

Abstract
Studies aimed at elucidating the unknown Mg2+ binding site in protein farnesyltransferase (FTase) are reported. FTase catalyzes the transfer of a farnesyl group to a conserved cysteine residue (Cys1p) on a target protein, an important step for proteins in the signal transduction pathways (e.g. Ras). Mg2+ ions accelerate the protein farnesylation reaction by up to 700-fold. The exact function of Mg2+ in catalysis and the structural characteristics of its binding remain unresolved to date. Molecular Dynamics (MD) simulations addressing the role of magnesium ions in FTase are presented, and relevant octahedral binding motifs for Mg2+ in wild type (WT) FTase and Dβ352A mutant are explored. Our simulations suggest that the addition of Mg2+ ions causes a conformational changes to occur in the FTase active site, breaking interactions known to keep FPP in its inactive conformation. Two relevant Mg2+ ion binding motifs were determined in WT FTase. In the first binding motif, WT1, the Mg2+ ion is coordinated to D352β, zinc-bound D297β, two water molecules, and one oxygen atoms from the α- and β-phosphates of farnesyl diphosphate (FPP). The second binding motif, WT2, is identical with the exception of the zinc-bound D297β being replaced by a water molecule in the Mg2+ coordination complex. In the Dβ352A mutant Mg2+ binding motif, D297β, three water molecules and one oxygen atom from the α- and β-phosphates of FPP complete the octahedral coordination sphere of Mg2+. Simulations of WT FTase, in which Mg2+ was replaced by water in the active site, re-created the salt bridges and hydrogen bonding patterns around FPP, validating these simulations. In all Mg2+ binding motifs, a key hydrogen bond was identified between a magnesium bound water and Cys1p, bridging the two metallic binding sites, and thereby, reducing the equilibrium distance between the reacting atoms of FPP Cys1p. The free energy profiles calculated for these systems provide a qualitative understanding of experimental results. They demonstrate that the two reactive atoms approach each other more readily in the presence of Mg2+ in WT FTase and mutant. The flexible WT2 model was found to possess the lowest barrier towards the conformational change, suggesting it is the prefered Mg2+ binding motif in WT FTase. In the mutant, the absence of D352β makes the transition towards a conformational change harder. Our calculations find support for the proposal that D352β performs a critical role in Mg2+ binding and Mg2+ plays an important role in the conformational transition step.
Protein farnesyltransferase (FTase) catalyzes the transfer of a farnesyl group to a cysteine residue (Cys1p) on a CaaX motif (a=aliphatic, X=Met) at or near the C-terminus of a protein substrate(1, 2) (see Figure 1). Farnesylation is a critical step in the posttranslational modification of the Ras family of proteins(3–7). Mutations in these proteins are associated with almost 50% of tumors in human-colon-carcinomas and 30% of human cancers. The potential to prevent unmitigated cell growth by inhibiting mutant Ras activity has made the FTase catalyzed farnesylation reaction the subject of numerous studies(2, 8–24). Several FTase inhibitors (FTIs) have entered different clinic trial phases(2, 25–31). FTase is a zinc metalloenzyme composed of two subunits, with the active site located in a funnel shaped cavity within the β subunit. Computational, kinetic, structural and mutagenesis investigations have revealed the nature of this active site. The two reacting atoms, namely C1 of the substrate farnesyldiphosphate (FPP) and S γ of Cys1p, are 7 Å (see Figure 2) apart in the crystal structures of the ternary complex (FTase-FPP-peptide substrate). The protein substrate coordinates to the zinc ion through S of Cys1p, forming a tetrahedral zinc complex along with the enzymatic residues D297β, C299β and H362β. The direct coordination of the reactive sulfur atom to the zinc ion, suggests that the zinc ion is involved in the catalytic reaction(32). The exact nature of the reaction pathway has been a source of debate. Recent kinetic isotope effects (KIE) experiments by the Fierke (33) and Distefano groups (20) have indicated that the reaction adopts a SN2-like associative pathway mechanism, with partial carbocation character (SN1-like) in the transition state structure.
Figure 1.

The FTase catalyzed farnesylation reaction.
Figure 2.
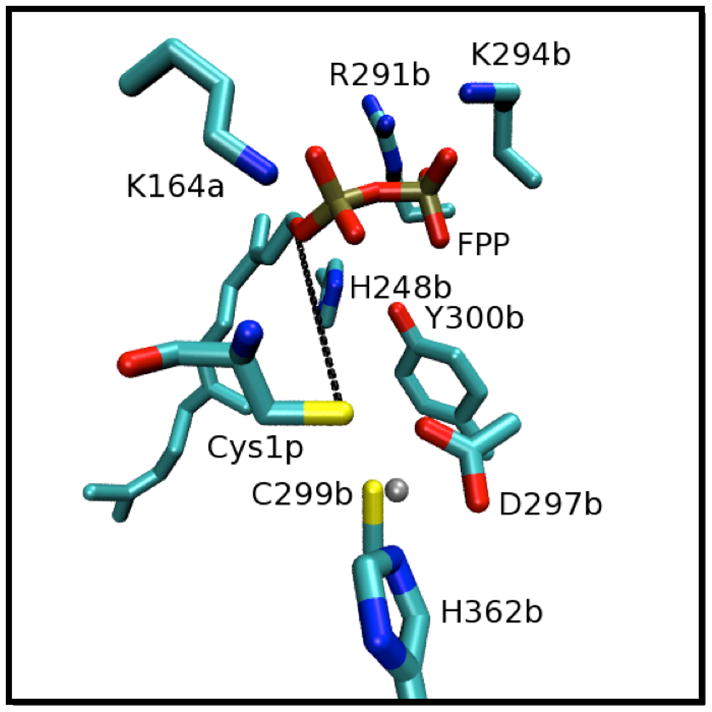
Snapshot of the FTase active site from the initial structure. Zinc is represented as a grey sphere. The black dashed line connects the two reactive atoms, C1 of FPP and S γ of the cysteine peptide (Cys1p) and is greater than 7Å in length. Important side chain amino acids are also labeled. Residues in the α and β subunit are labeled with a and b, e.g., K164α and K294β are labeled as K164a and K294b, separately.
