

Abstract
T cell epitopes derived from polymorphic proteins or from proteins encoded by alternative reading frames (ARFs) play an important role in (tumor) immunology. Identification of these peptides is successfully performed with mass spectrometry. In a mass spectrometry-based approach, the recorded tandem mass spectra are matched against hypothetical spectra generated from known protein sequence databases. Commonly used protein databases contain a minimal level of redundancy, and thus, are not suitable data sources for searching polymorphic T cell epitopes, either in normal or ARFs. At the same time, however, these databases contain much non-polymorphic sequence information, thereby complicating the matching of recorded and theoretical spectra, and increasing the potential for finding false positives. Therefore, we created a database with peptides from ARFs and peptide variation arising from single nucleotide polymorphisms (SNPs). It is based on the human mRNA sequences from the well-annotated reference sequence (RefSeq) database and associated variation information derived from the Single Nucleotide Polymorphism Database (dbSNP). In this process, we removed all non-polymorphic information. Investigation of the frequency of SNPs in the dbSNP revealed that many SNPs are non-polymorphic “SNPs”. Therefore, we removed those from our dedicated database, and this resulted in a comprehensive high quality database, which we coined the Human Short Peptide Variation Database (HSPVdb). The value of our HSPVdb is shown by identification of the majority of published polymorphic SNP- and/or ARF-derived epitopes from a mass spectrometry-based proteomics workflow, and by a large variety of polymorphic peptides identified as potential T cell epitopes in the HLA-ligandome presented by the Epstein–Barr virus cells.
Electronic supplementary material
The online version of this article (doi:10.1007/s00251-010-0497-1) contains supplementary material, which is available to authorized users.
Keywords: Mass spectrometry, Proteomics, Minor histocompatibility antigen, Alternative reading frame, Ligandome, Database
Introduction
T cell-mediated immunotherapy is an attractive treatment of cancer as it exploits the potential of cytolytic T cells to specifically recognize antigens that are selectively expressed on tumor cells (Storb 2003; Hambach and Goulmy 2005; Kessler and Melief 2007; Falkenburg et al. 2003; Bleakley and Riddell 2004; Eisenlohr 2007). The enormous specificity of T cells involved in killing tumor cells makes this kind of treatment very attractive. An excellent example is the powerful graft-versus-leukemia (GVL) effect witnessed after allogeneic hematopoietic stem cell transplantation. GVL is characterized by remission of a hematological malignancy coinciding with the in vivo expansion of tumor-specific T cells. These T cells react to a patient-specific epitope presented in human leukocyte antigen (HLA) molecules on tumor cells (Marijt et al. 2003; van Bergen et al. 2007). T cell epitopes are peptides with a length of generally 8–11 amino acids. T cells are capable of distinguishing epitopes differing by only one amino acid, caused by a single nucleotide difference between patient and donor (Spierings et al. 2007). T cell epitopes, identified to play a role in (tumor) immunology, may arise from regular reading frames, but can also be encoded by alternative reading frames (ARFs) (Ho et al. 2006). Given the need for therapeutically useful T cell epitopes, the identification of new epitopes is of unceasing importance. The identification of T cell epitopes has been achieved with an array of methods, among which mass spectrometry is one of the most prominent techniques (Engelhard 2007; Hillen and Stevanovic 2006; Nesvizhskii et al. 2007). Peptide identification by tandem mass spectrometry is most successfully applied in an ever increasing number of proteomics studies. In a typical high throughput proteomics/ligandomics setting (Oliveira et al. 2010), the experimentally determined tandem mass spectra are matched against a database of hypothetical spectra generated from known peptide sequences using search engines like Mascot (Perkins et al. 1999) and Sequest (Eng et al. 1994).
For mass spectrometry-based identification of epitopes from polymorphic proteins, like minor histocompatibility antigens (MiHA) and peptides arising from ARFs, the commonly used protein databases like UniProt (UniProt 2008), IPI (Kersey et al. 2004) and RefSeqP (Pruitt et al. 2007) are unsuitable data sources, since these display very incomplete information about polymorphisms. Most of the published polymorphic MiHA are, therefore, not present in the standard protein databases, used in mass spectrometry-based workflows. Several strategies have been employed to address this problem (MSIPI (Schandorff et al. 2007), PepHum (Edwards 2007)), each with its own merits and limitations, trying to find the right balance between database size and completeness. In addition, there is a wealth of ligand and/or epitope information databases (Salimi et al. 2010), but these are not applicable in mass spectrometry (MS)-based workflows. Knowing that customized search databases that provide detailed control over the search space can vastly outperform standard strategies (Reisinger and Martens 2009), we designed a database dedicated to MiHA, thereby improving the chance of their identification in a proteomics type of experimental set up.
