

Abstract
Objectives
Previous work in humans and in animal models supports a key role for histidyl-tRNA synthetase (HRS=Jo-1) in the pathogenesis of idiopathic inflammatory myopathy. While most investigations have focused on the ability of HRS to trigger adaptive immune responses, in vitro studies clearly indicate that HRS possesses intrinsic chemokine-like properties capable of activating the innate immune system. The purpose of this study was therefore to examine the ability of HRS to direct innate immune responses in a murine model of myositis.
Methods
Following intramuscular immunization with soluble HRS in the absence of exogenous adjuvant, selected strains of mice were evaluated at different time points for histopathologic evidence of myositis. ELISA-based assessment of autoantibody formation and CFSE proliferation studies provided complementary measures of B and T cell responses triggered by HRS immunization.
Results
Compared to appropriate control proteins, a murine HRS fusion protein induced robust, statistically significant muscle inflammation in multiple congenic strains of C57BL/6 and NOD mice. Time course experiments revealed that this inflammatory response occurred as early as 7 days post immunization and persisted for up to 7 weeks. Parallel immunization strategies in DO11.10/Rag2−/− and C3H/HeJ (TLR4−/−) mice indicated that the ability of murine HRS to drive muscle inflammation was not dependent on B cell receptor or T cell receptor recognition and did not require TLR4 signaling.
Conclusion
Collectively, these experiments support a model in which HRS can trigger both innate and adaptive immune responses which culminate in severe muscle inflammation that is the hallmark of idiopathic inflammatory myopathy.
INTRODUCTION
In idiopathic inflammatory myopathy (IIM), both muscle and extramuscular tissues are inappropriately targeted by a dysregulated immune response (1–3). Despite the wealth of data detailing the histopathologic characteristics of these disorders that include polymyositis (PM) and dermatomyostis (DM), much less is known regarding the precise mechanisms that initiate and perpetuate tissue damage. A number of elegant animal models have been described, but few replicate the systemic features of IIM or adequately explore potential antigenic triggers applicable to human disease (4, 5).
In fact, the striking autoantibody profiles characterizing subsets of IIM patients provide valuable clues to putative antigen triggers and clearly reflect an underlying antigen driven process. Supporting this contention, studies have shown that antibodies targeting histidyl-tRNA synthetase (HRS=Jo-1) undergo affinity maturation, parallel disease activity, and do not co-exist with other “myositis-specific” autoantibodies (6–11). Previous work examining human T cell responses in Jo-1 antibody positive myositis patients provides additional evidence that this stereotypical antibody response is driven by underlying antigen-specific T cells (12). Perhaps most convincing, however, is the combination of muscle and lung inflammation resulting from subcutaneous immunization of various congenic mice with emulsified murine HRS (mHRS) systemic features that partially replicate the anti-synthetase syndrome in humans (13).
While the aforementioned studies focus on the role of tRNA synthetases in triggering antigen-specific, adaptive immune responses, a growing body of literature has revealed that many of these autoantigens possess intrinsic cytokine or chemokine properties potentially contributing to innate immune activation. For example, Wakasugi and Schimmel showed that a cleavage product of tyrosyl-tRNA synthetase can function as a cytokine, at least in vitro (14). Extending this observation, Howard et. al. demonstrated that asparaginyl- and histidyl-tRNA synthetase possess independent chemokine activities (15). In the case of HRS, the authors elegantly demonstrated that the amino terminal portion of this molecule chemoattracts naive lymphocytes and immature dendritic cells through interactions with CCR5. Of note, the authors investigated the capacity of several other tRNA synthetases to exert chemokine-like effects, but found that this property was relatively specific for tRNA synthetases targeted by autoantibody responses in the setting of inflammatory myopathy and/or interstitial lung disease (15). Overall, this data raised the intriguing possibility that molecules such as HRS could play a role in coupling innate and adaptive immune responses contributing to the pathogenesis of IIM.
