1016/j.ajhg.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldschmidt R.B. A study of a spontaneous mutation. University of California Publications in Zoology. 1945;49:503–504. [Google Scholar]
- 4.Gubb D., Green C., Huen D., Coulson D., Johnson G., Tree D., Collier S., Roote J. The balance between isoforms of the prickle LIM domain protein is critical for planar polarity in Drosophila imaginal discs. Genes Dev. 1999;13:2315–2327. doi: 10.1101/gad.13.17.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Veeman M.T., Slusarski D.C., Kaykas A., Louie S.H., Moon R.T. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- 6.Deans M.R., Antic D., Suyama K., Scott M.P., Axelrod J.D., Goodrich L.V. Asymmetric distribution of prickle-like 2 reveals an early underlying polarization of vestibular sensory epithelia in the inner ear. J. Neurosci. 2007;27:3139–3147. doi: 10.1523/JNEUROSCI.5151-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 8.Mapp O.M., Wanner S.J., Rohrschneider M.R., Prince V.E. Prickle1b mediates interpretation of migratory cues during zebrafish facial branchiomotor neuron migration. Dev. Dyn. 2010;239:1596–1608. doi: 10.1002/dvdy.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tao H., Suzuki M., Kiyonari H., Abe T., Sasaoka T., Ueno N. Mouse prickle1, the homolog of a PCP gene, is essential for epiblast apical-basal polarity. Proc. Natl. Acad. Sci. USA. 2009;106:14426–14431. doi: 10.1073/pnas.0901332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murata T., Furushima K., Hirano M., Kiyonari H., Nakamura M., Suda Y., Aizawa S. ang is a novel gene expressed in early neuroectoderm, but its null mutant exhibits no obvious phenotype. Gene Expr. Patterns. 2004;5:171–178. doi: 10.1016/j.modgep.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Yagi T., Tokunaga T., Furuta Y., Nada S., Yoshida M., Tsukada T., Saga Y., Takeda N., Ikawa Y., Aizawa S. A novel ES cell line, TT2, with high germline-differentiating potency. Anal. Biochem. 1993;214:70–76. doi: 10.1006/abio.1993.1458. [DOI] [PubMed] [Google Scholar]
- 12.Wakeland E., Morel L., Achey K., Yui M., Longmate J. Speed congenics: a classic technique in the fast lane (relatively speaking) Immunol. Today. 1997;18:472–477. doi: 10.1016/s0167-5699(97)01126-2. [DOI] [PubMed] [Google Scholar]
- 13.Mares P., Kubova H. Electrical stimulation-induced models of seizures. In: Pitkanen A., Schwartzkroin P.A., Moshe S.L., editors. Models of Seizures and Epilepsy. Elsevier Academic Press; New York: 2006. [Google Scholar]
- 14.Mathies L.D., Kerridge S., Scott M.P. Role of the teashirt gene in Drosophila midgut morphogenesis: secreted proteins mediate the action of homeotic genes. Development. 1994;120:2799–2809. doi: 10.1242/dev.120.10.2799. [DOI] [PubMed] [Google Scholar]
- 15.Benzer S. From the gene to behavior. JAMA. 1971;218:1015–1022. [PubMed] [Google Scholar]
- 16.Ganetzky B., Wu C.F. Indirect Suppression Involving Behavioral Mutants with Altered Nerve Excitability in DROSOPHILA MELANOGASTER. Genetics. 1982;100:597–614. doi: 10.1093/genetics/100.4.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fergestad T., Olson L., Patel K.P., Miller R., Palladino M.J., Ganetzky B. Neuropathology in Drosophila mutants with increased seizure susceptibility. Genetics. 2008;178:947–956. doi: 10.1534/genetics.107.082115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bird T.D., Shaw C.M. Progressive myoclonus and epilepsy with dentatorubral degeneration: a clinicopathological study of the Ramsay Hunt syndrome. J. Neurol. Neurosurg. Psychiatry. 1978;41:140–149. doi: 10.1136/jnnp.41.2.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roszko I., Sawada A., Solnica-Krezel L. Regulation of convergence and extension movements during vertebrate gastrulation by the Wnt/PCP pathway. Semin. Cell Dev. Biol. 2009;20:986–997. doi: 10.1016/j.semcdb.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin Y.Y., Gubb D. Molecular dissection of Drosophila Prickle isoforms distinguishes their essential and overlapping roles in planar cell polarity. Dev. Biol. 2009;325:386–399. doi: 10.1016/j.ydbio.2008.10.042. [DOI] [PubMed] [Google Scholar]
