

Abstract
The aim of this study was to develop a new precise and accurate CZE-DAD method for honeybee venom analysis using cytochrome c as an internal standard. The 64.5 cm total length, 56 cm effective length, 75 μm ID, and 360 μm OD uncoated fused-silica capillary was used. The samples were injected into the capillary under a 50-mbar pressure for 7 s. There were 15 kV of electric field across the capillary applied. The current intensity was 26 μA. The separation was carried out at 25 °C. The analysis was run with the normal electrode polarity. The following steps and parameters were taken into account for the validation of the developed method: selectivity, precision, accuracy, linearity, limit of detection and limit of quantitation. All steps of the validation procedure proved that the developed analytical procedure was suitable for its intended purpose. Possibly this was the first study in which several honeybee venom components were separated and five of them were identified by capillary zone electrophoresis. In addition, the developed method was applied for quantitative analysis of 38 honeybee venom samples. The content (relative to the dry venom mass) of analyzed peptides in honeybee venom samples collected in 2002–2007 was as follows: apamine from 0.93% to 4.34% (mean, 2.85 ± 0.79%); mast cell degranulating peptide (MCDP) from 1.46% to 4.37% (mean, 2.82 ± 0.64%); phospholipase A2 from 7.41% to 20.25% (mean, 12.95 ± 3.09%); melittin from 25.40% to 60.27%, (mean, 45.91 ± 9.78%). The results were compared with the experimental data obtained for the same venom samples analyzed earlier by the HPLC method. It was stated that HPCE and HPLC data did not differ significantly and that the HPCE method was the alternative for the HPLC method. Moreover, using the results obtained principal component analysis (PCA) was applied to clarify the general distribution patterns or similarities of four major honeybee venom constituents collected from two different bee strains in various months and years. PCA has shown that the strain of bee appears to be the only criteria for bee venom sample classification. Strong correlations between apamine, MCDP, phospholipase A2, and melittin were confirmed. These correlations have to be taken into account in the honeybee venom standardization. The developed method due to its simplicity can be easily automated and incorporated into routine operations both in the bee venom identification, quality control, and standardization of the product.
Electronic supplementary material
The online version of this article (doi:10.1007/s00216-010-4627-2) contains supplementary material, which is available to authorized users.
Keywords: Bioanalytical methods, Biological samples, Capillary electrophoresis, Chemometrics/statistics, HPLC, Natural products
Introduction
Because of its popularity in folk medicine, honeybee venom has become the subject of intense pharmacological and biological investigations. Numerous studies have proven its effectiveness in treating pathological conditions such as arthritis [1], rheumatism, pain [2, 3], cancerous tumors [4, 5], and skin diseases. On the European and global market, there are some registered pharmaceutical formulations with honeybee venom (Forapin, Germany; Virapin, Slovakia; Apiven, France; Melivenon, Bulgaria; Apifor, Russia). However, there are no uniform guidelines for the standardization of these products. A significant number of papers dealing with honeybee venom chemistry were published and have demonstrated the variation of its chemical composition depending on different honeybee races and strains, place of origin, years and season of venom collection [6–9]. Therefore, in order to obtain official approval as a safe drug, honeybee venom needs chemical standardization that would guarantee its quality and repeatable pharmacological activity.
There are two approaches for standardization of complex natural products. If the pharmacological activity of the standardized product can be attributed to known constituents, they have to be quantified using appropriate method. If not all the active compounds are known or pharmacological activity of the product is caused by synergistic effect of all its constituents, some marker compounds must be used for standardization purposes. Since honeybee venom is a very complex mixture of chemical compounds which are still not sufficiently characterized, analyzing marker compounds seems to be necessary.
In order to determine the honeybee venom composition, several separation techniques have been applied. High-performance liquid chromatography (HPLC) methods allowed to separate and identify most of the bee venom components [10–15]. Thanks to chromatographic techniques, a variety of bee venom peptides including melittin, apamine, adolapin and the MCDP have been detected. Moreover, honeybee venom has been demonstrated to contain some enzymes (phospholipase A2 and hyaluronidase), biologically active amines (histamine and epinephrine) and non-peptide components (including lipids, carbohydrates and free amino acids).
