

Abstract
Familial Hypertrophic Cardiomyopathy (FHC) is a leading cause of sudden cardiac death among young athletes but the functional effects of the myofilament mutations during FHC-associated ischemia and acidosis, due in part to increased extravascular compressive forces and microvascular dysfunction, are not well characterized. We tested the hypothesis that the FHC-linked tropomyosin (Tm) mutation Tm-E180G alters the contractile response to acidosis via increased myofilament Ca2+ sensitivity. Intact papillary muscles from transgenic (TG) mice expressing Tm-E180G and exposed to acidic conditions (pH 6.9) exhibited a significantly smaller decrease in normalized isometric tension compared to non-transgenic (NTG) preparations. Times to peak tension and to 90% of twitch force relaxation in TG papillary muscles were significantly prolonged. Intact single ventricular TG myocytes demonstrated significantly less inhibition of unloaded shortening during moderate acidosis (pH 7.1) than NTG myocytes. The peak Ca2+ transients were not different for TG or NTG at any pH tested. The time constant of re-lengthening was slower in TG myocytes, but not the rate of Ca2+ decline. TG detergent-extracted fibers demonstrated increased Ca2+ sensitivity of force and maximal tension compared to NTG at both normal and acidic pH (pH 6.5). Tm phosphorylation was not different between TG and NTG muscles at either pH. Our data indicate that acidic pH diminished developed force in hearts of TG mice less than in NTG due to their inherently increased myofilament Ca2+ sensitivity, thus potentially contributing to altered energy demands and increased propensity for contractile dysfunction.
Keywords: tropomyosin, FHC, acidosis, calcium, contractility, myofilament calcium sensitivity
1. Introduction
Hypertrophic cardiomyopathy is a major cause of sudden cardiac death in the young but effective treatment has remained elusive [1, 2]. Inherited, or familial hypertrophic cardiomyopathy, FHC, may result from mutations in multiple sarcomeric proteins, including troponin T (TnT), troponin I (TnI), and tropomyosin (Tm) (for reviews see Morimoto, Alves [3, 4]). The pathophysiological phenotype of FHC varies and includes ventricular wall thickening, fibrosis, myocyte disarray, and contractile dysfunction. Moreover, myocardial ischemia is clinically associated with FHC but there is growing evidence that it also contributes significantly to disease progression in adult patients, including left ventricular remodeling, ventricular arrhythmias and contractile dysfunction [5–7]. In young FHC patients, apart from the ischemia and acidosis occurring with cardiac arrest, ischemia has recently been shown to be a precipitating factor in syncope and the onset of sudden death [8, 9].
Myocardial ischemia in FHC is attributable in part to inadequate tissue perfusion due to lower density and altered architecture of subendocardial arterioles and to elevated extramural compression leading to microvascular dysfunction (for review see Camici) [10–12]. Thus, the reduced capacity for coronary vasodilatation and oxygen delivery to the myocardium in FHC predisposes patients to ischemia and acidosis.
The FHC-associated mutation, Tm Glu180Gly (Tm-E180G), occurs in the region of Tm that interacts with TnT, potentially influencing the function of the troponin complex and thin filament activation (for review see Wolska & Wieczorek, 2003 and Jagatheesan, et al., 2010) [13, 14]. Expression of Tm-E180G in a transgenic mouse model causes increased myofilament Ca2+ sensitivity, left ventricular concentric hypertrophy, increased heart to body weight ratio, fibrosis and impaired ventricular relaxation [15, 16]. Thus mice expressing the Tm-E180G mutation provide an ideal model for testing the effects of acidosis on the contractile function of hypertrophic myocardium.
In this study we tested the hypothesis that the higher myofilament Ca2+ sensitivity in transgenic (TG) mice expressing Tm-E180G results in maintained contractility in acidosis, contributing to the deleterious effects of the disorder. Experiments were thus performed in papillary muscles, single ventricular myocytes and skinned fiber bundles isolated from TG and NTG mouse hearts.
2. Methods
2.1 Transgenic animals
Adult mice that express the FHC α-TM Glu180Gly (TG-Tm-E180G) mutation were generated as described previously in the FVB/N strain [16]. All experiments were conducted and care of animals provided in compliance with animal care policies according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85–23, revised 1985) and approved by the Institutional Animal Review Board of the University of Illinois at Chicago.
2.2 Isolation of mouse ventricular myocytes
Cells were isolated as previously described [17]. Hearts from heparinized (5000 U/kg) and anesthetized (sodium pentobarbital, 50 mg/kg) mice were quickly removed and put into ice cold, nominally Ca2+-free perfusion buffer (PB) of the following composition in mM: NaCl 113, KCL 4.7, Na2HPO4 0.6, MgSO4 1.2, phenol red 0.032, NaHCO3 12, KHCO3 10, taurine 30, HEPES 10, glucose 5.5 and BDM 10 (pH 7.4; 37° C). The aorta was cannulated and the heart mounted on a Langendorff perfusion system. Hearts were perfused for 4 min with Ca2+-free PB and subsequently for 8–12 minutes with digestion buffer (DB) containing PB and 12.5 μM Ca2+ together with 0.15 mg/ml Blendzyme 2 (Roche, Germany) and 0.14 mg/ml trypsin (Invitrogen, CA). Hearts were then removed and transferred to a dish containing DB, and the ventricles were cut into small pieces and gently triturated. At the end of the trituration period, the cell suspension was filtered through a mesh collector, placed into centrifuge tubes and the digestion process stopped with an equal volume of PB containing 12 μM Ca2+ plus 10% bovine calf serum (v/v). The cells were then permitted to settle gently under gravity for 5–7 minutes. The supernatant fraction was removed and the cells resuspended in fresh PB containing 12.5 μM Ca2+ plus 5% bovine calf serum (v/v). Cells were allowed to settle under gravity, the supernatant removed and the cells resuspended in fresh control solution (CS) of the following composition in mM: NaCl 133.5, KCL 4, MgSO4 1.2, NaH2PO4 1.2, HEPES 10, various Ca2+concentrations; first 200 μM Ca2+, followed by 500 μM and then 1 mM Ca2+. The cells were stored at room temperature (22–23° C) until used.