Mg2+ ions have been found to enhance the rate of product formation by ~700-fold without affecting the affinity of either the peptide or FPP(34). The exact function of Mg2+ ions has been a subject of recent interest, and is not entirely understood. A magnesium ion has been determined to be bound in the active site, though the exact nature of its binding remains to be resolved by structural analysis. Kinetic and mutagenesis experiments performed by Fierke and coworkers(34), in conjunction with crystallographic studies of FTase with Mn2+ ions carried out by Beese and coworkers(2), have provided insights into the characteristics of Mg2+ ion binding in the active site. It is widely accepted that bound Mg2+ forms an octahedral complex with negatively charged groups. In a proposed model, Mg2+ coordinates to a non-bridging phosphate oxygen of α- and β-phosphate in FPP, the carboxylate side chain of D352β, a carboxylic oxygen of zinc bound D297β, and a water molecule(34). D352β has been identified to be crucial for Mg2+ ion binding in the active site, with mutations (Dβ352A and Dβ352K) diminishing the rate enhancing ability of Mg2+ ions. It has been proposed that the formation of a D297β bridged bimetal catalytic site helps reduce the 7 Å distance gap between the two reacting atoms, bringing the enzyme into a pre-organized state for the chemical step. It is also thought that Mg2+ ions promote the farnesylation reaction by stabilizing the developing negative charge on the diphosphate (PPi) leaving group, and increasing the carbocation character of C1 in the chemical step.
Computational studies have aided in understanding the FTase catalyzed farnesylation reaction. Cui et al. have studied the substrate binding and conformational activation of FTase in the absence of Mg2+ ions using MD simulations and free energy sampling methods(17, 18). They found that the highly charged binding pocket kept FPP3− locked in an inactive conformation, and the monoprotonated diphosphate group (FPP2−) was the only from of FPP that could activate the conformational transition via rotation of the bond between the first two isoprenoid groups. The Ramos group’s computational efforts to explain the so-called “distance paradox”(35) agreed with the earlier Merz et al. study, that conformational activation is attributed to the isoprenoid-rotation mechanism. More recently, Klein and co-workers investigated the reaction pathway using a quantum mechanical/molecular mechanical (QM/MM) approach(19). Their results suggest that the reaction goes through “an associative pathway with dissociative characteristics”, in agreement with experimental observations. This study focused on the chemical step, and did not explore the prior conformational change in the system. However, the system investigated, FTase/FPP3− without Mg2+ is a system that is not capable of the necessary conformational change to bring the two reactive centers into contact. Moreover, it is a system that has not been observed experimentally because FPP3− under these conditions is likely protonated. Crucially, in all of the studies reported to date the role of Mg2+ has not been examined leaving a large gap in our understanding of FTase activity under physiological conditions. Nonetheless, previous studies have demonstrated the efficacy of computational methods in exploring FTase activity, and have opened the way to explore the catalytic reaction in the presence of Mg2+ ions.
In this paper we present a computational study aimed at locating the Mg2+ binding site in FTase. We use MD simulations and free energy calculation methods to elucidate the function of Mg2+ ions in the conformational transition step of the reaction. The role of D352β in binding a magnesium ion in the active site is also investigated in this study. We perform calculations on WT FTase and the Dβ352A single mutant with FPP3− and CVIM peptide as substrates, in the presence of an active site bound Mg2+ ion. The Dβ352A mutation was examined in order to reproduce and provide molecular-level insights into experimental results. We validate our simulations by replacing the active site bound Mg2+ ion with a water molecule, and compare the results from this simulation to previously published results. In the absence of crystal structures of FTase with bound Mg2+ ion in the active site, we generate starting structures using a methodology successfully utilized earlier by Merz(36) to identify CO2 binding sites in human carbonic anhydrase II. We start with several “educated” guesses as starting structures for WT and mutant FTase/FPP3−/Mg2+ complexes based on experimental data. We then identify relevant Mg2+ binding motifs and investigate them using MD simulations. We analyze our simulations for hydrogen-bonding patterns, and conformational changes across these motifs. We also calculate the potential of mean force (PMF) for the conformational change in FPP for every motif. The results from our investigations provide new insight into the role of Mg2+ ions in helping FPP undergo the conformational transition in the farnesylation reaction, and the associated experimentally observed rate enhancement. We identify a critical hydrogen-bonding pattern in WT FTase, and test the proposal for a D297β bridged bi-metallic catalytic site. These simulations provide a physical basis for the function of D352β in FTase, both with and without magnesium ions. Based on the results from our calculations, we propose a novel model for Mg2+ binding in the active site of FTase that can be experimentally tested.
The following section of this paper introduces the computational methods used to study the FTase reaction. In this section, we describe methods used to generate and investigate possible Mg2+ binding motifs. In Section 3, we present our analyses of the various systems. In the first part of this section, we present the results from modeling mutant and WT FTase with Mg2+ ions. The second part of this section analyzes the results from our simulation without bound magnesium in the active site. In the third part of the section, we present the results from our free-energy calculations. Our conclusions from this study are presented in Section 4. We explain the impact of mutations on the enzyme and, finally, we describe our proposed motif for Mg2+ binding in the FTase active site.
Methodology
1. Initial Structural Models
The starting structure of WT FTase was generated using the protocol from a previous study of the FTase reaction performed by Merz and coworkers(17). In brief, the reactive form of FTase/FPP complexed with the substrate peptide (acetyl-CVIM) was obtained by superimposing two crystal structures (PDB codes: 1QBQ and 1FT2). All crystal water molecules were retained. The AMBER force field ff99sb by Hornak et al. was applied to the system(37). Charged amino acid chains were modeled in their normal protonation states at physiological pH, and all histidine residues were treated as neutral. At physiological pH, Cys1p has been found to be deprotonated, and was modeled as a thiolate(38). It was coordinated to the zinc ion along with D297β, C299β and H362β from the protein. The zinc parameters previously derived by Cui et al. were used for all zinc complexes reported in this paper(39). The VDW parameters, ε=1.10 and r0=0.0125(40), were used for zinc. The AMBER 10 suite of programs(41) was chosen for all molecular mechanics and MD simulations. FPP was represented in its entirely deprotonated state, FPP3−, with 3 units of negative charge. The protonation of PPi leads to a loss of Mg2+ binding affinity(42), making FPP2− unimportant for the present study. The parameters used to model FPP were obtained from the generic AMBER force field (GAFF)(43) for small organic molecules. In addition, torsion parameters for the farnesyl group developed by Merz and coworkers(17), and diphosphate parameters developed by Carson and coworkers were used(44). The restrained electrostatic potential (RESP) charges for FPP3− were derived using the prescribed procedure for the AMBER force field(45, 46). In accordance with the procedure, a geometry optimization of FPP was performed using density functional theory at the B3LYP/6-31G* level with Gaussian 03(47). The electrostatic potential derived atomic point charges were then calculated at the HF/6-31G* level. Finally, the atomic charges were derived following a two-stage RESP fitting procedure.