To explore this hypothesis that HRS triggers both innate and adaptive immune responses leading to T cell-mediated tissue damage, the current study examines the impact of intramuscular (IM) administration of soluble mHRS without exogenous adjuvant. The results complement the in vitro studies of Howard et. al., demonstrating that mHRS is capable of directing exuberant T cell infiltration of muscle in multiple congenic strains of NOD and C57BL/6 mice. The inflammatory response occurs as early as 7 days following IM immunization with the amino-terminal fragment of mHRS (MA) and persists for at least 7 weeks. Replication of these findings in the TCR transgenic strain DO11.10/Rag2−/− provides compelling evidence that mHRS-induced T cell infiltration is mediated in part by mechanisms that are independent of antigen-specific T cell receptor (TCR) signals. Finally, the development of severe inflammation following IM immunization of the TLR4−/− strain C3H/HeJ with MA indicates that the capacity of mHRS to direct innate immune responses is not due to LPS contamination of recombinant antigens.
MATERIALS AND METHODS
Mice
Eight to ten week old male and female mice of the following strains (used in our previous model of HRS/CFA-induced muscle and lung inflammation) were employed in immunization protocols approved by the University of Pittsburgh Institutional Animal Care and Use Committee: C57BL/6 (B6), B6.G7 (NOD I-Ag7 MHC Class II locus crossed onto the C57BL/6 background), and NOD.Idd3/5 (C57BL/6 insulin dependent diabetes Idd3/5 non-MHC loci transgressed onto the NOD background). Additional strains used in the described experiments included DO11.10/Rag2−/− (Taconic, Germantown, NY) as well as C3H/HeJ (TLR4−/−; Jackson Laboratories, Bar Harbor, Maine) and C3H/HeOuJ (wild type (WT); Jackson Laboratories, Bar Harbor, Maine).
Generation of recombinant antigens
As previously described, the amino terminal fragment of mHRS was produced as a maltose binding protein fusion protein (MA/MBP=aa 1–151 of mHRS linked to the carboxy terminal end of MBP) by first subcloning the appropriate full length mHRS sequence into the bacterial expression vector pMALc2 (New England Biolabs, Ipswich, MA) and then introducing a premature stop codon after base pair 453 via in situ mutagenesis (Stratagene, La Jolla, CA) (13). MBP control protein was generated from the same vector system without additional sequence insertion. To minimize experimental variation related to potential fluctuations in protein quality, multiple lots of expressed proteins were purified with amylose resin per the manufacturer’s protocol (New England Biolabs, Ipswich, MA), dialyzed against PBS, and filter sterilized.
Immunization procedure
Experimental mice received intramuscular injections of soluble proteins (MA/MBP or MBP control proteins, 3–5 mg/ml) administered to both hamstrings in a total volume of 100 μl (50 μl/side). At designated time points, animals were sacrificed for harvesting of blood/serum, spleen, inguinal/para-aortic lymph nodes, quadriceps/hamstring muscles, liver, and kidneys.
Determination of serum antibody levels
IgG anti-MJo-1 and anti-MBP antibody levels in the sera of mice immunized with MA/MBP or MBP were measured using standard solid phase ELISA according to the following protocol. Ninety six-well microtiter plates (Nunc, Rochester, NY) were coated with recombinant MJo-1 (full length murine HRS, 0.1 μg/ml) or MBP (0.1 μg/ml) in carbonate buffer (50 mM NaHCO3/Na2CO3, pH 9.6) and incubated overnight at 4°C. Plates were then washed four times with PBS containing 0.05% Tween-20. After blocking wells with PBS containing 1% BSA, appropriately diluted serum samples (1:1000) from immunized mice were added for 2 hours. Following a 60 minute incubation with horseradish peroxidase-conjugated goat anti-mouse IgG (0.04 μg/ml, Santa Cruz Biotechnology, Santa Cruz, CA), enzymatic reaction was visualized using 3,3,5,5-Tetramethylbenzidine (TMB) (Sigma-Aldrich, St. Louis, MO) and subsequently terminated with 1 N H2SO4. Color development was measured at 450 nm by a Wallac 1420 multilabel counter (PerkinElmer, Wellesley, MA), and values were plotted as OD450 substrate antigen - OD450 no antigen.