High-performance capillary electrophoresis (HPCE) in contrast with HPLC has limited utilization for honeybee venom analysis. It was used only for determination of two main bee venom components (phospholipase A2 and melitin) [15, 16] and for determination of hyaluronidase activity in this product [17]. Moreover, the accuracy of HPCE methods for honeybee venom analysis presented in the literature has not been confirmed due to the lack of reference materials with certified values of bee venom components. Furthermore, their applications were limited to a few analyzed bee venom samples. Therefore, a new method with an internal standard for analyzing honeybee venom samples with high accuracy by capillary zone electrophoresis has been developed. Possibly, this was the first study in which several honeybee venom components were separated and five of them were identified by capillary zone electrophoresis. In addition, the developed method was applied for quantitative analysis of 38 honeybee venom samples. As a reference method, HPLC method presented in our earlier study was used. Moreover, using the results obtained Principal Component Analysis (PCA) was applied to clarify the general distribution patterns or similarities of four major honeybee venom constituents collected from two different bee strains in various months and years.
Experimental
Reagents and materials
Analytical standards: apamine (product number, A1289; lot number, 125K4119; purity, 99.7%), mast cell degranulating peptide (MCDP; product number, M8036; lot number, 92H03814; purity, 98%), phospholipase A2 (product number, P9279; lot number, 016K4145; activity, 1,769 units mg−1 solid), melittin (product number, M2272; lot number, 067K4136; purity, 89.4%), tertiapin (product number, T8316; lot number, 1406992) and cytochrome c from equine heart (product number, C7752; lot number, 098K7000; purity, 96.1%)—internal standard, were supplied by Sigma Chemicals Co. (St. Louis, MO, USA). Sodium hydroxide solutions (1 and 0.1 M) were purchased from Agilent Technologies (Palo Alto, CA, USA). Methanol (chromatographic grade, purity ≥99.8%) was supplied by Merck (Darmstadt, Germany). Potassium dihydrophosphate (analytical grade; purity, 98.0–100.5%) was purchased from Riedel-de Haën (Seelze, Germany). Orthophosphoric acid and 0.1 M hydrochloric acid were of analytical grade and were supplied by POCh (Gliwice, Poland). Deionized water was obtained by passing distilled water through a Millipore Simplicity UV water purification system (Waters Corporation, Milford, MA, USA). Membrane filters 0.45 μm were purchased from Agilent.
Samples of honeybee venom were collected from the apiary of the Department of Inorganic and Analytical Chemistry of Poznan University of Medical Sciences by stimulating the bees with electric current pulses. Venom-collecting frames were placed in the upper body of the hive in the middle space of the hive super. The duration of venom-collecting event was 2 h during full activity of bees. That schedule allowed to obtain the highest efficiency of bee venom milking. Twenty-eight samples were obtained from 2002 to 2007 during the whole beekeeping seasons from May until September. In 2006 and 2007, bee venom was collected from two different strains of carnica bee race (niemka—I strain, singer—II strain). Moreover, two archival samples originated from 1991-Georgia and eight archival samples originated from 1995-Poland (the archival samples were supplied by the Department of Pharmacology and Biotechnology, Research Institute of Medicinal Plants, Poznan, Poland). The samples were stored until analysis at 5 °C in darkness.
Preparation of standards and samples
The cytochrome c used as an internal standard (I.S.) was prepared in deionized water at the concentration of 25 μg mL−1 and then used as diluent of all samples.
The honeybee venom solutions were prepared by diluting 3 mg of that product in 10 mL of internal standard solution. The standard solutions of apamine, mast cell degranulating peptide, phospholipase A2, melittin, and tertiapin were prepared by dissolving them in I.S. solution (concentrations obtained are given in Table S1 (Electronic Supplementary Material)). All prepared solutions were subjected to sonication by ultrasonic bath—Intersonic (Olsztyn, Poland) for 5 min and then filtered through a 0.45-μm membrane filters. Preliminary experiments showed that the filtration process had no effect on the internal standard concentration and the other five compounds analyzed.
Instrumentation and methods
HPCE instrumentation and separation conditions
The experiments were carried out on an Agilent G1600 instrument (Agilent Technologies, Germany) equipped with a UV-DAD detector, set at 220 nm and controlled by the ChemStation software (Agilent Technologies).
The 64.5 cm total length, 56 cm effective length, 75 μm ID and 360 μm OD uncoated fused-silica capillary was used (CS-Chromatographie Service, Germany). A new capillary was conditioned by rinsing with sodium hydroxide (1 M) for 20 min, then with methanol and sodium hydroxide (0.1 M) for 10 min, respectively. Before each analysis, the capillary was rinsed with sodium hydroxide (1 M), methanol, deionized water, and running buffer for 1 min, respectively.
The samples were injected into the capillary under the 50 mbar pressure for 7 s. There were 15 kV of electric field across the capillary applied. The current intensity was 26 μA. The separation was carried out at 25 °C. The analysis was performed with the normal electrode polarity. The electroosmotic flow marked with dimethylsulfoxide occurred over 60 min of the analysis. All of the honeybee components were separated up to the 25th minute of analysis in such conditions.