2. Magnesium in FTase Modeling and MD
We examined the FPP binding pocket for aspartic acid residues proximal to PPi. D196α, D297β and D352β were identified as potential candidates for Mg2+ ion binding. Based on experimental and theoretical evidence, the sequence conserved D297β and D352β residues were recognized as the most possible Mg2+ binding ligands. We created “guess” starting structures for our simulations by positioning a magnesium ion at different sites within the binding pocket (see Figure 3). In these structures, the Mg2+ ion was positioned close to negatively charged PPi and one of the three candidate aspartic acid residues. Neighboring crystal water molecules completed the octahedral coordination of the Mg2+ ion. A total of 10 guess structures were produced. These systems are referred to as FTase/FPP3−/Mg2+ in the remainder of this text. The AMBER default VDW parameters were used for magnesium (ε=0.7926, r0=0.8947)(48). Attempts to find such binding structures using local enhanced sampling (LES) were unsuccessful. This was likely caused by the significant charge on the Mg2+ ion.
Figure 3.

Snapshots of the FTase active site and important hydrogen bond networks taken from different models. Mg2+ (in green) and Zn2+ (in grey) ligand environments are shown as black dashed lines while hydrogen bonds are labeled as red dashed lines. Top left: the 10 initial Mg2+ placements; top middle: WT1; top right: WT2; bottom left: WT3; bottom middle: MUT; bottom right: SWAP.
Each FTase/FPP3−/Mg2+ system was solvated in a periodically replicated truncated octahedral water box with each side of the box at least 10Å away from the closest solute atom. The three-site constraint TIP3P water model was employed(49), and counter ions were added to neutralize the net negative charge of the system. Long range electrostatic interactions were calculated by the particle mesh Ewald (PME)(50) method. An 8Å cutoff was used for the nonbonded interactions in real-space, along with a 1Å reciprocal space grid spacing. The SHAKE(51) algorithm was applied to constraint the length of covalent bonds in which hydrogen atoms were involved, and the translational movement of the system was removed. 5000 steps of steepest descent minimization was carried out with a weak harmonic positional restraint applied to the enzyme, substrate peptide and zinc complex, followed by a 5000-step conjugate gradient minimization. Next, the system was heated to 300K over 100ps of MD with 1fs time steps for a canonical ensemble (NVT). The temperature of the system was maintained using Langevin dynamics with a 1 ps−1 collision frequency. All restraints were released prior to data collection. A 6ns MD trajectory for an isobaric, isothermal ensemble (NPT) at 1atm was propagated with a 2fs timestep. Snapshots were saved every picosecond, and the resulting trajectories from the last 4ns of the simulation were examined to validate reasonable Mg2+ binding motifs. The D297β bridged Zn-Mg site binding motif featuring a bimetallic catalytic site, was artificially constructed by using an NMR restraint to force D297β to displace one of the Mg2+ bound water molecules in a D352β bound model. This motif was investigated using the scheme described above.
3. Dβ352A Mutation
We prepared the Dβ352A mutant form of FTase from the final snapshot of a successful D352β binding Mg2+ binding motif. All counter ions, and water molecules located more than 6Å away from the substrate FPP3− or Mg2+ were removed. D352β was modified as an Alanine, leaving D297β as the only neighboring aspartic acid residue. This system is referred to as MUT in this paper. The resulting MUT system was immersed in a truncated octahedral box and neutralized with counter-ions. The system was minimized to remove strain from the residue modification. The MUT system was then simulated using the same procedures described for WT FTase.
4. Mg2+/H2O Switch Simulation
We removed the bound Mg2+ ion from the active site of a successful FTase/FPP3−/Mg2+ simulation to further validate our Mg2+ bonding model. A straightforward method was adopted wherein the coordinates and velocity information of the bound Mg2+ ion and the oxygen atom of a bulk water molecule, denoted H2Oswap, were exchanged. The coordinates of the two H2Oswap hydrogen atoms were modified suitably, while their velocities were preserved. The remaining parts of the system were untouched. The resulting system is referred to as SWAP in the remainder of this paper. The SWAP system was studied with the same MD procedure used for WT FTase with active site bound Mg2+ ion. 4 ns of equilibration followed by 4 ns of NPT MD simulations were performed for this system.
5. Potential of Mean Force Studies
We calculated the potential of mean force (PMF) as a function of the distance between the two reacting centers, namely C1 of FPP and S γ of the target cysteine peptide for the various systems investigated in this study. In these calculations, the reactive atoms were “pulled” from the crystal structure distance of 8.0Å to 4.5Å. A PMF termination distance of 4.5Å was chosen based on the sum of the van der Waals (VDW) radii of sulfur and oxygen in the AMBER ff99SB force field since sampling below this distance would likely yield spurious results. We obtained the initial structures for the umbrella sampling(52) calculations from a MD scanning procedure driven by a large harmonic force. The reaction coordinate (RC) was divided into 15 windows spaced 0.25Å apart. A harmonic potential of 20kcal/(mol•Å2) was applied to each window during a 2ns simulation with a 2fs time step. Data was collected every 5 steps after 1ns of equilibration; therefore, 10,000 snapshots were saved for every window and at least 100,000 points were stored for the following analysis. Longer periods of sampling were performed in the regions near the intermediate or transition barrier to improve the overlap between adjacent windows. An increased force constant of 100kcal/(mol•Å2) was used with 29 biasing potentials for systems unable to sample the entire span of the RC. Grossfield’s weighted histogram analysis method (WHAM) (53) program was used to reconstruct the unbiased distribution of the reaction coordinate and then extracted the free energy profile.