Assessment of T cell proliferation
Spleens from immunized mice were mechanically disrupted and filtered through 70 micron mesh to generate whole splenocyte populations. After RBC lysis using a standardized buffer, cells were washed in PBS and then labeled with 0.4 μM CFSE (Molecular Probes, Eugene, OR) according to the manufacturer’s protocol. Parallel processing (excluding RBC lysis) of inguinal/para-aortic lymph nodes yielded separate populations of CFSE-labeled lymphocytes. In 48-well plates, 6 × 105 CFSE-labeled splenocytes or lymph node-derived lymphocytes/well were then combined with designated antigens in 400 μl of RPMI 1640 containing 10% FCS, sodium pyruvate, non-essential amino acids, 5 × 10−5 M 2-Mercaptoethanol, and penicillin/streptomycin. After 96 hours, cells were stained with Allophycocyanin (APC)-conjugated anti-Thy1.2 antibodies and fixed in 2% paraformaldehyde prior to flow cytometric assessment.
Histopathology and immunohistochemistry
Harvested organs were fixed in 10% formalin prior to paraffin embedding, sectioning, and staining with hematoxylin/eosin by the University of Pittsburgh’s Department of Transplant Immunology Core Histopathology Laboratory. Following these staining procedures, a blinded pathologist independently scored/confirmed the severity of inflammation in muscle tissue. Scores were based on the relative area and intensity of lymphocytic infiltration in multiple fields viewed under high power magnification, ranging from 0–3: 0 = no inflammation, 1 = minimal, 2 = moderate, 3 = severe.
For immunohistochemical assessment of cellular infiltrates, tissue sections from de-paraffinized muscle specimens were stained with a series of antibodies targeting different cell types according to a protocol developed in the University of Pittsburgh’s Department of Transplant Immunology Core Histopathology Laboratory. These antibodies consisted of anti-CD3 (Dako North America, Carpenteria, CA), anti-CD4 (Abcam, Cambridge, MA), anti-CD8 (Abcam, Cambridge, MA), anti-CD11c (Abcam, Cambridge, MA), anti-CD44 (Abcam, Cambridge, MA), anti-B220 (Serotec, Raleigh, NC), anti-F4/80 (Serotec, Raleigh, NC), and anti-CCR5 (Abcam, Cambridge, MA).
Statistical analysis
Severity scores for muscle inflammation were compared using the Mann-Whitney U-test, with significance based on a two-tailed p value <0.05.
RESULTS
Intramuscular immunization with soluble murine HRS induces myositis
To assess the role of HRS in directing T cell infiltration of muscle tissue in different genetic backgrounds, various congenic strains of C57BL/6 and NOD mice (previously studied in our HRS/CFA-induced model of the anti-synthetase syndrome) were immunized with soluble forms of recombinant mHRS or appropriate control proteins (without adjuvant) by IM injection. As shown in Figure 1, C57BL/6, B6.G7, and NOD.Idd3/5 mice all developed significant muscle inflammation 14–17 days following IM immunization with MA/MBP (MA/MBP=amino terminal fragment of mHRS (MA) fused to Maltose Binding Protein (MBP)). The degree of cellular infiltration was independent of gender (data not shown) and far exceeded that generated by administration of MBP control proteins (even in the case of NOD.Idd3/5 where background inflammation was variable), underscoring the antigen-specificity of this response. Careful review of the histopathologic findings demonstrated perivascular, perimysial, and endomysial infiltrates consisting primarily of CD3+ (both CD4+ and CD8+) T cells expressing the activation marker CD44 as well as the chemokine receptor CCR5. Interestingly, MA/MBP-induced mononuclear cell infiltrates persisted as long as 7 weeks following a single administration of soluble protein in both B6.G7 and NOD.Idd3/5 mice, though the difference in severity of muscle infiltration relative to MBP-immunized specimens was most significant in B6.G7 mice (MA/MBP (n=9) vs. MBP (n=8)--1.5 vs. 0.25, p=0.012).
Figure 1. Induction of myositis following intramuscular immunization of murine HRS.