- 21.Song J., Tanouye M.A. From bench to drug: human seizure modeling using Drosophila. Prog. Neurobiol. 2008;84:182–191. doi: 10.1016/j.pneurobio.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuebler D., Tanouye M. Anticonvulsant valproate reduces seizure-susceptibility in mutant Drosophila. Brain Res. 2002;958:36–42. doi: 10.1016/s0006-8993(02)03431-5. [DOI] [PubMed] [Google Scholar]
- 23.Lindsley . Carnegie Institution; Washington, DC: 1968. Genetic Variations of Drosophila melanogaster. p 472. [Google Scholar]
- 24.Lefevre G., Jr., Hanks G.D. Irradiation of Abruptex and apricot. Drosoph. Inf. Serv. 1952;26:108. [Google Scholar]
- 25.Lindsley G. Genetic Variations of D. melanogaster. Genetics. 1961;46:11. [Google Scholar]
- 26.Green M.M. Back mutation in Drosophila melanogaster. I. X-ray induced back mutations at the yellow, scute and white loci. Genetics. 1961;46:671–682. doi: 10.1093/genetics/46.6.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Escayg A., De Waard M., Lee D.D., Bichet D., Wolf P., Mayer T., Johnston J., Baloh R., Sander T., Meisler M.H. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am. J. Hum. Genet. 2000;66:1531–1539. doi: 10.1086/302909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chioza B., Nashef L., Asherson P., Makoff A. CACNA1A and P/Q-type calcium channels in epilepsy. Lancet. 2002;359:258. doi: 10.1016/S0140-6736(02)07416-0. [DOI] [PubMed] [Google Scholar]
- 29.Gökben S., Berdeli A., Serdaroğlu G. An inherited nonsense R1645X mutation in neuronal sodium channel alpha1-subunit gene in a Turkish patient with severe myoclonic epilepsy of infancy. Neuropediatrics. 2009;40:82–84. doi: 10.1055/s-0029-1234083. [DOI] [PubMed] [Google Scholar]
- 30.Krampfl K., Maljevic S., Cossette P., Ziegler E., Rouleau G.A., Lerche H., Bufler J. Molecular analysis of the A322D mutation in the GABA receptor alpha-subunit causing juvenile myoclonic epilepsy. Eur. J. Neurosci. 2005;22:10–20. doi: 10.1111/j.1460-9568.2005.04168.x. [DOI] [PubMed] [Google Scholar]
- 31.Ohmori I., Ouchida M., Miki T., Mimaki N., Kiyonaka S., Nishiki T., Tomizawa K., Mori Y., Matsui H. A CACNB4 mutation shows that altered Ca(v)2.1 function may be a genetic modifier of severe myoclonic epilepsy in infancy. Neurobiol. Dis. 2008;32:349–354. doi: 10.1016/j.nbd.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 32.Ganesh S., Delgado-Escueta A.V., Suzuki T., Francheschetti S., Riggio C., Avanzini G., Rabinowicz A., Bohlega S., Bailey J., Alonso M.E. Genotype-phenotype correlations for EPM2A mutations in Lafora's progressive myoclonus epilepsy: exon 1 mutations associate with an early-onset cognitive deficit subphenotype. Hum. Mol. Genet. 2002;11:1263–1271. doi: 10.1093/hmg/11.11.1263. [DOI] [PubMed] [Google Scholar]
- 33.Chan E.M., Young E.J., Ianzano L., Munteanu I., Zhao X., Christopoulos C.C., Avanzini G., Elia M., Ackerley C.A., Jovic N.J. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003;35:125–127. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- 34.Berkovic S.F., Dibbens L.M., Oshlack A., Silver J.D., Katerelos M., Vears D.F., Lüllmann-Rauch R., Blanz J., Zhang K.W., Stankovich J. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am. J. Hum. Genet. 2008;82:673–684. doi: 10.1016/j.ajhg.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medina M.T., Suzuki T., Alonso M.E., Durón R.M., Martínez-Juárez I.E., Bailey J.N., Bai D., Inoue Y., Yoshimura I., Kaneko S. Novel mutations in Myoclonin1/EFHC1 in sporadic and familial juvenile myoclonic epilepsy. Neurology. 2008;70:2137–2144. doi: 10.1212/01.wnl.0000313149.73035.99. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki T., Delgado-Escueta A.V., Aguan K., Alonso M.E., Shi J., Hara Y., Nishida M., Numata T., Medina M.T., Takeuchi T. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat. Genet. 2004;36:842–849. doi: 10.1038/ng1393. [DOI] [PubMed] [Google Scholar]