Results and Discussions
1. FTase/FPP3−/Mg2+ Modeling and Simulation
By manually placing Mg2+ into the FTase/FPP3− ternary complex at different locations, a series of Mg2+ ion binding motifs in the active site of WT FTase were generated. As described above, these binding motifs were then validated using MD simulations. In all, three possible Mg2+ binding models were detected for WT FTase, and are displayed in Figure 3. All three candidate amino acids, D196α, D352β and D297β were found involved in one or more of these schemes. A D297β bridged bimetallic catalytic site was observed in the first motif, in which the Mg2+ ion coordinated to two water molecules, an oxygen atom from both phosphate groups of FPP, a single carboxylic oxygen of D352β and a single carboxylic oxygen of D297β. In the second Mg2+ binding motif, the octahedral Mg2+ binding site involved two oxygen atoms from the FPP substrate with both the α- and β-phosphate groups contributing one oxygen atom each, a carboxylate oxygen of D352β and three water molecules. In the third motif, Mg2+ was octahedrally coordinated to an oxygen atom of the α-diphosphate of FPP, a carboxylate oxygen of D196α and four water molecules. These binding schemes, will be denoted as WT1 (D352β + D297β), WT2 (D352β), and WT3 (D196α), respectively in the remainder of this article.
The Mg2+ ion binding in the active site was found to be reasonable in all three cases via visual inspection. The bound Mg2+ ion in all motifs displayed octahedral coordination. Each motif has at least one aspartate side chain participating, and all of them were observed to have direct contacts with the diphosphate oxygens. We found these binding motifs to be stable over 6ns of MD simulations. The distance between the reacting atoms (the distance between Sγ and C1 = dRC) for the three schemes were calculated from their MD trajectories and this term is used through out the manuscript in this context. For the WT1 and WT2 binding motifs, the calculated average values of dRC indicated that the distance between the reacting atoms reduced to below 6.5Å, as compared to value of 7.2Å in the FTase/FPP2− case in the absence of magnesium ions. These results suggest that WT1 and WT2 are structurally more favored than FTase/FPP2− to move the system from the resting state to a reaction ready form with a dRC < 4.0Å. In contrast to WT1 and WT2, the computed dRC values for the WT3 binding motif showed the average distance between the reacting atoms to have significantly increased to ~10Å (~50% increase in length). The increased distance found in the WT3 binding motif makes it less likely to transition to the chemical step. In this motif, moreover, the involvement of only the α-phosphate oxygen is inconsistent with the model proposed by Fierke and coworkers(34, 42). Although the possibility of D196α being involved cannot be entirely ruled out, for the purposes of this study, we chose to pursue the more probable WT1 and WT2 binding motifs.
D297β and D352β are highly conserved residues, which implies an important role for them in FTase chemistry. Mutagenesis experiments have found D352β to be essential for the 700-fold increase in the rate of product formation associated with the presence of Mg2+(34). The results from our simulations support the assumption that the Mg2+ in the active site makes a contribution towards the conformational transition that closes the 7Å gap between the reacting atoms and stabilizes the developing negative charge on PPi in the chemical step. The involvement of both α- and β-phosphate oxygen atoms in Mg2+ ion coordination is consistent with the experimentally proposed model. We regards these motifs featuring the Mg2+-D352β interaction as our best estimate of the Mg2+ binding motifs in the WT FTase/FPP3−/Mg2+ system.
We observed similarities in the active sites of WT FTase across both motifs. The active site conformation and interaction patterns from simulations of WT1 and WT2 are shown in Figure 3. A key hydrogen bond was identified between Cys1p and a magnesium ion bound water molecule. The tetrahedral coordination of the zinc ion was well preserved in both systems. The bound Mg2+ ion was found to form strong electrostatic interactions with PPi, disrupting the previously existing R291β-FPP3− and K294β-FPP3− salt bridge interactions. Cui and Merz calculated a more than 5kcal/(mol•Å) free energy penalty for breaking these salt bridge interactions, making the conformational transition of FTase/FPP3− energetically unfavorable in the absence of Mg2+ ions(18). As a result of D352β approaching FPP3− to form the Mg2+ binding site, its adjacent residue, positively charged K353β, also moves towards PPi and forms a hydrogen bond to one of the β-phosphate oxygen atoms, further stabilizing the leaving group during the chemical step.
Y300β was observed to be important to the farnesylation reaction based on mutagenesis experiments, but was not found in our earlier MD simulations to form clear interactions with the resting state of FTase/FPP3−, while acting as a hydrogen bond donor to PPi in the FTase/FPP2− study. However, in this study, it moves away from the PPi and provides a hydrogen bond to the acetyl capping group, which can be viewed as further stabilizing the peptide substrate. On the other hand, another positively charged residue close to FPP3−, K164α, still forms a salt bridge interaction with the α-phosphate oxygen. It is noteworthy that in the previous FTase/FPP2− PMF study, the roles of K164α and Y300β were switched, with the lysine residue forming a hydrogen bond with the capping acetyl group, while the tyrosine residue formed a hydrogen bond with PPi. While their roles appear to exchange depending on the active site environment and position along the reaction coordinate, ultimately, these two groups help stabilize the peptide and the phosphate during the chemical step.
The two binding motifs differ in the function of zinc bound D297β. The WT1 binding motif is a D297β bridged bimetallic binding site, broadly similar to the model proposed by Fierke and co-workers(34). The two metal ions are 3.3Å apart, and the calculated equilibrium distance between the reacting atoms was 5.8Å, suggesting this binding scheme might be energetically favored over WT FTase/FPP2−/3− in the absence of Mg2+ ions. MD simulations of this motif found the distance between the reactive atoms to fluctuate over a small range. This suggests that the D297β bridge possibly constrains the system, making the active site less mobile. Such a constrained environment would make it harder for the reactive atoms to approach one another. The WT2 binding motif does not contain the D297β bridged bimetallic catalytic site. A water molecule replaces D297β in the magnesium coordination complex, allowing the positively charged zinc and magnesium ions to be separated by 5.1Å. The reactive atom distances from MD simulations of this model were found to fluctuate around 6.1Å. This suggests that the hydrogen bond observed between Cys1p and a Mg2+ ion bound water molecule functions as a substitute bridge between the two metal binding sites, and helps reduce the gap between the reacting partners. The larger fluctuation in the WT2 dRC suggests a more flexible binding site, which will allow the reactive atoms to reach a pre-organized state essential for the chemical step.