Panels A–C depict photomicrographs (100×) of hematoxylin and eosin-stained muscle tissue derived from MA/MBP- versus MBP-immunized mice of the indicated strains. Accompanying bar graphs represent the mean severity of muscle inflammation graded (in blinded fashion) on a scale from 0–3, as described in Materials and Methods. Error bars reflect SEM, while p values are derived from Mann U Whitney rank sum analysis. N equals the combined number of mice immunized with the indicated antigen in multiple experiments: C57BL/6 (B6), nMA/MBP=10 and nMBP=10; B6.G7, nMA/MBP=16 and nMBP=9; NOD.Idd3/5, nMA/MBP=17 and nMBP=10. In panel D, photomicrographs (400×) demonstrate immunohistochemical staining of inflammatory infiltrates resulting from MA/MBP immunization of NOD.Idd3/5 mice.
Antigen-specific B and T cell responses are triggered by intramuscular immunization with soluble murine HRS
Corresponding to these histopathologic changes in muscle tissue, ELISAs demonstrated class-switched antibody responses to MA/MBP (Figure 2A)—a surprising result given the absence of CFA or other exogenous adjuvants. Measurement of T cell responses to immunizing antigens through CFSE proliferation assays also indicated a level of antigen-specific priming, further highlighting the intrinsic immunogenicity of mHRS that could not simply be attributed to contaminants/impurities given the lack of similar responses to recombinant MBP derived from the same bacterial system (Figure 2B).
Figure 2. Development of autoantibodies and antigen-specific T cell proliferative responses following intramuscular administration of soluble HRS.

A) Dot plots depict relative anti-murine HRS (anti-MJo-1) antibody titers induced by immunization of the designated strains with MA/MBP or MBP. Individual data points represent mean OD450 values (measured by ELISA) generated by combining sera at 1:1000 dilution with wells containing recombinant MJo-1 (0.1 μg/ml). Immunizing antigens are indicated on the x-axis. Error bars are omitted given negligible SEMs. B) Individual histograms represent CFSE dilution of pooled C57BL/6 (B6) Thy1.2+ lymph node cells stimulated by the indicated antigens in 96 hour proliferation assays. While the upper panels correspond to LN-derived T cells obtained from MA/MBP-immunized mice, the lower panels reflect proliferation of LN-derived lymphocytes isolated from MBP-immunized mice. Percentages correspond to the proportion of leftward-shifted, proliferating T cells. No Ag=no antigen added, MBP=Maltose Binding Protein, MA/MBP=amino terminal fragment of murine HRS fused to MBP, ConA=concavalin A.
Myositis occurs within 7 days of intramuscular murine HRS immunization
Because the development of MA/MBP-induced T cell proliferation and class-switched antibody responses suggested that adaptive immunity might still contribute to the cellular infiltration of muscle tissue in this model of myositis, mice were then assessed at an earlier time point (7 days) following immunization. As shown in Figure 3A, MA/MBP immunization of NOD.Idd3/5 mice yielded significant, antigen-specific muscle inflammation (again consisting of CD3+CD44+CCR5+ T cells admixed with scattered F4/80+ macrophages and B220+ B cells, data not shown) after only 7 days—a time frame most consistent with innate, rather than (primary) adaptive, immune responses. However, MA/MBP-induced proliferation of draining lymph node-derived T cells (that occurred preferentially in MA/MBP-immunized mice, Figure 3B and data not shown) and the development of antigen-specific, class-switched antibody responses (Figure 3C) did not allow us to fully exclude a role for adaptive immunity in this process.
Figure 3. Development of muscle inflammation at early time points following intramuscular administration of murine HRS.

The bar graph in panel A corresponds to mean severity scores of muscle inflammation detected in muscle tissue 7 days following IM immunization of NOD.Idd3/5 mice with the indicated antigens (nMA/MBP=7, nMBP=4). Error bars and p-values are calculated as in Figure 1. While panel B demonstrates lymph node-derived T cell proliferation in response to the indicated antigens, panel C depicts anti-MJo-1 antibody formation promoted by IM immunization with MA/MBP (MA).