HPLC instrumentation and separation conditions
HPLC was used as a reference method. It was fully validated and presented in earlier report [12]. The SynChropack C8 6.5 μm, 4.6 × 100 mm column (Agilent) was applied. Bee venom was separated by linear gradient 5%B–80%B at 30 min (eluent A–0.1% TFA in water, eluent B–0.1% TFA in acetonitrile:water (80:20)). The flow rate of mobile phase was maintained at 1 mL/min, injection volume 40 μL, separation temperature 25 °C. The analysis was monitored at 220 nm. Applying this methodology, good separation of the several bee venom components was obtained, including: apamine (t R = 3.37 min), MCDP (t R = 4.47 min), phospholipase A2 (t R = 11.10 min) and mellitin (t R = 16.43 min). The internal standard was also applied (cytochrome c, t R = 12.48 min). This methodology allowed to separate at least nine honeybee venom components and four of them were quantified.
Results and discussion
The capillary electrophoresis method, used in this paper for the analysis of the bee venom samples, leads to the resolution of the several major peptide containing fractions. The separation of bee venom components was satisfactory and venom samples of different origin were analyzed and distinguished. The internal standard was used for the first time during electrophoretic studies of bee venom. Cytochrome c (t M = 15.30 min) was chosen as an internal standard. Cytochrome c is a protein which is not naturally present in bee venom and does not affect the separation process of other peptides components.
There were 38 honeybee venom samples analyzed and, as a result, five main peptide components of honeybee venom were identified and four of them were quantified (Fig. 1).
Fig. 1.

Electrophoretic patterns: a phosphate buffer, pH = 3.00 (base line); b honeybee venom sample. Phosphate buffer (pH 3.00). Voltage, 15 kV; injection, 7 s. Detection: 220.30 nm. Identified peaks: 1 MCDP (conc. = 8.8 μg/g), 2 cytochrome c (conc. = 25.0 μg/g), 3 apamine (conc. = 7.5 μg/g), 4 phospholipase A2 (conc. = 36.4 μg/g), 5 melittin (conc. = 159.0 μg/g)
Melittin (t M = 20.52 min), phospholipase A2 (t M = 19.31 min), mast cell degranulating peptide (t M = 12.04 min), and apamine (t M = 17.56 min) were the main peptide fractions of the bee venom which were identified and quantified. Moreover, there was also the fifth peptide component of the bee venom–tertiapin (t M = 13.29 min) identified. Tertiapin [18] is the trace peptide present in the honeybee venom dry mass, so it could be identified only in the high concentrated venom sample (about 3 mg/g). Such concentration was almost ten times greater than the concentration of the sample (about 300 μg/g) used for quantitative analysis of other peptide components and the quantitative analysis of tertiapin was ineffective.
Validation of the developed HPCE method and comparison with HPLC method
In order to confirm the experimental data obtained, the following steps and parameters were taken into account for the validation of the HPCE method: selectivity, precision, accuracy, linearity, limit of detection (LOD) and limit of quantitation (LOQ). All steps of the validation procedure proved that developed analytical procedure was suitable for its intended purpose. The HPLC method used during presented studies as a reference method was described previously [12].
It was stated that there were no interferences between the background electrolyte matrix and the bee venom components for both the HPCE and HPLC methods. The blank samples did not give any peaks near the migration and retention times of the standards. The peaks of standards were identical and had the same migration and retention times with those obtained from the bee venom samples. Since the developed methods are recommended for identification and assay tests, it was necessary to demonstrate that each peak on the electropherogram and chromatogram represents a single component. To avoid the possibility that the analyzed peaks overlap with each other, the peak purity was determined. This assessment is based on the comparison of peak spectra recorded during the electrophoretic or chromatographic analysis. The purity value for each spectrum is calculated using the average spectrum of the five selected spectra across the peak. If the peak spectra are not identical with the average spectrum, the peak contains a spectral impurity. The spectral impurity can be caused by one or more components, non-base-line separated peaks or by background absorption. The purity factor was determined for this purpose. At the extremes, a purity factor of 0 indicates no match between the spectra and 1,000 indicates identical spectra. Generally, values above 990 indicate that the spectra are similar. Values below 990 indicate that the spectra are different. According to such evaluations, all examined peaks responsive for analyzed compounds were demonstrated to be pure and could be attributed to only one component.
Precision of the method was estimated by establishing the injection repeatability (intra-run precision) and analysis repeatability (inter-run precision). The injection repeatability of the migration (t M) and retention times (t R) and the relative peak areas (A; peak area of an analyte × peak area of IS−1) were evaluated by variation coefficients obtained from five repetitions. In order to evaluate the analysis repeatability, the same concentration range of standards in the presence of internal standard was performed in the course of 3 days. The variation coefficients for analysis repeatability of related peak areas were much lower than 5% for chromatography method and lower than 13% in the case of electrophoretic method. The variation coefficients for retention times and migration times were lower than 1% and 8%, respectively.