2.3 Tension measurement in mouse isolated papillary muscles
Mice were anesthetized as described above and the heart was rapidly excised and perfused with a modified Krebs-Henseleit solution. Thin, unbranched and uniform papillary muscles with the tricuspid valve and a small part of the ventricle were carefully dissected from the right ventricle. The muscle preparation was mounted in an experimental chamber and perfused with a modified Krebs-Henseleit solution (0.2 mM Ca2+) at a flow of about 2.5 ml min−1. The muscle preparations were stimulated via platinum electrodes mounted along the sides of the muscle chamber. Stimulus strength was adjusted to 50% above threshold. The standard solution had the following composition in mM: NaCl 118.5, KCl 5.0, MgSO4 1.2, NaH2PO4 2.0, NaHCO2 26, D-glucose 10.0, and Ca2+ 1.0. During dissection of the papillary muscles [Ca2+]o was 0.2 mM and [K+]o was raised to 15 mM to stop spontaneous beating of the heart. The solutions were equilibrated with a 95% O2-5% CO2 (pH 7.4) or 85% O2-15% CO2 (pH 6.9) gas mixture by continuous bubbling at 25°C. The temperature in the muscle chamber was kept constant at 25°C by a heat exchanger at the inflow line and a circulating water bath. The remnant of the tricuspid valve served as a mounting point to the hook of a servo-controlled motor (Cambridge Technology), which was used to control the length of the muscle preparation. The basket connected to a force transducer was used as a second mounting point. Tension was measured using a force transducer (Fort10; World Precision Instruments) at a stimulation frequency of 0.25 Hz. After the initial equilibration [Ca2+]o was gradually increased to 1 mM and the muscle was stretched to generate 90% of maximum developed force.
2.4 Intracellular [Ca2+] and pHi measurement
Single, intact adult mouse ventricular myocytes were loaded with the Ca2+-indicator fura-2 by exposure for 20 min at room temperature to a loading solution consisting of CS with the addition of 1 mM Ca2+, bovine serum albumin (1 mg/ml) and 3 μM fura-2 (fura-2 AM; Invitrogen, Carlsbad, CA) made from a DMSO stock solution. Single myocytes suspended in CS containing 1.5 mM Ca2+ were transferred to a perfusion chamber mounted on the stage of an inverted Nikon microscope and cells were allowed to settle onto the glass. Cells were then continuously perfused with a solution containing (mM): NaCl 93, KCl 5, HEPES 10, MgCl2 1, CaCl2 1.5, Na2HCO3 20, Na2PO4 1, MgSO4 1.2, sodium acetate 20 and equilibrated with 5% CO2-95%O2 for pH 7.4. A minimum of 30 min wash in dye-free solution was allowed for de-esterification of the indicator. Cells were perfused with solution equilibrated with 15%CO2-85%O2 for pH 6.9, or solution with Na2HCO3 9.66 mM, equilibrated with 15%CO2-85%O2 for pH 7.1. Single cells were field-stimulated by applying a 4 ms square supra-threshold voltage pulse at 0.5 Hz to the cell bath through parallel platinum electrodes. Fura-2 fluorescence and shortening of cells were recorded simultaneously, as previously described [17–19]. Briefly, fura-2-loaded myocytes were alternately excited at wavelengths of 340 and 380 nm from a monochromator (Photon Technology International, Birmingham, New Jersey). The excitation light was transmitted to the cell through a 400 nm dichroic mirror. Emitted fluorescence was collected by the objective and transmitted to a multi-image module of the microscope where it was separated from long wavelength (>600 nm) using a 580 nm dichroic mirror and passed through a 510 ± 25 nm emission filter to a photomultiplier tube. Background fluorescence was recorded every day by measuring the auto-fluorescence of myocytes at both excitation wavelengths in the absence of dye loading, was stored in the acquisition software (Felix 32, Photon Technology International) and used for subtraction in subsequent recordings. Fluorescence signals were reported as background subtracted 340/380 ratio (fura-2 ratio). Cell shortening was recorded by illuminating the myocytes with red transmitted light (>600 nm). Output from the camera was split and sent to a chart recorder and to a video-edge detector (VED, Crescent Electronics, Sandy, UT) [20]. The cell length and fluorescent signals were recorded simultaneously on an acquisition computer for later analysis off-line.
To measure intracellular pH, the cells were loaded with 9 μM BCECF (2′,7′-bis-(2-carboxyethyl)-5,6-carboxyfluorescein-AM, Invitrogen, Carlsbad, CA) loading solution for 10 min. Cells were then continuously perfused with the pH 7.4 extracellular solution described above. A minimum of 30 min wash in dye-free solution was allowed for de-esterification of the indicator. BCECF-loaded myocytes were alternately excited at 450 and 490 nm, and fluorescence was monitored using the same emission filter. The excitation light was transmitted to the cell through a 510 nm dichroic mirror. Emitted light was separated from long wavelength (>600 nm) using a 580 nm dichroic mirror, passed to an emission monochromator and then to a photomultiplier tube. Background fluorescence was measured every day by recording the auto-fluorescence of myocytes at both excitation wavelengths in the absence of dye loading, was stored in the acquisition software (Felix 32, Photon Technology International) and used for subtraction in subsequent recordings. In vivo calibration was performed using a modified procedure as previously described [21, 22]. Following recording of BCECF fluorescence from intact myocytes in the pH 7.4 and 6.9 solutions described above, cells were perfused with a nigericin-containing solution of the following composition in mM: KCl 140, MgSO4 1.2, KH2PO4 1.2, HEPES 10, and 10 μM nigericin (Invitrogen, Carlsbad, CA), adjusted to either pH 7.4 or 6.9 with KOH. Fluorescence was collected at 6 sec intervals throughout the treatment. The cells were perfused with the pH 7.4 nigericin-containing solution for at least 5 min to allow complete permeabilization and equilibration. When the fluorescence was stable, the solution was changed to pH 6.9 then returned to 7.4. Fluorescence values were linearly related to pH and used to calibrate intracellular pH to fluorescence in intact cells.