Murine HRS can trigger myositis independently of BCR and TCR signaling
To better delineate the relative contributions of innate and adaptive immune responses triggered by soluble mHRS administration, we performed IM immunizations in DO11.10/Rag2−/− (OVA-specific TCR transgenic) mice to eliminate TCR-based recognition of MA or the fusion partner, MBP. Despite the abrogation of antigen-specific B and T cell responses, Figure 4 clearly demonstrates that MA/MBP immunization of DO11.10/Rag2−/− mice results in significant muscle inflammation possessing an endomysial and perimysial distribution pattern similar to that seen in congenic strains of NOD and C57BL/6 mice—findings that support a key role for innate immune responses in this antigen (HRS)-induced model of myositis.
Figure 4. Murine HRS induces muscle inflammation in DO11.10/Rag2−/− mice.
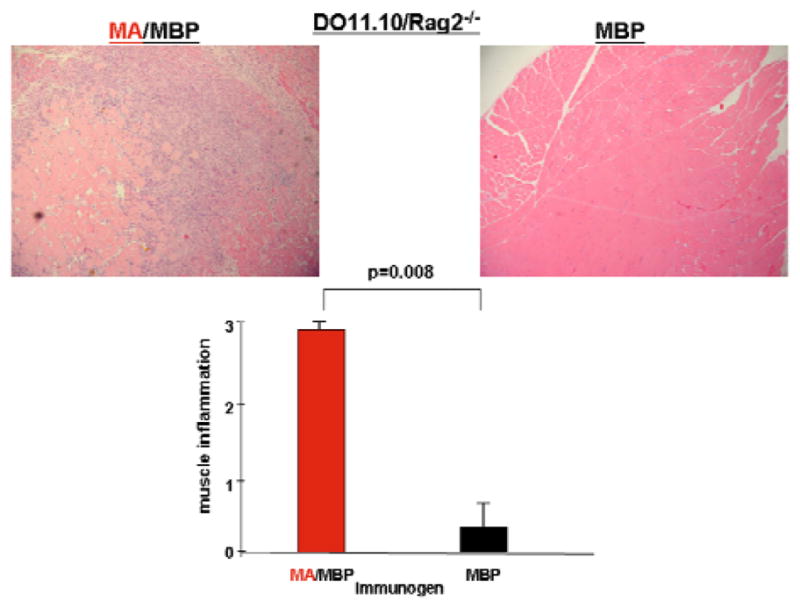
Photomicrographs (100x) depict representative H&E-stained muscle specimens derived from DO11.10/Rag2−/− mice 17 days following IM immunization of the hamstring musculature with MA/MBP or MBP alone. The accompanying bar graph indicates mean severity scores for mice immunized with MA/MBP (n=5) or MBP (n=5).
TLR4 signaling is not required for murine HRS-induced myositis
Although the muscle inflammation stemming from IM immunization of DO11.10/Rag2−/−mice provided compelling evidence that MA/MBP-induced myositis is not dependent on B cell receptor (BCR) or TCR generated signals, the precise mechanisms/pathways fueling mHRS-triggered muscle inflammation could not be ascertained from these collective data. To more fully assess the potential impact of factors such as protein-bound LPS in this model of myositis, we therefore compared the ability of MA/MBP to induce muscle inflammation in the TLR4−/− strain C3H/HeJ and its wild type counterpart, C3H/HeOuJ. As shown in Figure 5A, MA/MBP clearly retains the ability to induce extensive cellular infiltration of muscle tissue, even after elimination of TLR4 signaling in C3H/HeJ mice. Comprised predominantly of T lymphocytes (CD44+CCR5+) with relatively small numbers of macrophages, dendritic cells, and B cells (Figure 5B), the infiltrate in C3H/HeJ mice is virtually indistinguishable from that seen in C3H/HeOuJ mice—both in terms of severity and distribution. Of note, the titers of class-switched antibodies induced by MA/MBP immunization are markedly reduced in C3H/HeJ mice (Figure 5C), suggesting that antibody formation in wild type control (C3H/HeOuJ) mice may be facilitated by LPS and occur independently of T cell help. By extension, the development of myositis in C3H/HeJ mice lacking significant levels of anti-HRS antibodies effectively uncouples MA/MBP-induced muscle inflammation from adaptive humoral/cellular immune responses (as well as LPS-related effects), strongly supporting the findings in DO11.10/Rag2−/− mice.
Figure 5. Induction of myositis is not dependent on TLR4 signaling.