The accuracy of the HPCE procedure was ascertained from assessing agreement between the measured and theoretical concentration of analyzed samples before and after loading with standard solutions (recovery). In the case of the HPCE method, the accuracy was established using three different procedures. In the first one, the amounts of the analyzed bee venom components were approximately 80%, 100%, and 120% of the total amounts of these determined in the venom (Table S2 Electronic Supplementary Material). In the second procedure, the bee venom sample was loaded up to 125%, 150%, and 175% of the content of the particular components in the venom sample (Table S2 Electronic Supplementary Material). In these two procedures, each sample solution was analyzed in the replication of five (n = 5). In the last procedure, the mixture with the precisely known concentrations of the analyzed standards was prepared and then recovery was established. The sample was injected ten times into the capillary (Table S2 Electronic Supplementary Material). Although some of the established recoveries exceed the range 80–110%, the mean recoveries for all three procedures are in that range and are acceptable according to the guidelines of AOAC. Given the fact that honeybee venom is a highly complex biological matrix, the accuracy for the method is very good.
The accuracy for the HPLC method was investigated using almost the identical procedures, with only one exception: samples loaded up to 125%, 150%, and 175% of the content of the particular components in the venom sample were not investigated.
The results obtained proved very good accuracy of the HPCE and HPLC methods as recoveries were on the acceptable levels.
Linearity and operating range
The linearity range for the HPLC method was established for the standards in the following ranges of concentrations: apamine 3–15 μg g−1, MCDP 6–20 μg g−1, phospholipase A2 15–75 μg g−1, and melittin 100 – 300 μg g−1.
The linearity of the HPCE method was measured in the following range of the standards concentrations: apamine 1.8–20.1 μg g−1, MCDP 4.9–32 μg g−1, phospholipase A2 9.9–103.2 μg g−1, and melittin 27.6–306.3 μg g−1.
The linearity of the HPLC and HPCE methods was confirmed by linear correlation coefficients (R). The obtained values of R were close to 1 in both analytical methods used (Table S3 Electronic Supplementary Material).
The concentration of the honeybee venom samples used during the HPLC and HPCE analysis were always about 300 μg g−1.
The LOD and LOQ for the apamine, MCDP, phospholipase A2, and melittin were calculated according to International Conference on Harmonization guidelines:
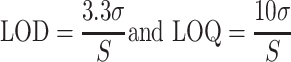 |
where σ denotes the standard deviation of the response, S denotes the slope of the calibration curve.
The LOD and LOQ parameters obtained for both HPLC and HPCE methods are presented in the Table 1. Values of these parameters are comparable. Only in the case of phospholipase A2 the calculated LOD and LOQ were slightly higher for the HPCE method (Table 1).
Table 1.
Limit of detection (LOD) and limit of quantitation (LOQ) for MCDP, apamine, phospholipase A2 and melittin [μg g−1] determined for HPCE and HPLC
| Standard | HPCE | HPLC | ||
|---|---|---|---|---|
| LOD | LOQ | LOD | LOQ | |
| MCDP | 1.7 | 5.2 | 1.3 | 4.1 |
| Apamine | 1.2 | 3.5 | 0.6 | 1.8 |
| Phospholipase A2 | 8.3 | 25.2 | 3.2 | 9.8 |
| Melittin | 4.6 | 13.8 | 4.8 | 14.4 |
The parameters calculated during the validation process showed that the analysis utilizing new developed HPCE method and HPLC method described in our previous study gave the comparable results.
Application of the developed method
The 38 different samples of the honeybee venom were analyzed using HPCE method developed in this study. Each of the analyzed honeybee venom samples was different because of strains of the bees, year and season of the venom collection, and the place of origin (Poland, Georgia). The analysis of each sample was carried out in five replicates and mean values with standard deviations were calculated (Table 2 and 3). In Table 3, the archival honeybee venom (collected in the years 1991 and 1995) samples are presented. These samples were originated from the apiary in Georgia (1991) and from the apiary located in the southern Poland (1995). The HPCE and HPLC results obtained for these honeybee venom samples were not involved during the statistical calculations (ANOVA and PCA) performed for the honeybee venom samples collected in the years 2002 and 2005–2007. They were analyzed in the respect of the content of the main peptides only.
Table 2.