2.5 Force measurement of skinned fiber bundles
Measurements of the force-calcium relationship were carried out on skinned fiber bundles prepared from the left ventricular papillary muscles of adult mice [23]. Mice 4–6 months old were anesthetized as described above and hearts were quickly removed and put into a cold high relaxing (HR) solution of the following composition in mM: KCl 53, EGTA 10, Mops 20, free Mg2+ 1, MgATP2− 5, creatine phosphate 12, and 10 i.u. ml−1 creatine phosphokinase. The pH of the solution was adjusted to 7.0 and 6.5. The ionic strength of all solutions was 150 mM. Papillary muscles from the left ventricle were dissected out, and small fiber bundles approximately 150–200 μm in width and 4–5 mm long were prepared. Fiber bundles were mounted between a micromanipulator and a force transducer with cellulose-acetate glue. Fibers were skinned in HR solution containing 1% Triton X-100 for 30 min. A resting sarcomere length was established from laser diffraction patterns and set at 2.0 μm. Isometric tension was recorded at room temperature on a chart recorder. After skinning, the fibers were initially washed in HR solution and their thickness was measured. Thereafter the fibers were exposed to solutions of varying Ca2+ concentration ([Ca2+]o range from 10−8 to 10−4.5 M). All solutions contained the protease inhibitors pepstatin A (2.5 μg ml−1), leupeptin (1 μg ml−1) and phenylmethylsulphonyl fluoride (PMSF, 50 μM). To determine the maximum stress developed by fiber bundles, we measured the maximum calcium-activated isometric force (at free [Ca2+] of 10−4.5) and divided by the cross-sectional area, as described previously [24]. The thickness of the fiber bundles was measured in two perpendicular planes at three points (the center and two distal ends) using a mirror and graticule fitted to the eyepiece of the microscope. The mean radius from three points was calculated and used to determine the cross-sectional area based on an elliptical shape of the fibers in cross-section.
2.6 Gel electrophoresis and Western blotting
The level of Tm phosphorylation was determined using a modified glycerol-PAGE method previously described by Fewell et al. [25]. Following measurement of force generation papillary muscle samples were frozen in liquid N2. The samples were thawed in TCA/TE buffer. DTT (10 mM) was added to this solution and samples were homogenized. After homogenization, samples were centrifuged at 12,000 rpm for 4 min and the supernatant discarded. The pellet was washed in ether 4 times and was then resuspended in sample buffer containing 10 mM DTT, 20 mM Tris base (pH 6.8), 9 M urea and 20 mM EDTA and loaded in a gel with 12% bis-acrylamide (1:19), 40% glycerol, 20 mM Tris base (pH 8.6) and 23 mM glycine (pH 8.6). The stacking gel contained 5% bis-acrylamide (1:19), 16% glycerol, 20 mM Tris base (pH 6.8) and 3.15 M urea. Gels were run for 2h at 40 V after the samples entered the separating gel. Proteins were transferred to a 0.22 μm nitrocellulose membrane and the membrane was blocked in 5% nonfat milk in Tris buffered saline (TBS), washed and incubated overnight in a polyclonal antibody against phospho-α Tm (p-α Tm). After washing, the membrane was incubated in a horseradish peroxidase-coupled anti-goat antibody (1:3000) for 40 min followed by ECL detection. Two bands were detected, an upper band representing endogenous Tm, and a lower band representing expressed Tm-E180G. The amount of p-α Tm in the samples was determined by densitometry (Personal densitometer, Amersham Bioscences) using DataQuant software and was adjusted for the total Tm in the sample.
2.7 Data presentation and statistical analysis
Results are reported as means ± standard error of the mean (S.E.M.) for the indicated number of preparations (n). Statistical significance was evaluated using a one-way ANOVA test.
3. Results
3.1 Force generation was maintained during acidosis in TG-Tm-E180G papillary muscle
In order to determine the effect of the Tm-E180G mutation on the decrease in force generation that is associated with acidosis, we measured isometric force development in intact papillary muscles from mouse right ventricles. Figure 1 shows representative examples of original force recordings in normal (pH 7.4) or acidic (pH 6.9) conditions followed by recovery to normal pH in NTG (A) and TG-Tm-E180G (B) papillary muscles. In both NTG and TG-Tm-E180G papillary muscles acidosis caused a transient drop in force followed by a sustained phase, however in the TG-Tm-E180G preparation the sustained phase was less inhibited. Figure 2 summarizes the changes in tension in NTG and TG-Tm-E180G papillary muscles in normal and acidic conditions. Acidosis reduced normalized tension in NTG preparations to 47.1 ± 5.0% of normal (n = 9) but in TG-Tm-E180G preparations to 81.6 ± 2.3% of normal (n = 9, p < 0.01) (Fig. 2A). Tension recovered to 95.0 ± 5.4% of normal in NTG muscles upon return to pH 7.4 and to 96.8 ± 4.7% of normal in TG-Tm-E180G muscles. There was no difference in the force normalized to cross-sectional area for NTG (8.8 ± 1.6 mN/mm2) compared to TG-TmE180G muscles (10.0 ± 2.5 mN/mm2, p > 0.05). Thus acidosis inhibited isometric force generation (Fig. 2B) by 52.9 ± 5.0% in NTG preparations but by only 18.4 ± 2.3% in TG-Tm-E180G (p < 0.01). The time to peak contraction was not affected by acidosis in either NTG or TG-Tm-E180G papillary muscles, however, it was significantly longer in TG-Tm-E180G muscles than in NTG muscles in all conditions tested. The time to 90% of twitch force relaxation (RT90) was also not altered by acidosis in NTG or TG-Tm-E180G muscles, but was significantly prolonged in muscles expressing TG-Tm-E180G compared to NTG in all conditions (Table 1). Thus the kinetics of contraction and relaxation were slower in TG-Tm-E180G papillary muscles than in NTG independent of pH, but the developed tension was significantly less inhibited by acidosis.
Figure 1.

Representative recordings of isometric force in intact papillary muscles during normal (pH 7.4) and acidic (pH 6.9) conditions in (A) NTG and (B) TG-Tm-E180G mouse preparations. See text and methods for details.
Figure 2.

Summary of (A) developed tension, (B) inhibition of tension by acidosis in NTG and TG-Tm-E180G preparations during normal and acidic conditions. Data are presented as means ± S.E.M. * Significantly different from NTG.
Table 1.