In Panel A, photomicrographs (100×) correspond to H&E-stained muscle tissue obtained from C3H/HeOuJ (WT) versus C3H/HeJ (TLR4−/−) mice 17 days following IM immunization with MA/MBP. The bar graph indicates the relative severity of muscle inflammation resulting from IM immunization with MA/MBP (C3H/HeOuJ (n=10), C3H/HeJ (n=20)) or MBP (C3H/HeJ (n=5)). Immunohistochemical staining (400×) with antibodies targeting CD3, B220, F4/80, CD44, CCR5, and CD11c reveals the relative number as well as activation state of T cells, B cells, macrophages, and dendritic cells comprising inflammatory infiltrates of muscle tissue obtained from MA/MBP-immunized C3H/HeJ mice (Panel B). Finally, the ELISA data in panel C demonstrate relative anti-MJo-1 antibody formation in C3H/HeOuJ versus C3H/HeJ mice immunized with MA/MBP. Data points correspond to mean OD450 values generated by 1:1000 dilutions of sera from individual mice.
DISCUSSION
Based on intramuscular immunization with a soluble derivative of mHRS, the reported experiments demonstrate that this putative autoantigen can trigger innate immune responses which bypass BCR and TCR signaling to generate significant muscle inflammation in a novel model of IIM. This model is highly reproducible in multiple strains of mice, providing additional evidence of innate immune mechanisms that are less dependent than adaptive immune responses on genetic background or the specificity of TCR/BCR recognition. As shown in the time course experiments, the inflammation resulting from IM administration of soluble MA/MBP consists predominantly of CD44+CCR5+ T cells, occurs as early as 7 days post immunization, and persists up to 7 weeks (longer time points not assessed)--suggesting that initial innate immune responses may eventually give way to adaptive immune responses capable of sustaining long term muscle inflammation. Additional support for the role of HRS-triggered innate immune responses comes from the experiments involving DO11.10/Rag2−/− mice given the development of MA/MBP-induced myositis in the setting of a highly restricted, OVA-specific BCR/TCR repertoire that precludes TCR recognition of HRS. Complementing these findings, the muscle inflammation resulting from MA/MBP immunization of C3H/HeJ mice demonstrates that this model of myositis is not dependent on LPS/TLR4-mediated signals.
Overall, these observations are consistent with in vitro studies showing that human HRS exerts chemotactic effects on immature dendritic cells and naïve lymphocytes (note that in addition to abundant CCR5+ lymphocytes, small numbers of CD11c+ dendritic cells appear in muscle inoculated with MA/MBP; Figure 1, Figure 5, and data not shown). In their seminal study focusing on chemotactic properties of various tRNA synthetases, Howard et. al. provide convincing evidence that human HRS interacts with CCR5 (15). More firmly establishing the role of CCR5 in our model would require immunization of CCR5−/− mice or in vivo application of CCR5 antagonists. Although CCR5 knockouts exist in several C57BL/6 derivative strains, the use of affinity purified, bacterially produced fusion proteins that associate with LPS could preclude definitive conclusions regarding the precise role of CCR5 (by generating background inflammation through additional/alternative signaling pathways mediated by TLR4). Employing baculovirus-derived proteins would circumvent this problem, but purification of stable mHRS from baculovirus-infected Sf9/Sf21 cells has not yet been possible.
Regardless of the exact mechanism(s) by which recombinant mHRS triggers innate immune responses and subsequent lymphocytic muscle infiltration, the model presented herein is clearly antigen-driven. Coupled with the reproducibility of significant muscle inflammation following a single immunization of soluble MA/MBP, this factor sets this model apart from other systems that are not antigen-induced and often involve fairly minimal, non-lymphocytic infiltrates. At the same time, this technique of myositis induction through IM immunization of soluble protein complements findings from our previous adjuvant-based model in which subcutaneous administration of mHRS/CFA emulsions generates a combination of muscle and lung inflammation that replicates features of the anti-synthetase syndrome (13). Compared to the adjuvant-based model, the current system of IM immunization actually results in significantly greater muscle inflammation suggesting that full expression of disease requires not only a breach of B and T cell tolerance, but also adequate antigen exposure that can promote innate as well as adaptive immune responses against HRS.