In contrast to the model proposed by Fierke and co-workers, D352β does not donate both oxygen atoms from its carboxylate group to Mg2+ in either binding motif for WT FTase. Instead only one oxygen atom participates. In order to test the possibility of the Mg2+-D352β double interaction scheme, tests were conducted that forced Mg2+ to establish a second interaction to both D352β carboxylate oxygen atoms by applying a harmonic force between the two atoms; however, a stable structure was not observed.
2. Dβ352A Mutation and Mg2+/H2O Swap Simulation
In the experimental mutagenesis study of Dβ352A in the presence of Mg2+ ions, a ~30-fold difference in the product formation rate constant was observed. With Mg2+ losing one of its favored coordinating ligands, D352β, another candidate aspartic acid, D297β, can be assumed to replace it in the magnesium coordination shell. A second series of MD simulations were carried out to study the impact of this mutation and account for the nearly 2kcal/mol increase in the free energy barrier. As previously described, the WT2 motif was used as the starting structure for this mutation. A new Mg2+ binding scheme was observed and was verified by MD simulations. The new binding motif was found similar to the previous case of WT1. In this new motif, a water molecule replaces D352β to complete the Mg2+ coordination sphere (see Figure 3) and D297β serves as a bridge residue connecting the two metal binding sites together into what appears as a bimetallic site. The two metal cations are separated by 3.4Å, similar to the binding motif observed in WT1, but considerably shorter than before the mutation (5.1Å for WT2). It is interesting that Mg2+ binds to the sequence conserved D297β in the absence of D352β.
Inside the catalytic pocket, several key hydrogen bonds are well preserved, including those between K164α and the α-phosphate of FPP, K353β and β-phosphate, and Y300β and the capping acetyl group. To further stabilize PPi, several water molecules surrounding it were found to form hydrogen bonds with the free β-phosphate oxygen. H248β was observed to form a hydrogen bond with Y300β instead of PPi. Interestingly, in addition to the D297β bridged bimetallic site, two Mg2+ bound water molecules are now found to interact with the sulfur atom of the peptide via a hydrogen bond, which helps shortens dRC. The equilibrium value of dRC, however, increased from 6.1 in WT2 to 6.5Å, but was still considerably shorter than that of the resting state of FTase/FPP2−/3− without Mg2+. Based on the different equilibrium values of dRC in the different FTase/FPP systems, the difficulty of undergoing the conformational step prior to the chemical steps was predicted to follow the order of: FTase/FPP2− > MUT > WT1/WT2. The real trend, however, cannot be determined without carrying out a series of PMF studies for each of the systems using hybrid QM/MM methods covering the entire length of the RC from ~8.0Å to the covalent bonding distance of ~1.8Å. Such a study is currently underway in our laboratory.
A third set of MD simulations was carried out on the SWAP system. In this system, the active site bound Mg2+ ion was exchanged with a bulk solvent water molecule in order to study the impact of losing Mg2+ in the active site of FTase. The ideal result would be to relax to the FTase/FPP3− starting structure in the absence of Mg2+. As expected, a binding pocket similar to FTase/FPP3− was obtained (see Figure 3). The key interactions observed in the resting state of FTase/FPP3- were largely restored, including the hydrogen bond between H248β and the diphosphate, the salt bridge between K294β and the β-phosphate oxygen, and the double interaction between R291β and an α-phosphate oxygen and the diphosphate bridging oxygen. These interactions have been shown to “lock” FPP3− in its conformational state, and prevent it from undergoing a transition in its conformation prior to the chemical step. At the same time, D352β was found to move away from the FPP3− substrate. In the absence of Mg2+, the Cys1p residue was found to move away, leaving an equilibrium dRC of ~7.4Å, similar to the global minimum found for FTase/FPP3− by Cui and Merz(18).
3. Potential of Mean Force (PMF) Studies
Computing the free energy has not been a straightforward task in molecular modeling and simulations. The main difficulty comes from the inability of general MD simulations to overcome free energy barriers and adequately sample the phase space in regions such as transition or intermediate states. The implementation of special techniques, for instance, umbrella sampling, allows for the study of the free energy surface along a specified RC (DC1-Sγ). 15 simulations were carried out with anchor points placed between 8.0 and 4.5Å for WT2, MUT and SWAP systems. To efficiently sample the RC for WT1, a stronger bias was employed and trajectories were propagated at 29 anchor points.
We found that the inclusion of Mg2+ into the active site altered the free energy profile. For WT1, the global minimum shifted from 7.4Å in FTase/FPP3− to 5.8Å, decreasing the distance between the two reacting atoms by 1.8Å. The free energy profile, however, suggests that this might not be the correct binding motif for Mg2+ in the active form of FTase. With steep increases in free energy found on both side of this equilibrium point, the system appears locked into this minimum position, with the reactive atoms unlikely to get closer. This in association with small fluctuations observed in the distance between the two reactive atoms during MD simulations, lead to the conclusion that the bridged bimetallic Zn2+-D297β-Mg2+ active site in WT1 is very rigid. In WT2, we found the global minimum shifted to 6.1Å, corresponding to a larger distance (by 0.3Å) between the reacting atoms compared to WT1. Also, a second minimum corresponding to an intermediate state was found at around 5.0Å, in a location similar to that for FTase/FPP2−. The intermediate itself has a free energy approximately 0.5kcal/mol higher than that of the resting state, and the free energy barrier for the conformational transition is about 0.8kcal/mol, slightly lower than that observed for the FTase/FPP2− system. With such a low transition barrier and a flat energy profile, the approach of the two reacting centers is facilitated. This is supported by dRC distributions (see SI) extracted from the last 4ns of the MD simulation; another observation supported by the dRC distribution map is the existence of a second minimum under 5.5Å in addition to the global minimum. Moreover, the free energy penalty for further shortening dRC beyond the intermediate state is significantly reduced from ~1.7kcal/mol (in FTase/FPP2−) to ~1.0kcal/mol in WT2, meaning the two reactive atoms can more readily approach each other in this relatively flexible active site model. However, due to the possible strong VDW clashes at distances close to 4.5Å, this finding needs to be further explored by QM/MM PMF’s for distances shorter than 4.5Å.