The content ([%] ± SD) of apamine, MCDP, phospholipase A2 and melittin in bee venom samples from the years: 2006 (bee strain 1 and 2), 2007 (bee strain 1 and 2), 2002, and 2005
| Sample number | Apamine | MCDP | Phospholipase A2 | Melittin | Month | Year | Bee strain |
|---|---|---|---|---|---|---|---|
| 1 | 0.93 ± 0.06 | 1.84 ± 0.18 | 7.41 ± 0.68 | 26.11 ± 1.13 | May | 2006 | 1 |
| 2 | 1.69 ± 0.18 | 1.80 ± 0.10 | 8.92 ± 0.49 | 31.22 ± 1.32 | June | ||
| 3 | 2.95 ± 0.36 | 3.04 ± 0.40 | 10.62 ± 0.46 | 35.47 ± 0.97 | July | ||
| 4 | 2.37 ± 0.26 | 2.92 ± 0.36 | 10.26 ± 0.75 | 38.69 ± 1.46 | August | ||
| 5 | 1.43 ± 0.20 | 2.20 ± 0.32 | 8.30 ± 0.41 | 32.17 ± 1.13 | September | ||
| 6 | 2.69 ± 0.27 | 2.86 ± 0.09 | 12.16 ± 1.13 | 47.44 ± 3.19 | May | 2 | |
| 7 | 2.71 ± 0.08 | 2.79 ± 0.04 | 12.59 ± 1.26 | 50.70 ± 3.72 | June | ||
| 8 | 3.56 ± 0.27 | 3.15 ± 0.14 | 16.12 ± 1.19 | 58.80 ± 1.06 | July | ||
| 9 | 2.84 ± 0.32 | 2.75 ± 0.22 | 12.65 ± 0.94 | 47.67 ± 2.59 | August | ||
| 10 | 1.73 ± 0.21 | 2.96 ± 0.57 | 11.30 ± 1.05 | 43.46 ± 2.25 | September | ||
| 11 | 2.42 ± 0.33 | 2.83 ± 0.03 | 9.75 ± 0.46 | 49.87 ± 1.25 | May | 2007 | 1 |
| 12 | 1.29 ± 0.08 | 1.52 ± 0.06 | 8.95 ± 0.39 | 29.98 ± 0.51 | June | ||
| 13 | 2.15 ± 0.10 | 2.77 ± 0.09 | 11.17 ± 1.00 | 56.75 ± 0.39 | July | ||
| 14 | 4.34 ± 0.26 | 4.50 ± 0.15 | 18.14 ± 1.61 | 94.23 ± 6.14 | August | ||
| 15 | 3.69 ± 0.33 | 3.57 ± 0.21 | 16.22 ± 0.53 | 61.87 ± 1.56 | September | ||
| 16 | 3.38 ± 0.40 | 3.57 ± 0.21 | 16.42 ± 0.23 | 59.93 ± 0.56 | May | 2 | |
| 17 | 3.71 ± 0.29 | 2.98 ± 0.16 | 16.61 ± 1.05 | 53.11 ± 1.74 | June | ||
| 18 | 3.25 ± 0.39 | 3.15 ± 0.35 | 20.25 ± 3.58 | 54.65 ± 5.03 | July | ||
| 19 | 3.68 ± 0.70 | 2.81 ± 0.21 | 14.51 ± 2.27 | 65.52 ± 6.30 | August | ||
| 20 | 2.79 ± 0.20 | 2.80 ± 0.07 | 11.28 ± 1.26 | 44.39 ± 1.15 | May | 2002 | - |
| 21 | 2.36 ± 0.19 | 2.94 ± 0.16 | 11.45 ± 0.53 | 50.64 ± 0.66 | June | ||
| 22 | 2.44 ± 0.18 | 3.16 ± 0.10 | 11.88 ± 0.42 | 44.05 ± 0.31 | July | ||
| 23 | 2.78 ± 0.16 | 3.00 ± 0.24 | 11.78 ± 0.91 | 51.07 ± 1.75 | August | ||
| 24 | 2.84 ± 0.21 | 3.34 ± 0.07 | 15.53 ± 1.11 | 49.36 ± 0.61 | September | ||
| 25 | 2.84 ± 0.19 | 3.15 ± 0.65 | 12.21 ± 1.08 | 50.14 ± 1.98 | May | 2005 | - |
| 26 | 4.12 ± 0.24 | 4.04 ± 0.14 | 15.09 ± 0.76 | 52.94 ± 0.84 | June | ||
| 27 | 3.26 ± 0.25 | 2.68 ± 0.08 | 16.55 ± 1.00 | 39.60 ± 1.04 | July | ||
| 28 | 3.13 ± 0.22 | 3.56 ± 0.10 | 12.58 ± 0.75 | 55.23 ± 1.31 | August |
Table 3.