When evaluated in the context of other in vitro and in vivo models of myositis pathogenesis, the results from this study support a coordinated pathway in which muscle damage, MHC Class I overexpression, endoplasmic reticulum stress, and release of soluble HRS combine to generate lymphocytic invasion of muscle (15–17). As shown by the schematic in Figure 6, some form of mechanical, infectious, or immunologic insult to muscle likely initiates the process by stimulating MHC Class I expression as well as the release of soluble proteins that include HRS. Perhaps through interaction with CCR5, HRS is then able to recruit naive, polyclonal T cells and immature dendritic cells (iDCs). Whether these cells form local lymphoid aggregates or migrate to regional lymph nodes is unclear, but subsequent DC maturation and presentation of HRS via MHC Class II promote development of HRS-specific CD4+ effector T cells. In turn, this population of HRS-specific T cells provides help to autoantibody producing B cells as well as CD8+ T cells mediating myocytotoxicity. In the proposed model, the CD8+ cytotoxic T cells need not have the same antigen specificity as the CD4+ T cells targeting HRS, particularly if such T cell subpopulations reside in the same local microenvironment. Critical to this model, however, is the HRS-mediated coupling of innate and adaptive immune responses that may also extend to extramuscular tissues (such as the lung) targeted in the anti-synthetase syndrome.
Figure 6. Hypothesized role of HRS in the pathogenesis of idiopathic inflammatory myopathy.

Combining data from the literature as well as current findings, this schematic demonstrates the putative role of HRS in bridging innate and adaptive immune responses that ultimately result in myocytotoxicity. As shown in the upper left portion of this diagram, muscle damage leads to upregulated surface expression of MHC Class I as well as release of soluble HRS that can recruit immature dendritic cells (iDCs) and naive lymphocytes via CCR5 (innate immunity). Subsequent maturation of DCs that have incorporated HRS allows efficient presentation of HRS peptides in the context of MHC Class II and enhanced co-stimulation, promoting activation of HRS-specific CD4+ T cells (in situ or in draining/regional lymph nodes). This subset of HRS-specific CD4+ T cells can then provide help to autoantibody-producing B cells and CD8+ T cells mediating myocytotoxicity in the adaptive arm of HRS-targeted immune responses.
More detailed assessment of this model and the relative contributions of innate and adaptive immunity will clearly require further studies examining the progression of disease from focal muscle inflammation to systemic organ involvement. Beyond extension of the time course and the use of various knockout strains, future experiments may require novel methods of antigen delivery or overexpression in extramuscular tissues given the critical role of antigen exposure in triggering many of the referenced immune cascades. Defining these complexities in IIM has obvious clinical implications given the need to establish more focused, pathway-specific therapeutic targets in this often devastating disease. The current system of HRS-induced myositis represents an important first step in this process, providing compelling evidence that this antigen has intrinsic immunostimulatory properties potentially contributing to multiple facets of IIM pathogenesis.
Acknowledgments
The authors take full responsibility for the contents of this paper, which do not represent the views of the Department of Veterans Affairs or the United States Government. The work encompassed by this manuscript was supported in part by NIH R21 AR056279 (DPA).