Summary of time to peak developed tension and time to 90% of relaxation (RT90) in NTG and TG-Tm-E180G preparations during normal and acidic conditions.
| Time to peak (ms) | RT90 (ms) | |||||
|---|---|---|---|---|---|---|
| pH 7.4 | pH 6.9 | pH 7.4 | pH 7.4 | pH 6.9 | pH 7.4 | |
| NTG | 121.5 ± 5.6 | 114.3 ± 5.1 | 115.0 ± 4.8 | 170.3 ± 22.6 | 186.6 ± 30 | 160.5 ± 15.8 |
| TG-Tm-E180G | 144.4 ± 5.8* | 139.3 ± 4.7* | 133.5 ± 4.1* | 274.1 ± 16.5* | 271.0 ± 16.1* | 267.0 ± 14.0* |
Data are presented as means ± S.E.M.
Significantly different from NTG.
3.2 Single cell shortening was less inhibited by acidosis in TG-Tm-E180G myocytes
Changes in contractility are governed in part by alterations in the functional state of the myofilaments but also by the characteristics of the intracellular Ca2+ transient. Expression of the Tm-E180G mutation results in increased myofilament Ca2+ sensitivity, but the effects of the mutation on intracellular Ca2+ fluxes and their response to acidosis are not known. Thus we examined the relationship between shortening and the Ca2+ transient during normal and acidic conditions in NTG and TG-Tm-E180G ventricular myocytes using video edge detection and fura-2 fluorescence. Figure 3 shows original recordings of shortening and fura-2 fluorescence in a representative NTG myocyte (A) and TG-Tm-E180G myocyte (B) at external pH 7.4 (normal), pH 7.1 (moderate acidosis) and pH 6.9 (acidosis). At normal pH conditions the NTG myocyte shortened to a lesser extent than the TG-Tm-E180G, but the Ca2+ transient peaks were nearly the same. In moderate acidosis shortening was inhibited to a greater degree in NTG than in TG-Tm-E180G, but the Ca2+ peaks were not different compared to each other or to normal pH. At the most acidic pH tested, 6.9, shortening was almost completely suppressed in both the NTG and TG-Tm-E180G myocytes but the Ca2+ transients were still relatively unaffected.
Figure 3.

Representative recordings of unloaded cellular shortening and Ca2+ transients from single, isolated (A) NTG and (B) TG-Tm-E180G mouse ventricular myocytes at pH 7.4, 7.1 and 6.9. (C) Representative recordings of intracellular pH (pHi) from isolated NTG and TG-Tm-E180G myocytes. See text and methods for details.
To establish that the blunted inhibition of shortening in the TG-Tm-E180G myocytes compared to NTG was not due to a differential change in intracellular pH in response to the extracellular pH conditions, we measured intracellular pH using the fluorescent indicator BCECF. Figure 3C shows the change in intracellular pH during perfusion with acidic extracellular solution of representative NTG and TG-Tm-E180G myocytes. Intracellular pH was determined from the nigericin-calibrated BCECF fluorescence. In the pH 7.4 extracellular solution, the intracellular pH in NTG cells was 7.10 ± 0.03 (n = 12 cells from 4 hearts) and in TG-Tm-E180G was 7.14 ± 0.02 (n = 12 cells from 4 hearts, p > 0.05). In the same cells bathed in the pH 6.9 solution, the intracellular pH in NTG was 6.91 ± 0.03, and in TG-Tm-E180G was 6.95 ± 0.02 (p > 0.05). Thus myocytes expressing the Tm-E180G mutation had the same change in internal pH in response to acidic conditions as NTG controls.
The Tm-E180G-induced changes in single-cell shortening are summarized in Figure 4. Unloaded cellular shortening (A) at normal pH in NTG cells was 3.4 ± 0.5% of total length (n = 11 cells from 6 hearts) and in TG-Tm-E180G cells was 7.2 ± 0.7% (n = 16 cells from 7 hearts, p < 0.01). In moderate acidosis shortening in NTG cells was 1.4 ± 0.1% of total length, and in TG-Tm-E180G cells was 4.1 ± 0.4% of total length (p < 0.01). Thus moderate acidosis inhibited shortening (B) in NTG cells by 56.2 ± 4.6% compared to normal pH, significantly more than in TG-Tm-E180G cells (40.6 ± 4.5%, p < 0.05). In acidosis (pH 6.9), shortening in NTG was 0.4 ± 0.1% of total length and in TG-Tm-E180G was 1.3 ± 0.3% (p > 0.05). Thus at the most acidic pH shortening was equally inhibited in NTG (87.4 ± 3.1%) and in TG-Tm-E180G cells (83.6 ± 3.3%, p > 0.05). The kinetics of cellular re-lengthening and the Ca2+ transient decay were evaluated by a mono-exponential fit and are presented in Table 2. The times of cellular re-lengthening in TG-Tm-E180G cells were significantly slower than in NTG at any pH tested, although the Ca2+ decay rates were not different.
Figure 4.

Summary of (A) unloaded cellular shortening as a percent of overall cell length, (B) inhibition of shortening by moderate acidosis (pH 7.1) and acidosis (pH 6.9). Data are presented as means ± S.E.M. * Significantly different from NTG.
Table 2.
Summary of the peak amplitude of the fura-2 fluorescence ratio, the time constant of re-lengthening and the time constant of fura-2 fluorescence decay in NTG and TG-Tm-E180G myocytes at pH 7.4, 7.1 and 6.9
| Fura-2 Ratio (340/380) | Time constant of Fura-2 decay (ms) | Time constant of re-lengthening (ms) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| pH 7.4 | pH 7.1 | pH 6.9 | pH 7.4 | pH 7.1 | pH 6.9 | pH 7.4 | pH 7.1 | pH 6.9 | |
| NTG | 0.79 ± 0.04 | 0.84 ± 0.04 | 0.85 ± 0.05 | 278.5 ± 10.4 | 295.5 ± 16.7 | 321.4 ± 20 | 111.4 ± 6.4 | 126.0 ± 6.7 | 146.9 ± 6.2 |
| TG-Tm-E180 G | 0.72 ± 0.03 | 0.77 ± 0.04 | 0.78 ± 0.04 | 311.1 ± 12.6 | 314.2 ± 16.6 | 352.3 ± 17.3 | 163.7 ± 9.4* | 181.8 ± 11.5* | 212.4 ± 17.1* |
Data are presented as means ± S.E.M.
Significantly different from NTG.