Our approach is based on the coding potential of the human genome, including its documented variations, as described in the RefSeq database. We chose RefSeq because it contains minimal redundancy, while still retaining splice variants, incorporates single nucleotide polymorphism (SNP) data from Single Nucleotide Polymorphism Database (dbSNP) (Sherry et al. 2001), which are richly annotated. We have created a database that contains all possible short peptides in different reading frames from a non-redundant mRNA set, combined with the known and annotated variations/SNPs. In this process, we removed all non-polymorphic information. Investigation of the frequency of SNPs in the dbSNP revealed that many of these SNPs are non-polymorphic “SNPs”. Therefore, we removed those from our dedicated database as well, and this resulted in a high quality comprehensive polymorphic peptide database. Centered on the amino acid polymorphisms of non-synonymous SNPs, our dedicated Human Short Peptide Variation Database (HSPVdb) outperforms existing databases in MS/MS-based T cell epitope identification.
The value of our HSPV database is shown by identification of the majority of published polymorphic SNP- and/or ARF-derived epitopes from a mass spectrometry-based proteomics workflow, as well as by a large variety of polymorphic peptides identified as potential T cell epitopes in the HLA-ligandome presented by EBV cells.
Materials and methods
Database preparation
The HSPVdb consists of peptides derived from genomic sequence variations. The database only contains peptides of seven amino acids or longer. The RefSeq database release 32 was downloaded from the NCBI FTP site and indexed using our local SRS installation (Etzold et al. 1996), (http://srs.bioinformatics.nl). The human mRNA subsection of RefSeq was extracted by selecting records with molecule type “mRNA” and organism source “Homo sapiens”. The resulting list of RefSeq records was subsequently processed using a series of Perl scripts.
To create the peptides derived from genomic sequence variations, we made use of the variation annotations that were added to RefSeq by the dbSNP staff. Variations found in the 5′ and 3′ UTRs were purposely included to allow detection of T cell epitopes derived from ARFs. For each annotated variation, the nucleotide sequences corresponding to the different alleles were generated. Instead of duplicating the complete mRNA sequence for each allele, we took a fragment starting from 30 nucleotides upstream and ending 32 nucleotides downstream of the variation. The three forward reading frames of each allele were translated to amino acid sequences. This typically results in three peptide sequences of 20 amino acids. Translation ignored the presence or absence of start codons. Codons that could not be translated to a single amino acid due to ambiguous nucleotides were translated to a stop codon. The amino acid translation was split on stop codons to get peptides derived from a continuous reading frame. Only the peptides, including the variation were kept in the database. To minimize redundancy, a translation for an allele was only included when the variation gives rise to a change in amino acid sequence (non-synonymous SNPs). This part of the database is optimized for finding peptides in the size range between 8 and 11 amino acids, but databases containing other peptide lengths can be produced at will. The database presented here consists of 20-mer peptides.
Each peptide sequence that was created, was stored as a separate database record and annotated with the ID of the originating mRNA sequence and the location of its encoding reading frame. If the RefSeq entry contains a coding sequence (CDS), the protein identifier and the position of that CDS on the mRNA with corresponding protein identifier, were added as annotation to the database record. For variations, we included the corresponding dbSNP identifiers, the positions of the variations, the nature of the amino acid changes and the percentage heterozygosity. If a variation causes an amino acid substitution, a SAP (single amino acid polymorphism), the possible amino acids were listed. Insertions or deletions were annotated as “in/del”. The resulting database was stored as a flat file in FASTA format for mass spectrometry-based proteomics purposes. This HSPVdb is fully dedicated to finding polymorphic epitopes. To reduce the size of this database, all duplicate amino acid sequences were deleted. These peptides contain both polymorphisms for each position, thereby describing all possible SNP information.
Subsets of the HSPV database were created based on reported heterozygosity. Three heterozygosity categories were defined: 0/1, unknown, all others. Additionally, for all categories ARFs were either included or left out.
Peptides for which the encoding DNA sequence is not part of the RefSeq-annotated open reading frame are labeled as alternative reading frame or ARF peptides. These include CDS that are in a different reading frame and sequences that are located up- or downstream of the annotated open reading frame.
SNP genotyping assays
Genomic DNA was isolated from 192 HLA A*0201-positive patient and donor samples (peripheral blood mononuclear or bone marrow cells) by the Gentra Systems PUREGENE genomic isolation kit (Biocompare, San Francisco, CA). SNPs rs4848158, rs61378134, rs36023150, rs11540526, rs11554279, rs35958189, rs56013141, rs11541290, rs34422048, rs11541416, rs28659989, rs2070159, rs4261080, rs11557142, rs11555631, rs11479605, rs11541519, rs5030742, rs11548263 were analyzed using a KASPar assay with allele-specific primers labeled with VIC and FAM dyes, (KBioScience, Hoddesdon, UK). Genotyping was performed according to manufacturer’s instructions.
Illumina custom array was used for genotyping rs10960, rs1143138, rs12986002, rs34669146, rs1047844, rs11266765, rs11539866, rs11541416, rs11541519, rs11542419, rs11542836, rs11544489, rs11545551, rs11548082, rs11553285, rs11553982, rs11554156, rs11554279, rs11555631, rs11557142, rs11558570, rs13202878, rs17848351, rs17851857, rs17853301, rs17853718, rs1803181, rs2070159, rs2261324, rs28934887, rs28935171, rs28940302, rs3180961, rs34136999, rs34418712, rs3962697, rs4848158, rs5030742, rs6112008, rs6686209, rs6794514
Genotyping was performed according to manufacturer’s instructions.