The content ([%] ± SD) of apamine, MCDP, phospholipase A2 and melittin in archival bee venom samples from Georgia (1991) and archival samples from Poland (1995)
| Sample number | Apamine | MCDP | Phospholipase A2 | Melittin | Month | Year | Bee strain |
|---|---|---|---|---|---|---|---|
| 29 | 2.06 ± 0.25 | 2.51 ± 0.32 | 11.04 ± 0.49 | 42.04 ± 1.42 | – | 1991 | - |
| 30 | 3.82 ± 0.16 | 3.30 ± 0.17 | 15.66 ± 0.89 | 46.22 ± 1.70 | – | ||
| 31 | 1.18 ± 0.32 | 1.89 ± 0.26 | 9.38 ± 1.48 | 30.56 ± 1.71 | – | 1995 | - |
| 32 | 1.75 ± 0.20 | 2.06 ± 0.38 | 9.72 ± 1.48 | 17.85 ± 2.59 | – | ||
| 33 | 1.44 ± 0.09 | 2.36 ± 0.20 | 10.96 ± 0.44 | 32.12 ± 1.97 | – | ||
| 34 | 2.78 ± 0.21 | 2.86 ± 0.12 | 13.56 ± 0.69 | 37.16 ± 0.56 | – | ||
| 35 | 2.02 ± 0.43 | 2.69 ± 0.29 | 11.29 ± 1.34 | 47.18 ± 4.74 | – | ||
| 36 | 1.93 ± 0.11 | 2.77 ± 0.31 | 12.00 ± 2.00 | 41.58 ± 2.41 | – | ||
| 37 | 1.20 ± 0.07 | 2.03 ± 0.42 | 8.96 ± 0.79 | 29.24 ± 1.74 | – | ||
| 38 | 3.30 ± 0.37 | 4.67 ± 0.87 | 16.66 ± 3.00 | 60.77 ± 8.05 | – |
The content (relative to the dry venom mass) of analyzed peptides in honeybee venom samples collected in 2002–2007 was as follows: apamine from 0.93% to 4.34% (mean, 2.85% ± 0.79%); MCDP from 1.46% to 4.37% (mean, 2.82% ± 0.64%); phospholipase A2 from 7.41% to 20.25% (mean, 12.95% ± 3.09%); melittin from 25.40% to 60.27%, (mean, 45.91% ± 9.78%). For the HPLC method, the content relative to the dry venom mass of apamine ranged from 1.75% to 3.82% (mean, 2.64% ± 0.44%), MCDP 1.69% to 4.04% (mean, 3.19% ± 0.47%), phospholipase A2 6.66% to 17.17% (mean, 13.04% ± 2.22%) and mellitin 30.29% to 63.28% (mean: 54.08% ± 8.26%).
In order to compare results obtained in this study by using HPCE method with results obtained in the previous work by using HPLC method [12], statistical analysis was performed. Mean values of the content of four components (apamine, MCDP, phospholipase A2, and melittin) in bee venom samples collected from 2002 to 2007 were taken into consideration. In order to verify the null hypothesis that the variances of mean values of a single component are the same, an F test of the equality of two variances was used. At level α = 0.05 with f 1 = n1 − 1 and f 2 = n 2 − 1df, two variances of all four components differ significantly. This is in agreement with the results of precision study of HPLC and HPCE methods, which showed that the precision differed between them. Next, a T test without homogeneity of variance was applied. According to the null hypothesis there is no difference between two means of a single component. At level α = 0.05 with f 1 = n 1 − 1 and f 2 = n 2 − 2df two means of all four components do not differ significantly.
In order to check the significant differences between independent variables (the content of analyzed components), the data from analyzed samples were subjected to statistical analysis—factorial ANOVA. Three factors were considered to the calculations: the line of bees, month and year of venom collection. Statistical analysis showed that there were no significant differences in the content of apamine and mast cell degranulating peptide. The content of phospholipase A2 and melittin depended on line of the bees. Additionally, significant year-to-year variation was been recorded for melittin (results of statistical analysis are available on request).
Moreover, the HPCE analyses of archival honeybee venom samples from 1991 and 1995 showed some differences in the content of analyzed constituents. The content of apamine was from 1.18% to 3.82% (mean, 2.15% ± 0.89%). MCDP content varied from 1.89% to 4.67% (mean, 2.71% ± 0.81%). Phospholipase A2 content came within a range of 8.96% to 16.66% (mean, 11.92% ± 2.61%) and melittin content was between 17.85% to 60.77% (mean, 38.47% ± 11.88%). The obtained results (Table 3) showed that the archival honeybee samples from the years 1991 and 1995 were very similar to those collected in the years 2002 and 2005–2007. The content of the four main honeybee venom peptides (apamine, MCDP, phospholipase A2, and mellitin) were found to be on the same level as in the case of the samples collected almost 10 years later. It can be stated that the storage conditions of the bee venom samples described in the experimental chapter were adequate to prevent them from the decomposition process. Such conclusion requires further systematic investigations to be proved, but seems to be very important information during the standardization and quality control process of the natural products.