3.3 TG-Tm-E180G fiber bundles maintain increased myofilament Ca2+ sensitivity and maximal developed tension in acidosis
To evaluate the effect of the Tm-E180G mutation on the thin filament response to acidosis, we examined the force-calcium relationship using skinned fiber bundles from NTG and TG-Tm-E180G mice. These preparations expose the myofilaments directly to the solutions mimicking the interior of the cell, thus the normal condition was set at pH 7.0 and acidic at pH 6.5. Figure 5A shows that at normal pH the TG-Tm-E180G myofilaments were significantly more Ca2+ sensitive than NTG myofilaments. Acidosis caused a significant decrease of the myofilament Ca2+ sensitivity in both NTG and TG-Tm-E180G preparations. Figure 5B shows that at normal pH the pCa50 was 5.75 ± 0.02, n = 9 in NTG myofilaments and 5.99 ± 0.02, n = 6, (p < 0.01) in TG-Tm-E180G. In acidic conditions the pCa50 in NTG was 5.11 ± 0.02 p < 0.01 and in TG-Tm-E180G was 5.35 ± 0.02 (p < 0.01). There was no difference in the Hill coefficient between NTG and TG-Tm-E180G preparations at any pH tested. Figure 5C shows that maximal force was inhibited by the shift to pH 6.5 to a much greater degree in NTG preparations (28.2 ± 2.7%) than in TG-Tm-E180G preparations (9.2 ± 3.5%, p < 0.01).
Figure 5.
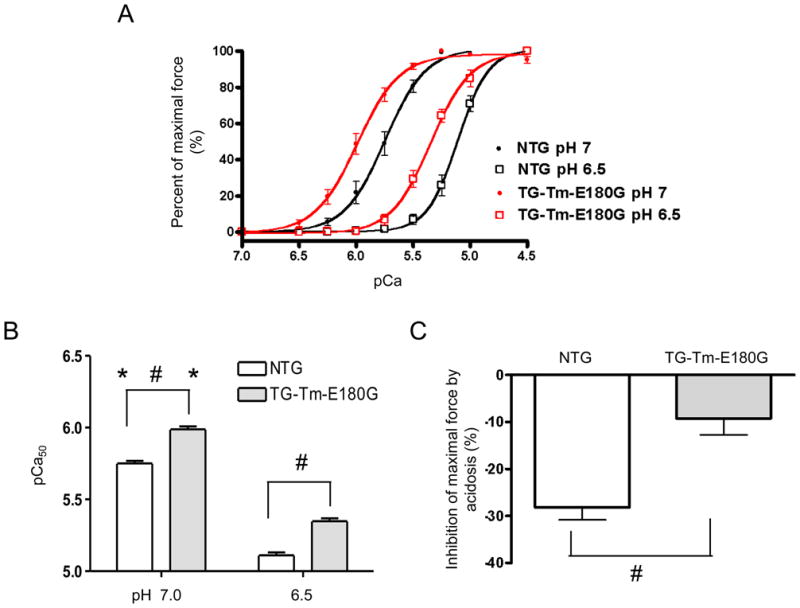
(A) Normalized force-pCa relationship of detergent-extracted NTG and TG-Tm-E180G cardiac fiber bundles during normal (pH 7.0) and acidic (pH 6.5) conditions. (B) pCa50 values, and (C) inhibition of the maximum developed tension in NTG and TG-Tm-E180G preparations at both pH conditions. Data are presented as means ± S.E.M. * Significantly different within each group from pH 6.5 to pH 7.0. # Significantly different from NTG.
3.4 Phosphorylation of Tm was not affected by acidic conditions
Since Tm phosphorylation at Ser 283 was reduced in mouse hearts expressing another Tm mutation (Tm-E54K) linked to dilated cardiomyopathy and is associated with decreased myofilament Ca2+ sensitivity [26, 27], we evaluated the phosphorylation state of Tm in samples from the NTG and TG-Tm-E180G papillary muscles used in the force-generation experiments. Figure 6A shows a representative Western blot of p- Tm from NTG and TG-Tm-E180G papillary muscle samples prepared at pH 7.4 and 6.9. Lanes with samples from NTG muscles had a single band of endogenous Tm. The TG-Tm-E180G lanes had an upper band of endogenous Tm and a lower band of expressed TG-Tm-E180G Tm. Figure 6B shows that the Tm phosphorylation level in NTG preparations was not different in acidic conditions (576 ± 49 a.u., n = 3) compared to normal (673 ± 116 a.u., n = 4, P > 0.05). Furthermore, the total Tm phosphorylation of combined endogenous and mutant Tm in the TG-Tm-E180G preparations was not different from total Tm phosphorylation in NTG in normal conditions (507 ± 51 a.u., n = 3, n.s.) or during acidosis (575 ± 120 a.u., n = 3, n.s.). Thus the increased myofilament Ca2+ sensitivity associated with the Tm-E180G mutation does not appear to be related to the phosphorylation state of Tm at Ser 283.
Figure 6.

(A) Representative Western blot probed with an antibody specific for phospho- Tm and (B) summary of the total Tm phosphorylation in NTG and TG-Tm-E180G preparations at both pH conditions. Data are presented as means ± S.E.M.
4. Discussion
The major findings of this study are that muscle preparations from hearts expressing the mutation Tm-E180G linked to FHC are less sensitive to acidic conditions than preparations from normal NTG heart. During acidosis the developed force and extent of cell shortening decreased less in Tm-E180G compared to controls, in agreement with the blunted depression of myofilament Ca2+ sensitivity. The smaller change in contractility in acidosis occurred with no change in the steady-state peak Ca2+ transient or in the level of Tm phosphorylation.
4.1 Tm-E180G and maladaptation to acidosis in FHC
Although increased myofilament sensitivity to Ca2+ and the ability to maintain force in acidosis is in some situations beneficial and acts to sustain proper blood pressure and cardiac output, this functional change can be detrimental in the setting of FHC. We have previously shown that ssTnI, the isoform type that is transiently expressed in neonatal hearts, renders the heart less sensitive to the ischemic and acidic conditions encountered, for example, during delivery and birth [28]. However, in the case of the neonate, the increase in myofilament Ca2+ sensitivity and resistance to acidosis due to the expression of ssTnI is not associated with any pathological change such as hypertrophy, myocyte disarray or fibrosis. Conversely, although expression of Tm-E180G in cardiac muscle maintains Ca2+ sensitivity and contractility in acidosis, it effectively increases the energetic demand on tissue that is already compromised by increased muscle mass and dysfunctional microvascular perfusion. Myocardial ischemia is known to occur in patients with FHC, and is emerging as an important and previously undervalued factor contributing to the onset of cardiac arrest in young patients, and to the progression of the disease [5, 9]. Thus we propose that the maintained contractility in acidosis of myocardium expressing the Tm-E180G mutation is maladaptive, as it potentially sustains oxygen demand in tissue that is predisposed to ischemia.