The PMF calculated for the D352A mutant found a global minimum corresponding to a reactive atom distance of ~6.6Å, 0.5Å longer than that of WT2. From the MD simulation, the equilibrium dRC distance was found to oscillate around 6.6Å. An intermediate state was not observed, and a much larger free energy penalty, up to ~6kcal/mol, was observed close to 4.5Å. Experimental measurements suggested an approximately 30-fold difference in the rate constant of product formation meaning a 2kcal/mol higher free energy barrier than FTase/FPP3−/Mg2+. Assuming that the difference can be entirely ascribed to the conformational step, our observation of a 5kcal/mol energy difference (6kcal/mol - ~1kcal/mol) is too large. The difference can be made up in the chemistry steps, but this has yet to be determined. The free energy profile also shows that all regions between 5.5Å and the resting state are accessible with only a ~1kcal/mol free energy difference. Regardless, we find that the Dβ352A has a significant effect on the conformational step and it is left to be determined how it affects subsequent chemical steps.
Finally, a fourth PMF study was carried out on the SWAP system that aimed to reproduce the free energy profile obtained from the wild type FTase/FPP3− system in the absence of Mg2+. As expected, a similar free energy profile to the case of FTase/FPP3− without Mg2+ was obtained. Comparing our plot (see Figure 4) to that published by Cui and Merz (denoted as FTase/FPP3−/CVIM), excellent agreement is found: both the 7.5Å global minimum and the free energy jump beyond that point are well reproduced. Overall, the agreement is quite good with only minor differences observed. Hence, the present study recapitulates our earlier effort and further confirms the reasonableness of our active site model for Mg2+ bound to FTase.
Figure 4.

Calculated free energy profiles for the WT1, WT2, MUT and SWAP systems.
Conclusion
We modeled and validated several FTase active site magnesium binding schemes using MD simulations and classical PMF studies with umbrella sampling. Three possible active site models were observed for wild type FTase/FPP3−/Mg2+ (see Figure 3), but only one was consistent with all available experimental information. In this motif, WT2, Mg2+ adopts an octahedral coordination formed with an oxygen atom from both α- and β-diphosphate, a carboxylate oxygen from D352β and three water molecules (see Figure 3). The involvement of zinc ion bound D297β, creating a bridged bimetallic catalytic site is not found to be a reasonable option for Mg2+ binding in FTase/FPP3-/Mg2+. An interesting hydrogen bond formed between a magnesium ion bound water molecule and Cys1p was detected, which contributes to the reduction of dRC. Dβ352A/FPP3−/Mg2+ was also simulated and a single active site model emerged for this system as well (see Figure 3). Free energy calculations show that Mg2+ ions allow the reacting atoms to approach each other more readily. Our simulations show that WT FTase/FPP3−/Mg2+ and the MUT systems both have resting states (6.1Å and 6.5Å, respectively) with a significantly shortened equilibrium dRC relative to FTase/FPP3- (7.4Å) but they experience different free energy profiles beyond this point. With a free energy difference of less than 1kcal/mol from the resting state to the lower end of our PMF study at 4.5Å, the whole RC range is accessible for the WT FTase/FPP3−/Mg2+ system. This ready accessibility, over the entire RC range supports the isoprene-rotation mechanism for the conformational transition. On the other hand, a nearly 6kcal/mol free energy penalty from the resting state to 4.5Å makes the approach of FPP3− to the target cysteine residue much more difficult for the mutant.
Introducing Mg2+ into the FTase/FPP3− active site is probably the only way in which FPP3− can approach the peptide substrate bound to the zinc ion. The highly charged binding pocket has been shown to freeze FPP in its conformation in FTase/FPP3− in the absence of Mg2+, preventing the transition required before the chemical step. With Mg2+ included, the resting state is significantly perturbed and the free energy barrier for the conformational transition is dramatically reduced. Binding of Mg2+ in FTase/FPP3− pulls the diphosphate away from the positive charged pocket observed in the FTase active site, especially R291β and K294β. Mg2+ compensates for the lost salt bridge interactions by coordinating to both the α- and β-diphosphate oxygen atoms. K164α is ‘locked’ into an interaction with an oxygen from the α-phosphate even with Mg2+ binding to FTase, regardless whether D352β was mutated or not, in accordance with the experimental evidence that this interaction is important for stabilizing the diphosphate leaving group during the chemical step. When Mg2+ was swapped out of the active site with a bulk water molecule we were able observe the relaxation back to the resting state for FTase/FPP3− in the absence of Mg2+ with a dRC minimum of 7.5Å. Moreover, key hydrogen bonds between the diphosphate and residues like H248β, R291β and K294β were reformed, implying that Mg2+ is indeed the sole reason for the conformational transition.
Through the use of classical simulations we have identified the elusive Mg2+ binding state in FTase/FPP3−, thereby, setting the stage for further experimental and theoretical studies aimed at further understanding the catalytic mechanism of this fascinating and important enzyme.
Supplementary Material
Acknowledgments
The authors want to thank Dr. Guanglei Cui and Dr. Bing Wang for numerous helpful discussions. We thank the NIH (GM044974) for funding this research. We also acknowledge the University of Florida High-Performance Computing Center for providing computational resources and support that have contributed to the research results reported within this paper. URL: http://hpc.ufl.edu.
Abbreviations
- FTase
farnesyltransferase
- WT
wild type
- FPP
farnesyl diphosphate
- FTI
farnesyltransferase inhibitor
- KIE
kinetic isotope effect
- QM/MM
quantum mechanical/molecular mechanical
- MD
molecular dynamics
- GAFF
General AMBER force field
- RESP
restrained electrostatic potential
- LES
local enhanced sampling
- PME
particle mesh ewald
- PMF
potential of mean force
- RC
reaction coordinate
- VDW
van der Waals
- WHAM
weighted histogram analysis method
Footnotes
Supporting Information Available
The PDB files of WT and MUT FTase systems, along with several other helpful figures. This material is available free of charge online at http://pubs.acs.org.