- 37.Alakurtti K., Virtaneva K., Joensuu T., Palvimo J.J., Lehesjoki A.E. Characterization of the cystatin B gene promoter harboring the dodecamer repeat expanded in progressive myoclonus epilepsy, EPM1. Gene. 2000;242:65–73. doi: 10.1016/s0378-1119(99)00550-8. [DOI] [PubMed] [Google Scholar]
- 38.Koskiniemi M., Donner M., Majuri H., Haltia M., Norio R. Progressive myoclonus epilepsy. A clinical and histopathological study. Acta Neurol. Scand. 1974;50:307–332. [PubMed] [Google Scholar]
- 39.Kaasik A., Kuum M., Aonurm A., Kalda A., Vaarmann A., Zharkovsky A. Seizures, ataxia, and neuronal loss in cystatin B heterozygous mice. Epilepsia. 2007;48:752–757. doi: 10.1111/j.1528-1167.2007.00985.x. [DOI] [PubMed] [Google Scholar]
- 40.Bingham S.M., Sittaramane V., Mapp O., Patil S., Prince V.E., Chandrasekhar A. Multiple mechanisms mediate motor neuron migration in the zebrafish hindbrain. Dev. Neurobiol. 2010;70:87–99. doi: 10.1002/dneu.20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borovina A., Superina S., Voskas D., Ciruna B. Vangl2 directs the posterior tilting and asymmetric localization of motile primary cilia. Nat. Cell Biol. 2010;12:407–412. doi: 10.1038/ncb2042. [DOI] [PubMed] [Google Scholar]
- 42.Cao Y., Park A., Sun Z. Intraflagellar transport proteins are essential for cilia formation and for planar cell polarity. J. Am. Soc. Nephrol. 2010;21:1326–1333. doi: 10.1681/ASN.2009091001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Copp A.J., Greene N.D. Genetics and development of neural tube defects. J. Pathol. 2010;220:217–230. doi: 10.1002/path.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim S.K., Shindo A., Park T.J., Oh E.C., Ghosh S., Gray R.S., Lewis R.A., Johnson C.A., Attie-Bittach T., Katsanis N., Wallingford J.B. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science. 2010;329:1337–1340. doi: 10.1126/science.1191184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mirzadeh Z., Han Y.G., Soriano-Navarro M., García-Verdugo J.M., Alvarez-Buylla A. Cilia organize ependymal planar polarity. J. Neurosci. 2010;30:2600–2610. doi: 10.1523/JNEUROSCI.3744-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreau M.M., Piguel N., Papouin T., Koehl M., Durand C.M., Rubio M.E., Loll F., Richard E.M., Mazzocco C., Racca C. The planar polarity protein Scribble1 is essential for neuronal plasticity and brain function. J. Neurosci. 2010;30:9738–9752. doi: 10.1523/JNEUROSCI.6007-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rocha P.P., Scholze M., Bleiss W., Schrewe H. Med12 is essential for early mouse development and for canonical Wnt and Wnt/PCP signaling. Development. 2010;137:2723–2731. doi: 10.1242/dev.053660. [DOI] [PubMed] [Google Scholar]
- 48.Song H., Hu J., Chen W., Elliott G., Andre P., Gao B., Yang Y. Planar cell polarity breaks bilateral symmetry by controlling ciliary positioning. Nature. 2010;466:378–382. doi: 10.1038/nature09129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tissir F., Goffinet A.M. Planar cell polarity signaling in neural development. Curr. Opin. Neurobiol. 2010;20:572–577. doi: 10.1016/j.conb.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 50.Panáková D., Werdich A.A., Macrae C.A. Wnt11 patterns a myocardial electrical gradient through regulation of the L-type Ca(2+) channel. Nature. 2010;466:874–878. doi: 10.1038/nature09249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carreira-Barbosa F., Concha M.L., Takeuchi M., Ueno N., Wilson S.W., Tada M. Prickle 1 regulates cell movements during gastrulation and neuronal migration in zebrafish. Development. 2003;130:4037–4046. doi: 10.1242/dev.00567. [DOI] [PubMed] [Google Scholar]