The Ca2+ sensitivity, or pCa50, of the Tm-E180G-expressing myofilaments was depressed in response to acidosis to a similar degree as in controls although the maximal developed force was not similarly depressed. When expressed as EC50 the Ca2+ sensitivity of the Tm-E180G myofilaments was significantly less changed by acidosis than controls. Konhilas et al, 2002 showed that with ssTnI substitution into myofilaments the ΔEC50 changed significantly but the ΔpCa50 did not, demonstrating that although the two values describe the same concentration of Ca2+, their interpretation can be different [29]. Thus the reduction of Ca2+ sensitivity in the Tm-E180G myofilaments by acidosis is less than in controls, consistent with maintained contractility and the smaller depression of maximum tension observed in Figure 5C.
4.2 Modification of myofilament protein interactions by Tm-E180G
Expression of the Tm-E180G mutation alters myofilament function in the absence of a change in Tm phosphorylation level. The Tm glutamate to glycine mutation occurs in the region of interaction with TnT, potentially altering binding with TnT, interactions within the Tm dimers, and the cooperativity of the myofilaments [13, 15]. The Tm-E180G mutation destabilizes the coiled-coil structure, thus increasing Tm flexibility in the surrounding regions. This mutation also decreases the affinity of Tm for F-actin by more than 2-fold whether or not the troponin complex (Tn) is present, consistent with the increased Ca2+ sensitivity of developed tension and slowed contraction and relaxation kinetics we observed [30, 31]. Although Tm does not interact directly with cTnC, it appears that the presence of a mutation in the region of Tm interaction with cTnT changes the manner of Tm movement in coordination with the Tn complex to increase the force developed for a particular level of Ca2+-dependent activation. Previous work from our laboratory indicates the Tm-E180G mutation causes a secondary alteration in the interaction of TnC and TnI, which may contribute to the increased myofilament Ca2+ sensitivity [32]. Moreover, recent electron microscopy reconstruction studies indicate that structural interaction between the regulatory C-terminal domain of TnI and Tm is a significant mechanism that regulates the binding of Tm to actin and the actin-myosin interaction [33]. Effects of the mutation on the interaction of Tm with TnI may be of particular functional significance, as the role of TnI isoform is crucial in the differential response to pH observed in skeletal versus cardiac muscle [28, 34, 35]. This evidence supports a reduction of steric interference as a potential mechanism responsible for the effects of Tm-E180G on myosin-crossbridge interaction and the functional consequences we have reported here.
4.3 Tm-E180G and the intracellular Ca2+ response to acidosis
Acidosis affects myocardial contractility through altered cellular Ca2+ fluxes as well as by modifying the functional properties of myofilament proteins. The early drop in developed force at the onset of acidosis is generally attributed to depressed myofilament Ca2+ sensitivity, not the peak Ca2+ levels that may either remain unchanged or decrease [36, 37]. However, the initial phase of force decline is often followed by a slow recovery that is attributed to increased cytosolic Ca2+. The apparent Ca2+ recovery may be due to a combination of inhibited RyR function, higher diastolic Ca2+ and slowed reuptake of Ca2+ to the SR [38, 39]. We observed a small decrease in the steady-state TG-Tm-E180G peak Ca2+ transient level compared to NTG, but this was not significant at any pH examined. Furthermore, an increase in peak Ca2+ would be expected if this were primary to maintained contractility. Thus the effect of the Tm-E180G mutation on the contractile response to acidosis appears to be mainly due to the smaller depression of myofilament Ca2+ sensitivity rather than through altered intracellular Ca2+.
4.4 Ventricular arrhythmias in FHC
Sudden death in FHC is associated with ischemia and the initiation of sustained ventricular arrhythmias but the mechanisms of the progression to cardiac arrest are unclear. Localized regions of ischemia and scarring in hypertrophic left ventricle predispose the tissue to electrical instability and re-entry consistent with microvascular dysfunction [40, 41]. On the level of the myofilaments, there is evidence indicating arrhythmogenic Ca2+ waves arise at sites of functional non-uniformity in rat cardiac trabeculae, caused by local mechanical or ischemic damage [42]. Additional recent evidence further indicates that myofilament Ca2+ sensitization alone can increase the likelihood of cardiac arrhythmias through increased dispersion of the action potential along the tissue [43]. Thus in an environment of localized ischemia, the increased myofilament Ca2+ sensitivity due to Tm-E180G expression has the potential to enhance the development of an arrhythmic substrate. Further studies are required to determine the effects of acidosis on the development of arrhythmias that are beyond the scope of this work. It appears likely that enhanced sensitivity to Ca2+ may also produce an arrhythmic substrate in hearts expressing Tm-E180G.
5. Summary
In the current study we have shown that the TG-Tm-E180G mutation alters the contractile response to acidosis mainly through resistance to depression of myofilament Ca2+ sensitivity, rather than through alteration of the Ca2+ transient. We have established that papillary muscles from mouse hearts expressing the Tm-E180G mutation maintain tension in acidosis through a mechanism independent of Tm phosphorylation. We propose that the increased myofilament Ca2+ sensitivity in TG-Tm-E180G hearts reduces the ability of acidosis to decrease tension in an already compromised environment, contributing to maladaptation and progression of the disease.
Acknowledgments
This work was supported by grants from NIH R01: HL79032 (BMW), HL64035 (RJS, BMW), HL22231 (RJS), HL81680 (DFW).