References
- 1.Zhang FL, Casey PJ. Protein prenylation: Molecular mechanisms and functional consequences. Ann Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 2.Long SB, Casey PJ, Beese LS. Reaction path of protein farnesyltransferase at atomic resolution. Nature. 2002;419:645–650. doi: 10.1038/nature00986. [DOI] [PubMed] [Google Scholar]
- 3.Roskoski R. Protein prenylation: A pivotal posttranslational process. Biochem Bioph Res Co. 2003;303:1–7. doi: 10.1016/s0006-291x(03)00323-1. [DOI] [PubMed] [Google Scholar]
- 4.Adjei AA. Blocking oncogenic ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 5.Resh MD. Regulation of cellular signalling by fatty acid acylation and prenylation of signal transduction proteins. Cell Signal. 1996;8:403–412. doi: 10.1016/s0898-6568(96)00088-5. [DOI] [PubMed] [Google Scholar]
- 6.Seabra MC. Membrane association and targeting of prenylated ras-like gtpases. Cell Signal. 1998;10:167–172. doi: 10.1016/s0898-6568(97)00120-4. [DOI] [PubMed] [Google Scholar]
- 7.Sinensky M. Recent advances in the study of prenylated proteins. Bba-Mol Cell Biol L. 2000;1484:93–106. doi: 10.1016/s1388-1981(00)00009-3. [DOI] [PubMed] [Google Scholar]
- 8.Wu Z, Demma M, Strickland CL, Radisky ES, Poulter CD, Le HV, Windsor WT. Farnesyl protein transferase: Identification of k164 alpha and y300 beta as catalytic residues by mutagenesis and kinetic studies. Biochemistry. 1999;38:11239–11249. doi: 10.1021/bi990583t. [DOI] [PubMed] [Google Scholar]
- 9.Hightower KE, Casey PJ, Fierke CA. Farnesylation of nonpeptidic thiol compounds by protein farnesyltransferase. Biochemistry. 2001;40:1002–1010. doi: 10.1021/bi002237d. [DOI] [PubMed] [Google Scholar]
- 10.Pickett JS, Bowers KE, Hartman HL, Fu HW, Embry AC, Casey PJ, Fierke CA. Kinetic studies of protein farnesyltransferase mutants establish active substrate conformation. Biochemistry. 2003;42:9741–9748. doi: 10.1021/bi0346852. [DOI] [PubMed] [Google Scholar]
- 11.Dunten P, Kammlott U, Crowther R, Weber D, Palermo R, Birktoft J. Protein farnesyltransferase: Structure and implications for substrate binding. Biochemistry. 1998;37:7907–7912. doi: 10.1021/bi980531o. [DOI] [PubMed] [Google Scholar]
- 12.Long SB, Casey PJ, Beese LS. Cocrystal structure of protein farnesyltransferase complexed with a farnesyl diphosphate substrate. Biochemistry. 1998;37:9612–9618. doi: 10.1021/bi980708e. [DOI] [PubMed] [Google Scholar]
- 13.Long SB, Casey PJ, Beese LS. The basis for k-ras4b binding specificity to protein farnesyl-transferase revealed by 2 angstrom resolution ternary complex structures. Structure. 2000;8:209–222. doi: 10.1016/s0969-2126(00)00096-4. [DOI] [PubMed] [Google Scholar]
- 14.Long SB, Hancock PJ, Kral AM, Hellinga HW, Beese LS. The crystal structure of human protein farnesyltransferase reveals the basis for inhibition by caax tetrapeptides and their mimetics. Proc Natl Acad Sci USA. 2001;98:12948–12953. doi: 10.1073/pnas.241407898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park HW, Boduluri SR, Moomaw JF, Casey PJ, Beese LS. Crystal structure of protein farnesyltransferase at 2.25 angstrom resolution. Science. 1997;275:1800–1804. doi: 10.1126/science.275.5307.1800. [DOI] [PubMed] [Google Scholar]
- 16.Strickland CL, Windsor WT, Syto R, Wang L, Bond R, Wu Z, Schwartz J, Le HV, Beese LS, Weber PC. Crystal structure of farnesyl protein transferase complexed with a caax peptide and farnesyl diphosphate analogue. Biochemistry. 1998;37:16601–16611. doi: 10.1021/bi981197z. [DOI] [PubMed] [Google Scholar]
- 17.Cui G, Wang B, Merz KM., Jr Computational studies of the farnesyltransferase ternary complex part i: Substrate binding. Biochemistry. 2005;44:16513–16523. doi: 10.1021/bi051020m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui G, Merz KM., Jr Computational studies of the farnesyltransferase ternary complex part ii: The conformational activation of farnesyldiphosphate. Biochemistry. 2007;46:12375–12381. doi: 10.1021/bi701324t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho M-H, Vivo MD, Peraro MD, Klein ML. Unraveling the catalytic pathway of metalloenzyme farnesyltransferase through qm/mm computation. J Chem Theory Comput. 2009;5:1657–1666. doi: 10.1021/ct8004722. [DOI] [PubMed] [Google Scholar]
- 20.Lenevich S, Xu JH, Hosokawa A, Cramer CJ, Distefano MD. Transition state analysis of model and enzymatic prenylation reactions. J Am Chem Soc. 2007;129:5796. doi: 10.1021/ja069119j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sousa SF, Fernandes PA, Ramos MJ. Molecular dynamics simulations on the critical states of the farnesyltransferase enzyme. Bioorgan Med Chem. 2009;17:3369–3378. doi: 10.1016/j.bmc.2009.03.055. [DOI] [PubMed] [Google Scholar]
- 22.Hougland JL, Lamphear CL, Scott SA, Gibbs RA, Fierke CA. Context-dependent substrate recognition by protein farnesyltransferase. Biochemistry. 2009;48:1691–1701. doi: 10.1021/bi801710g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hougland JL, Hicks KA, Hartman HL, Kelly RA, Watt TJ, Fierke CA. Identification of novel peptide substrates for protein farnesyltransferase reveals two substrate classes with distinct sequence selectivities. J Mol Biol. 2010;395:176–190. doi: 10.1016/j.jmb.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowers KE, Fierke CA. Positively charged side chains in protein farnesyltransferase enhance catalysis by stabilizing the formation of the diphosphate leaving group. Biochemistry. 2004;43:5256–5265. doi: 10.1021/bi049822p. [DOI] [PubMed] [Google Scholar]
- 25.Taveras AG, Kirschmeier P, Baum CM. Sch-66336 (sarasar (r)) and other benzocycloheptapyridyl farnesyl protein transferase inhibitors: Discovery, biology and clinical observations. Curr Top Med Chem. 2003;3:1103–1114. doi: 10.2174/1568026033452104. [DOI] [PubMed] [Google Scholar]