- 52.Takeuchi M., Nakabayashi J., Sakaguchi T., Yamamoto T.S., Takahashi H., Takeda H., Ueno N. The prickle-related gene in vertebrates is essential for gastrulation cell movements. Curr. Biol. 2003;13:674–679. doi: 10.1016/s0960-9822(03)00245-8. [DOI] [PubMed] [Google Scholar]
- 53.Okuda H., Miyata S., Mori Y., Tohyama M. Mouse Prickle1 and Prickle2 are expressed in postmitotic neurons and promote neurite outgrowth. FEBS Lett. 2007;581:4754–4760. doi: 10.1016/j.febslet.2007.08.075. [DOI] [PubMed] [Google Scholar]
- 54.Long J.M., LaPorte P., Paylor R., Wynshaw-Boris A. Expanded characterization of the social interaction abnormalities in mice lacking Dvl1. Genes Brain Behav. 2004;3:51–62. doi: 10.1046/j.1601-183x.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 55.Eickholt B.J., Towers G.J., Ryves W.J., Eikel D., Adley K., Ylinen L.M.J., Chadborn N.H., Harwood A.J., Nau H., Williams R.S.B. Effects of valproic acid derivatives on inositol trisphosphate depletion, teratogenicity, glycogen synthase kinase-3beta inhibition, and viral replication: a screening approach for new bipolar disorder drugs derived from the valproic acid core structure. Mol. Pharmacol. 2005;67:1426–1433. doi: 10.1124/mol.104.009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelly K.M., Gross R.A., Macdonald R.L. Valproic acid selectively reduces the low-threshold (T) calcium current in rat nodose neurons. Neurosci. Lett. 1990;116:233–238. doi: 10.1016/0304-3940(90)90416-7. [DOI] [PubMed] [Google Scholar]
- 57.Shimada Y., Yonemura S., Ohkura H., Strutt D., Uemura T. Polarized transport of Frizzled along the planar microtubule arrays in Drosophila wing epithelium. Dev. Cell. 2006;10:209–222. doi: 10.1016/j.devcel.2005.11.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse video (the embryo is oriented in a lateral view) consisting of pseudocolored ratio images derived from image pairs (340 and 380 nm excitation wavelengths) collected in 15 s intervals over a 50 min period. Low Ca2+ is represented by blue, and high Ca2+ is represented by yellow and red.
Time-lapse video (the embryo is oriented in a lateral view) consisting of pseudocolored ratio images derived from image pairs (340 and 380 nm excitation wavelengths) collected in 15 s intervals over a 50 min period. Low Ca2+ is represented by blue, and high Ca2+ is represented by yellow and red.
Time-lapse video (the embryo is oriented in a lateral view) consisting of pseudocolored ratio images derived from image pairs (340 and 380 nm excitation wavelengths) collected in 15 s intervals over a 50 min period. Low Ca2+ is represented by blue, and high Ca2+ is represented by yellow and red.
Time-lapse video (the embryo is oriented in a lateral view) consisting of pseudocolored ratio images derived from image pairs (340 and 380 nm excitation wavelengths) collected in 15 s intervals over a 50 min period. Low Ca2+ is represented by blue, and high Ca2+ is represented by yellow and red.
pksple1 homozygous flies (left) display a severe bang-sensitive phenotype; most flies remain at the bottom of the vial throughout the testing period. Wild-type flies are shown on the right.
pksple1 homozygous flies (left) climb normally as compared to wild-type flies (right) prior to bang testing.
pksple1 homozygous flies (left) climb normally as compared to wild-type flies (right) after bang testing.
pksple1 heterozygous (right) flies display delayed climbing and increased seizures as compared to wild-type flies (left).
pksple1 heterozygous (right) flies display delayed climbing off the bottom and increased seizures as compared to wild-type flies (left). Note that several pksple1 heterozygous flies at the bottom of the vial are on their backs and shaking.