Footnotes
Disclosures
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212–8. doi: 10.1016/s0735-1097(00)01003-2. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Roberts WC, Epstein SE. Sudden death in hypertrophic cardiomyopathy: a profile of 78 patients. Circulation. 1982;65:1388–94. doi: 10.1161/01.cir.65.7.1388. [DOI] [PubMed] [Google Scholar]
- 3.Morimoto S. Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res. 2008;77:659–66. doi: 10.1093/cvr/cvm084. [DOI] [PubMed] [Google Scholar]
- 4.Alves ML, Gaffin RD, Wolska BM. Rescue of familial cardiomyopathies by modifications at the level of sarcomere and Ca2+ fluxes. J Mol Cell Cardiol. 48:834–42. doi: 10.1016/j.yjmcc.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maron MS, Olivotto I, Maron BJ, Prasad SK, Cecchi F, Udelson JE, et al. The case for myocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:866–75. doi: 10.1016/j.jacc.2009.04.072. [DOI] [PubMed] [Google Scholar]
- 6.Olivotto I, Cecchi F, Camici PG. Coronary microvascular dysfunction and ischemia in hypertrophic cardiomyopathy. Mechanisms and clinical consequences. Ital Heart J. 2004;5:572–80. [PubMed] [Google Scholar]
- 7.Cannon RO, 3rd, Rosing DR, Maron BJ, Leon MB, Bonow RO, Watson RM, et al. Myocardial ischemia in patients with hypertrophic cardiomyopathy: contribution of inadequate vasodilator reserve and elevated left ventricular filling pressures. Circulation. 1985;71:234–43. doi: 10.1161/01.cir.71.2.234. [DOI] [PubMed] [Google Scholar]
- 8.Dilsizian V, Bonow RO, Epstein SE, Fananapazir L. Myocardial ischemia detected by thallium scintigraphy is frequently related to cardiac arrest and syncope in young patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1993;22:796–804. doi: 10.1016/0735-1097(93)90193-5. [DOI] [PubMed] [Google Scholar]
- 9.Basso C, Thiene G, Corrado D, Buja G, Melacini P, Nava A. Hypertrophic cardiomyopathy and sudden death in the young: pathologic evidence of myocardial ischemia. Hum Pathol. 2000;31:988–98. doi: 10.1053/hupa.2000.16659. [DOI] [PubMed] [Google Scholar]
- 10.Knaapen P, Germans T, Camici PG, Rimoldi OE, ten Cate FJ, ten Berg JM, et al. Determinants of coronary microvascular dysfunction in symptomatic hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2008;294:H986–93. doi: 10.1152/ajpheart.00233.2007. [DOI] [PubMed] [Google Scholar]
- 11.Schwartzkopff B, Mundhenke M, Strauer BE. Alterations of the architecture of subendocardial arterioles in patients with hypertrophic cardiomyopathy and impaired coronary vasodilator reserve: a possible cause for myocardial ischemia. J Am Coll Cardiol. 1998;31:1089–96. doi: 10.1016/s0735-1097(98)00036-9. [DOI] [PubMed] [Google Scholar]
- 12.Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830–40. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 13.Wolska BM, Wieczorek DM. The role of tropomyosin in the regulation of myocardial contraction and relaxation. Pflugers Arch. 2003;446:1–8. doi: 10.1007/s00424-002-0900-3. [DOI] [PubMed] [Google Scholar]
- 14.Jagatheesan G, Rajan S, Wieczorek DF. Investigations into tropomyosin function using mouse models. J Mol Cell Cardiol. 2010;48:893–8. doi: 10.1016/j.yjmcc.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prabhakar R, Boivin GP, Grupp IL, Hoit B, Arteaga G, Solaro JR, et al. A familial hypertrophic cardiomyopathy α-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J Mol Cell Cardiol. 2001;33:1815–28. doi: 10.1006/jmcc.2001.1445. [DOI] [PubMed] [Google Scholar]
- 16.Prabhakar R, Petrashevskaya N, Schwartz A, Aronow B, Boivin GP, Molkentin JD, et al. A mouse model of familial hypertrophic cardiomyopathy caused by a α-tropomyosin mutation. Mol Cell Biochem. 2003;251:33–42. [PubMed] [Google Scholar]
- 17.Dias FA, Walker LA, Arteaga GM, Walker JS, Vijayan K, Pena JR, et al. The effect of myosin regulatory light chain phosphorylation on the frequency-dependent regulation of cardiac function. J Mol Cell Cardiol. 2006;41:330–9. doi: 10.1016/j.yjmcc.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 18.Wolska BM, Kitada Y, Palmiter KA, Westfall MV, Johnson MD, Solaro RJ. CGP-48506 increases contractility of ventricular myocytes and myofilaments by effects on actin-myosin reaction. Am J Physiol. 1996;270:H24–32. doi: 10.1152/ajpheart.1996.270.1.H24. [DOI] [PubMed] [Google Scholar]
- 19.Wolska BM, Solaro RJ. Method for isolation of adult mouse cardiomyocytes for studies of contraction and microfluorimetry. Am J Physiol. 1996;271:H1250–H5. doi: 10.1152/ajpheart.1996.271.3.H1250. [DOI] [PubMed] [Google Scholar]
- 20.Steadman BW, Moore KB, Spitzer KW, Bridge JH. A video system for measuring motion in contracting heart cells. IEEE Trans Biomed Eng. 1988;35:264–72. doi: 10.1109/10.1375. [DOI] [PubMed] [Google Scholar]
- 21.Miyata H, Hayashi H, Suzuki S, Noda N, Kobayashi A, Fujiwake H, et al. Dual loading of the fluorescent indicator fura-2 and 2,7-biscarboxyethyl-5(6)-carboxyfluorescein (BCECF) in isolated myocytes. Biochem Biophys Res Commun. 1989;163:500–5. doi: 10.1016/0006-291x(89)92165-7. [DOI] [PubMed] [Google Scholar]
- 22.Wolska BM, Averyhart-Fullard V, Omachi A, Stojanovic MO, Kallen RG, Solaro RJ. Changes in thyroid state affect pHi and Nai+ homeostasis in rat ventricular myocytes. J Mol Cell Cardiol. 1997;29:2653–63. doi: 10.1006/jmcc.1997.0495. [DOI] [PubMed] [Google Scholar]
- 23.Wolska BM, Keller RS, Evans CC, Palmiter KA, Phillips RM, Muthuchamy M, et al. Correlation between myofilament response to Ca2+ and altered dynamics of contraction and relaxation in transgenic cardiac cells that express β-tropomyosin. Circ Res. 1999;84:745–51. doi: 10.1161/01.res.84.7.745. [DOI] [PubMed] [Google Scholar]