Based on the obtained results it can be stated that both HPCE and HPLC methods are suitable for the analysis of honeybee venom components.
To clarify the general distribution patterns or similarities of bee venom collected from two strains of honeybee in various months and years, principal component analysis was utilized.
For the analysis of correlations between the four constituents of honeybee venom (apamine, MCDP, phospholipase A2, melittin) the sample numbers 1 to 19 (Table 2) were arranged in a matrix and standardized. These samples, unlike the rest, were collected in two different years from two different bee strains. The first principal component (PC1) explained 90.84% of the total variance (Fig. 2). This component had eigenvalue greater than 1, which contributed more to the variance than the original variables (Table S4 Electronic Supplementary Material). The first PC (PC1) correlated with all of the analyzed components. Eigenvalues of the other principal components did not exceed 1 and thus they seemed to be sampling noise. The score plot of PC1 vs. PC2 for analyzed samples of bee venom is shown in Fig. 3. Most of the samples are distributed along PC1 in accordance with the bee strain (1 or 2).There is no such division between the samples according to the month or year of bee venom collection. Therefore, bee strain appears to be the only criteria for sample classification suggesting that genetic differences between two bee strains influence the composition of honeybee venom.
Fig. 2.

The Scree test for the data set
Fig. 3.

The score plot of PC1 vs. PC2 for bee venom samples (number 1 to 19)
In order to check whether any strong linear relations between analyzed constituents of bee venom existed, the correlation matrix for all variables was computed (Table S5 Electronic Supplementary Material). Very strong correlations between all analyzed variables were confirmed (r > 0.8).
Because honeybee venom is a complex natural product, the content of its constituents can vary depending on multiple factors. Our statistical analysis showed that there were differences in the composition of analyzed samples. The mutual relations between apamine, MCDP, phospholipase A2 and melittin were demonstrated. Therefore, in the standardization process beside the quantitative analysis of the main bee venom constituents using HPLC or HPCE, statistical analysis needs to be applied. Statistical tools such as: ANOVA or PCA should confirm the relevant quality of the sample which can be used as a safe raw material for drug formulation.
Conclusions
The investigations of honeybee venom presented in this paper allowed to develop the new precise and accurate HPCE method for determination of four major constituents (apamine, MCDP, phospholipase A2, and melittin) of this product. The results were compared with the experimental data obtained for the same venom samples analyzed earlier by the HPLC method. It was stated that HPCE and HPLC data did not differ significantly and that the HPCE method was the alternative for the HPLC method. The advantage of the HPCE method is that the consuming of the analytical reagents and costs of the analysis are considerably lower in comparison with the HPLC method.
Statistical analysis (PCA) has shown that the strain of bee appears to be the only criteria for bee venom sample classification. Moreover, strong correlations between apamine, MCDP, phospholipase A2, and melittin were confirmed. These correlations have to be taken into account in the honeybee venom standardization. On the basis of these considerations, it can be stated that in order to identify trends in the relationship between the composition of the samples analyzed and the factors influencing composition (origin, bee strain, season of bee venom collection), all the data should be subjected to chemometric analysis (PCA, ANOVA).