Funding
Eun Ha Kang: The Myositis Association (TMA) Fellowship Grant
Dana P. Ascherman: NIH R21 AR056279
ABBREVIATIONS
- IIM
idiopathic inflammatory myopathy
- PM
polymyositis
- DM
dermatomyositis
- HRS
histidyl-tRNA synthetase
- MA/MBP
amino terminal 151 amino acid fragment of murine Jo-1 fused to MBP
- MBP
maltose binding protein
References
- 1.Greenberg SA. Inflammatory myopathies: disease mechanisms. Curr Opin Neurol. 2009;22:516–23. doi: 10.1097/WCO.0b013e3283311ddf. [DOI] [PubMed] [Google Scholar]
- 2.Suber TL, Casciola-Rosen L, Rosen A. Mechanisms of disease: autoantigens as clues to the pathogenesis of myositis. Nat Clin Pract Rheumatol. 2008;4:201–9. doi: 10.1038/ncprheum0760. [DOI] [PubMed] [Google Scholar]
- 3.Pignone A, Fiori G, Del Rosso A, Generini S, Matucci-Cerinic M. The pathogenesis of inflammatory muscle diseases: on the cutting edge among the environment, the genetic background, the immune response and the dysregulation of apoptosis. Autoimmun Rev. 2002;1:226–32. doi: 10.1016/s1568-9972(02)00055-1. [DOI] [PubMed] [Google Scholar]
- 4.Katsumata Y, Ascherman DP. Animal models in myositis. Curr Opin Rheumatol. 2008;20:681–5. doi: 10.1097/BOR.0b013e328310e0ac. [DOI] [PubMed] [Google Scholar]
- 5.Nagaraju K, Plotz PH. Animal models of myositis. Rheum Dis Clin North Am. 2002;28:917–33. doi: 10.1016/s0889-857x(02)00026-1. [DOI] [PubMed] [Google Scholar]
- 6.Martin A, Shulman MJ, Tsui FW. Epitope studies indicate that histidyl-tRNA synthetase is a stimulating antigen in idiopathic myositis. FASEB J. 1995;9:1226–33. doi: 10.1096/fasebj.9.12.7672516. [DOI] [PubMed] [Google Scholar]
- 7.Miller FW, Twitty SA, Biswas T, Plotz PH. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest. 1990;85:468–75. doi: 10.1172/JCI114461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller FW, Waite KA, Biswas T, Plotz PH. The role of an autoantigen, histidyl-tRNA synthetase, in the induction and maintenance of autoimmunity. Proc Natl Acad Sci U S A. 1990;87:9933–7. doi: 10.1073/pnas.87.24.9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raben N, Nichols R, Dohlman J, McPhie P, Sridhar V, Hyde C, Leff R, Plotz P. A motif in human histidyl-tRNA synthetase which is shared among several aminoacyl-tRNA synthetases is a coiled-coil that is essential for enzymatic activity and contains the major autoantigenic epitope. J Biol Chem. 1994;269:24277–83. [PubMed] [Google Scholar]
- 10.Stone KB, Oddis CV, Fertig N, Katsumata Y, Lucas M, Vogt M, Domsic R, Ascherman DP. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum. 2007;56:3125–3131. doi: 10.1002/art.22865. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida S, Akizuki M, Mimori T, Yamagata H, Inada S, Homma M. The precipitating antibody to an acidic nuclear protein antigen, the Jo-1, in connective tissue diseases. A marker for a subset of polymyositis with interstitial pulmonary fibrosis. Arthritis Rheum. 1983;26:604–11. doi: 10.1002/art.1780260505. [DOI] [PubMed] [Google Scholar]
- 12.Ascherman DP. The role of Jo-1 in the immunopathogenesis of polymyositis: current hypotheses. Curr Rheumatol Rep. 2003;5:425–30. doi: 10.1007/s11926-003-0052-2. [DOI] [PubMed] [Google Scholar]
- 13.Katsumata Y, Ridgway W, Oriss T, Gu X, Chin D, Wu Y, Fertig N, Oury T, Vandersteen D, Clemens P, et al. Species-specific immune responses generated by histidyl-tRNA synthetase immunization are associated with muscle and lung inflammation. Journal of Autoimmunity. 2007;29:174–186. doi: 10.1016/j.jaut.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wakasugi K, Schimmel P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science. 1999;284:147–51. doi: 10.1126/science.284.5411.147. [DOI] [PubMed] [Google Scholar]
- 15.Howard OM, Dong HF, Yang D, Raben N, Nagaraju K, Rosen A, Casciola-Rosen L, Hartlein M, Kron M, Yiadom K, et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. 2002;196:781–91. doi: 10.1084/jem.20020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henriques-Pons A, Nagaraju K. Nonimmune mechanisms of muscle damage in myositis: role of the endoplasmic reticulum stress response and autophagy in the disease pathogenesis. Curr Opin Rheumatol. 2009;21:581–7. doi: 10.1097/BOR.0b013e3283319265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, Danning C, Wada R, Thompson C, Bahtiyar G, et al. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci U S A. 2000;97:9209–14. doi: 10.1073/pnas.97.16.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]