- 26.Doll RJ, Kirschmeier P, Bishop WR. Farnesyltransferase inhibitors as anticancer agents: Critical crossroads. Curr Opin Drug Disc. 2004;7:478–486. [PubMed] [Google Scholar]
- 27.Huang CY, Rokosz L. Farnesyltransferase inhibitors: Recent advances. Expert Opin Ther Pat. 2004;14:175–186. [Google Scholar]
- 28.Khuri FR, Glisson BS, Kim ES, Statkevich P, Thall PF, Meyers ML, Herbst RS, Munden RF, Tendler C, Zhu YL, Bangert S, Thompson E, Lu C, Wang XM, Shin DM, Kies MS, Papadimitrakopoulou V, Fossella FV, Kirschmeier P, Bishop WR, Hong WK. Phase i study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in solid tumors. Clin Cancer Res. 2004;10:2968–2976. doi: 10.1158/1078-0432.ccr-03-0412. [DOI] [PubMed] [Google Scholar]
- 29.Rao S, Cunningham D, de Gramont A, Scheithauer W, Smakal M, Humblet Y, Kourteva G, Iveson T, Andre T, Dostalova J, Illes A, Belly R, Perez-Ruixo JJ, Park YC, Palmer PA. Phase iii double-blind placebo-controlled study of farnesyl transferase inhibitor r115777 in patients with refractory advanced colorectal cancer. J Clin Oncol. 2004;22:3950–3957. doi: 10.1200/JCO.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 30.Lane KT, Beese LS. Thematic review series: Lipid posttranslational modifications. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type i. J Lipid Res. 2006;47:681–699. doi: 10.1194/jlr.R600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Zhang YH, Cao R, Yin F, Hudock MP, Guo RT, Krysiak K, Mukherjee S, Gao YG, Robinson H, Song Y, No JH, Bergan K, Leon A, Cass L, Goddard A, Chang TK, Lin FY, Van Beek E, Papapoulos S, Wang AHJ, Kubo T, Ochi M, Mukkamala D, Oldfield E. Lipophilic bisphosphonates as dual farnesyl/geranylgeranyl diphosphate synthase inhibitors: An x-ray and nmr investigation. J Am Chem Soc. 2009;131:5153–5162. doi: 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang CC, Hightower KE, Fierke CA. Mechanistic studies of rat protein farnesyltransferase indicate an associative transition state. Biochemistry. 2000;39:2593–2602. doi: 10.1021/bi992356x. [DOI] [PubMed] [Google Scholar]
- 33.Pais JE, Bowers KE, Fierke CA. Measurement of the alpha-secondary kinetic isotope effect for the reaction catalyzed by mammalian protein farnesyltransferase. J Am Chem Soc. 2006;128:15086–15087. doi: 10.1021/ja065838m. [DOI] [PubMed] [Google Scholar]
- 34.Pickett JS, Bowers KE, Fierke CA. Mutagenesis studies of protein farnesyltransferase implicate aspartate beta 352 as a magnesium ligand. J Biol Chem. 2003;278:51243–51250. doi: 10.1074/jbc.M309226200. [DOI] [PubMed] [Google Scholar]
- 35.Sousa SF, Fernandes PA, Ramos MJ. Theoretical studies on farnesyltransferase: The distances paradox explained. Proteins-Structure Function and Bioinformatics. 2007;66:205–218. doi: 10.1002/prot.21219. [DOI] [PubMed] [Google Scholar]
- 36.Merz KM., Jr Co2 binding to human carbonic anhydrase-ii. J Am Chem Soc. 1991;113:406–411. [Google Scholar]
- 37.Viktor Hornak RA, Okur Asim, Strockbine Bentley, Roitberg Adrian, Simmerling Carlos. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins: Struct, Funct, Bioinf. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hightower KE, Huang CC, Casey PJ, Fierke CA. H-ras peptide and protein substrates bind protein farnesyltransferase as an ionized thiolate. Biochemistry. 1998;37:15555–15562. doi: 10.1021/bi981525v. [DOI] [PubMed] [Google Scholar]
- 39.Cui G, Wang B, Merz KM., Jr Computational studies of the farnesyltransferase ternary complex part i: Substrate binding. Biochemistry. 2005;44:16513. doi: 10.1021/bi051020m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merz KM, Jr, Murcko MA, Kollman PA. Inhibition of carbonic-anhydrase. J Am Chem Soc. 1991;113:4484–4490. [Google Scholar]
- 41.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KM, Jr, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. Amber. Vol. 10. University of California; San Francisco: 2008. [Google Scholar]
- 42.Saderholm MJ, Hightower KE, Fierke CA. Role of metals in the reaction catalyzed by protein farnesyltransferase. Biochemistry. 2000;39:12398–12405. doi: 10.1021/bi0011781. [DOI] [PubMed] [Google Scholar]
- 43.Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 44.Meagher KL, Redman LT, Carlson HA. Development of polyphosphate parameters for use with the amber force field. J Comput Chem. 2003;24:1016–1025. doi: 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- 45.Cieplak P, Cornell WD, Bayly C, Kollman PA. Application of the multimolecule and multiconformational resp methodology to biopolymers: Charge derivation for DNA, rna, and proteins. J Comput Chem. 1995;16:1357–1377. [Google Scholar]
- 46.Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The resp model. Journal of Physical Chemistry. 1993;97:10269–10280. [Google Scholar]
- 47.Frisch MJT, GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, revision c.02. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 48.Aqvist J. Ion water interaction potentials derived from free-energy perturbation simulations. J Phys Chem. 1990;94:8021–8024. [Google Scholar]
- 49.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 50.Darden T, York D, Pedersen L. Particle mesh ewald - an n.Log(n) method for ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 51.Allen MP, Tildesley DJ. Computer simulation of liquids. Clarendon Press; Oxford, U.K: 1987. [Google Scholar]
- 52.Torrie GM, Valleau JP. Non-physical sampling distributions in monte-carlo free-energy estimation - umbrella sampling. J Comput Phys. 1977;23:187–199. [Google Scholar]
- 53.Grossfield A. Wham: The weighted histogram analysis method. 1.6d [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.