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 61 kb)
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Park HJ, Lee SH, Son DJ, Oh KW, Kim KH, Song HS, Kim GJ, Oh GT, Yoon DY, Hong JT. Antiarthritic effect of bee venom: inhibition of inflammation mediator generation by suppression of NF-kappaB through interaction with the p50 subunit. Arthritis Rheum. 2004;50:3504–3515. doi: 10.1002/art.20626. [DOI] [PubMed] [Google Scholar]
- 2.Kwon YB, Ham TW, Kim HW, Roh DH, Yoon SY, Han HJ, Yang IS, Kim KW, Beitz AJ, Lee JH. Water soluble fraction (<10 kDa) from bee venom reduces visceral pain behavior through spinal alpha 2-adrenergic activity in mice. Pharmacol Biochem Behav. 2005;80:181–187. doi: 10.1016/j.pbb.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 3.Kim HW, Kwon YB, Ham TW, Roh DH, Yoon SY, Lee HJ, Han HJ, Yang IS, Beitz AJ, Lee JH. Acupoint stimulation using bee venom attenuates formalin-induced pain behavior and spinal cord fos expression in rats. J Vet Med Sci. 2003;65:349–355. doi: 10.1292/jvms.65.349. [DOI] [PubMed] [Google Scholar]
- 4.Russell PJ, Hewish D, Carter T, Sterling-Levis K, Ow K, Hattarki M, Doughty L, Guthrie R, Shapira D, Pl M, Werkmeister JA, Kortt AA. Cytotoxic properties of immunoconjugates containing melittin-like peptide 101 against prostate cancer: in vitro and in vivo studies. Cancer Immunol Immunother. 2004;53:411–421. doi: 10.1007/s00262-003-0457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Putz T, Ramoner R, Gander H, Rahm A, Bartsch G, Thurnher M. Antitumor action and immune activation through cooperation of bee venom secretory phospholipase A2 and phosphatidylinositol-(3, 4)-bisphosphate. Cancer Immunol Immunother. 2006;55:1374–1383. doi: 10.1007/s00262-006-0143-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kokot ZJ, Matysiak J, Kłos J, Kędzia B, Hołderna-Kędzia E. Application of principal component analysis for evaluation of chemical and antimicrobial properties of honeybee (Apis mellifera) venom. J Apicult Res. 2009;48:168–175. doi: 10.3896/IBRA.1.48.3.04. [DOI] [Google Scholar]
- 7.Leluk J, Schmidt J, Jones D. Comparative studies on the protein composition of hymenopteran venom reservoirs. Toxicon. 1989;27:105–114. doi: 10.1016/0041-0101(89)90410-8. [DOI] [PubMed] [Google Scholar]
- 8.Schumacher MJ, Schmidt JO, Egen NB, Dillon KA. Biochemical variability of venoms from individual European and africanized honeybees (Apis mellifera) J Allergy Clin Immunol. 1992;90:59–65. doi: 10.1016/S0091-6749(06)80011-4. [DOI] [PubMed] [Google Scholar]
- 9.Palma MS, Brochetto-Braga MR. Biochemical variability between venoms from different honey-bee (Apis mellifera) races. Comparative Biochemistry and Physiology C-Pharmacology,Toxicology and Endocrinology. 1993;106:423–427. doi: 10.1016/0742-8413(93)90157-G. [DOI] [Google Scholar]
- 10.Szokan G, Horvath J, Almas M, Saftics G, Palocz A. Liquid chromatographic analysis and separation of polypeptide components from honeybee venoms. J Liq Chromatogr. 1994;17(16):3333–3349. doi: 10.1080/10826079408013516. [DOI] [Google Scholar]
- 11.Gauldie J, Hanson JM, Rumjanek FD, Shipolini RA, Vernon CA. The peptide components of bee venom. Eur J Biochem. 1976;61:369–376. doi: 10.1111/j.1432-1033.1976.tb10030.x. [DOI] [PubMed] [Google Scholar]
- 12.Kokot ZJ, Matysiak J. Simultaneous determination of major constituents of honeybee venom by LC-DAD. Chromatographia. 2009;69:1401–1405. doi: 10.1365/s10337-009-1052-9. [DOI] [Google Scholar]
- 13.Rader K, Wildfeuer A, Wintersberger F, Bossinger P, Mucke HW. Characterization of bee venom and its main components by high-performance liquid chromatography. J Chromatogr. 1987;408:341–348. doi: 10.1016/S0021-9673(01)81820-3. [DOI] [PubMed] [Google Scholar]
- 14.Banks BE, Dempsey CE, Pearce FL, Vernon CA, Wholley TE. New methods of isolating been venom peptides. Anal Biochem. 1981;116:48–52. doi: 10.1016/0003-2697(81)90320-1. [DOI] [PubMed] [Google Scholar]
- 15.Pacakova V, Stulik K, Hau P, Jelinek J, Vins I, Sykora D. Comparison of high-performance liquid chromatography and capillary electrophoresis for determination of some bee venom components. J Chromatogr A. 1995;700:187–193. doi: 10.1016/0021-9673(94)01170-J. [DOI] [Google Scholar]
- 16.Pacakova V, Stulik K. Validation of a method for determination of phospholipase A2 and melittin in bee venom preparations by capillary electrophoresis. J AOAC Int. 2000;83:549–554. [PubMed] [Google Scholar]
- 17.Pattanaargson S, Roboz J, Roboz J. Determination of hyaluronidase activity in venoms using capillary electrophoresis. Toxicon. 1996;34:1107–1117. doi: 10.1016/0041-0101(96)00083-9. [DOI] [PubMed] [Google Scholar]
- 18.Xu X, Nelson JW. Solution structure of tertiapin determined using nuclear magnetic resonance and distance geometry. Proteins. 1993;17:124–137. doi: 10.1002/prot.340170203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 61 kb)