- 24.Evans CC, Pena JR, Phillips RM, Muthuchamy M, Wieczorek DF, Solaro RJ, et al. Altered hemodynamics in transgenic mice harboring mutant tropomyosin linked to hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2000;279:H2414–23. doi: 10.1152/ajpheart.2000.279.5.H2414. [DOI] [PubMed] [Google Scholar]
- 25.Fewell JG, Osinska H, Klevitsky R, Ng W, Sfyris G, Bahrehmand F, et al. A treadmill exercise regimen for identifying cardiovascular phenotypes in transgenic mice. Am J Physiol. 1997;273:H1595–605. doi: 10.1152/ajpheart.1997.273.3.H1595. [DOI] [PubMed] [Google Scholar]
- 26.Warren CM, Arteaga GM, Rajan S, Ahmed RP, Wieczorek DF, Solaro RJ. Use of 2-D DIGE analysis reveals altered phosphorylation in a tropomyosin mutant (Glu54Lys) linked to dilated cardiomyopathy. Proteomics. 2008;8:100–5. doi: 10.1002/pmic.200700772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajan S, Ahmed RP, Jagatheesan G, Petrashevskaya N, Boivin GP, Urboniene D, et al. Dilated cardiomyopathy mutant tropomyosin mice develop cardiac dysfunction with significantly decreased fractional shortening and myofilament calcium sensitivity. Circ Res. 2007;101:205–14. doi: 10.1161/CIRCRESAHA.107.148379. [DOI] [PubMed] [Google Scholar]
- 28.Wolska BM, Vijayan K, Arteaga GM, Konhilas JP, Phillips RM, Kim R, et al. Expression of slow skeletal troponin I in adult transgenic mouse heart muscle reduces the force decline observed during acidic conditions. J Physiol. 2001;536:863–70. doi: 10.1111/j.1469-7793.2001.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konhilas JP, Irving TC, Wolska BM, Jweied EE, Martin AF, Solaro RJ, et al. Troponin I in the murine myocardium: influence on length-dependent activation and interfilament spacing. J Physiol. 2003;547:951–61. doi: 10.1113/jphysiol.2002.038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golitsina N, An Y, Greenfield NJ, Thierfelder L, Iizuka K, Seidman JG, et al. Effects of two familial hypertrophic cardiomyopathy-causing mutations on alpha-tropomyosin structure and function. Biochemistry. 1997;36:4637–42. doi: 10.1021/bi962970y. [DOI] [PubMed] [Google Scholar]
- 31.Golitsina N, An Y, Greenfield NJ, Thierfelder L, Iizuka K, Seidman JG, et al. Effects of two familial hypertrophic cardiomyopathy-causing mutations on alpha-tropomyosin structure and function. Biochemistry. 1999;38:3850. doi: 10.1021/bi9950701. [DOI] [PubMed] [Google Scholar]
- 32.Burkart EM, Arteaga GM, Sumandea MP, Prabhakar R, Wieczorek DF, Solaro RJ. Altered signaling surrounding the C-lobe of cardiac troponin C in myofilaments containing an α-tropomyosin mutation linked to familial hypertrophic cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2003;35:1285–93. doi: 10.1016/s0022-2828(03)00240-2. [DOI] [PubMed] [Google Scholar]
- 33.Galinska-Rakoczy A, Engel P, Xu C, Jung H, Craig R, Tobacman LS, et al. Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J Mol Biol. 2008;379:929–35. doi: 10.1016/j.jmb.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engel PL, Kobayashi T, Biesiadecki B, Davis J, Tikunova S, Wu S, et al. Identification of a region of troponin I important in signaling cross-bridge dependent activation of cardiac myofilaments. J Biol Chem. 2006 doi: 10.1074/jbc.M512337200. [DOI] [PubMed] [Google Scholar]
- 35.Urboniene D, Dias FA, Pena JR, Walker LA, Solaro RJ, Wolska BM. Expression of slow skeletal troponin I in adult mouse heart helps to maintain the left ventricular systolic function during respiratory hypercapnia. Circ Res. 2005;97:70–7. doi: 10.1161/01.RES.0000173849.68636.1e. [DOI] [PubMed] [Google Scholar]
- 36.Allen DG, Orchard CH. The effects of changes of pH on intracellular calcium transients in mammalian cardiac muscle. J Physiol. 1983;335:555–67. doi: 10.1113/jphysiol.1983.sp014550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi HS, Trafford AW, Orchard CH, Eisner DA. The effect of acidosis on systolic Ca2+ and sarcoplasmic reticulum calcium content in isolated rat ventricular myocytes. J Physiol. 2000;529(Pt 3):661–8. doi: 10.1111/j.1469-7793.2000.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hulme JT, Orchard CH. Effect of acidosis on Ca2+ uptake and release by sarcoplasmic reticulum of intact rat ventricular myocytes. Am J Physiol. 1998;275:H977–87. doi: 10.1152/ajpheart.1998.275.3.H977. [DOI] [PubMed] [Google Scholar]
- 39.Fabiato A, Fabiato F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiac and skeletal muscles. J Physiol. 1978;276:233–55. doi: 10.1113/jphysiol.1978.sp012231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozhevnikov D, Caref EB, El-Sherif N. Mechanisms of enhanced arrhythmogenicity of regional ischemia in the hypertrophied heart. Heart Rhythm. 2009;6:522–7. doi: 10.1016/j.hrthm.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 41.Hedman A, Hartikainen J, Vanninen E, Laitinen T, Jaaskelainen P, Laakso M, et al. Inducibility of life-threatening ventricular arrhythmias is related to maximum left ventricular thickness and clinical markers of sudden cardiac death in patients with hypertrophic cardiomyopathy attributable to the Asp175Asn mutation in the α-tropomyosin gene. J Mol Cell Cardiol. 2004;36:91–9. doi: 10.1016/j.yjmcc.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 42.Ter Keurs HE, Wakayama Y, Miura M, Shinozaki T, Stuyvers BD, Boyden PA, et al. Arrhythmogenic Ca2+ release from cardiac myofilaments. Prog Biophys Mol Biol. 2005 doi: 10.1016/j.pbiomolbio.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 43.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, et al. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]