Sample preparation for test set
Peptide synthesis
Peptides were synthesized by standard Fmoc chemistry on a Syro II peptide synthesizer as described previously (Hiemstra et al. 1997). The integrity of the peptides was checked by reversed-phase high-performance liquid chromatography (HPLC) and mass spectrometry.
Liquid chromatography–mass spectrometry
The peptides studied are listed in Table 1. These are minor histocompatibility antigens as identified by different research groups around the world. A more complete listing of MiHA can be found at http://www.lumc.nl/dbminor. To perfectly mimic the conditions used in a normal mass spectrometry-based HLA-ligand identification process, all peptides included in Table 1 were measured by on-line chromatography/mass spectrometry (see below), and tandem mass spectra were recorded of their singly, doubly, and triply charged form. Subsequently, a selection of relevant charge states was made for each peptide, and charge states with a substantial contribution to the overall intensity only were used to construct a Mascot generic file (MGF) containing 31 tandem mass spectra, see Table 2.
Table 1.
Overview of known MiHA used as a test set in this study. It displays the epitope name and the HLA-molecule it is presented in. In addition, its immunogenicity is indicated together with the gene name and the polymorphisms are indicated. aNames according to http://www.lumc.nl/dbminor
| Epitope namea / HLA | Sequence | Remarks | Gene | polymorphic AA | dnSNP entry |
|---|---|---|---|---|---|
| HA1 / A2 | VLHDDLLEA | immunogenic | HMHA1 | VL[R/H]DDLLEA | rs1801284 |
| HA2 / A2 | YIGEVLVSV | immunogenic | MYO1G | YIGEVLVS[V/M] | rs61739531 |
| HA3 / A1 | VTEPGTAQY | immunogenic | AKAP13 | V[M/T]EPGTAQY | rs2061821 |
| HA8 / A2 | RTLDKVLEV | immunogenic | KIAA0020 | [R/P]TLDKVLE[V/I] | rs2270891 |
| HA1 / B60 | KECVLHDDL | immunogenic | HMHA1 | KECVL[R/H]DDL | rs1801284 |
| LB-ADIR-1 F / A2 | SVAPALALFPA | immunogenic; ARF in 5’UTR | TOR3A (ADIR) | SVAPALAL[F/S]PA | rs2296377 |
| LB-ADIR-1 S / A2 | SVAPALALSPA | allelic counterpart | |||
| CTSHr / A31 | ATLPLLCAR | immunogenic | CTSH | ATLPLLCA[G/R] | rs2289702 |
| CTSHr / A33 | WATLPLLCAR | immunogenic | CTSH | WATLPLLCA[G/R] | rs2289702 |
| ACC1y / A24 | DYLQYVLQI | immunogenic | BCL2A1 | DYLQ[C/Y]VLQI | rs1138357 |
| ACC1c / A24 | DYLQCVLQI | immunogenic | |||
| ACC1c+ cystinylated | DYLQCVLQI | immunogenic | |||
| HB1h / B44 | EEKRGSLHVW | immunogenic | HMHB1 | EEKRGSL[H/Y]VW | rs161557 |
| ACC2d / B44 | KEFEDDIINW | immunogenic | BCL2A1 | KEFED[G/D]IINW | rs3826007 |
| ACC2g / B44 | KEFEDGIINW | allelic counterpart | |||
| LB-ECGF1-1 H/B7# | RPHAIRRPLAL | immunogenic; ARF | TYMP (ECGF1) | RP[H/R]AI[R/C]RPLAL | no entry; rs1061205 |
Table 2.
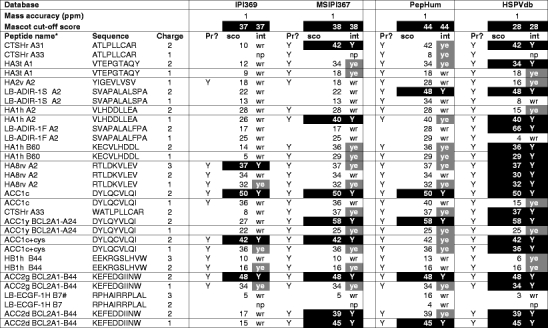
Summary of the searches with the test set of known MiHA against the IPI, MSIPI, PepHum, and HSPV database. The peptide names and sequences are given together with the charge of the precursor, submitted to tandem mass spectrometry. For each database, three columns are displayed: (1) whether the peptide is present in the database (Pr?), followed by (2) the mascot ion score assigned to the tandem mass spectrum (black filling if the mascot ion score is above the threshold of the search), and (3) the evaluation, i.e., was the tandem mass spectrum matched to the correct peptide (black filling and (Y) if correct, and above the mascot threshold (cut-off score), gray filling if correct and below (ye) the mascot threshold. In short, the blacker the better. The HSPVdb scores very well, due to its reduced format in combination with a high density of relevant SNP information. Wr wrong interpretation of MS2 spectrum; np no matching/no proposal from mascot search. aNames according to http://www.lumc.nl/dbminor. #Charge state 4+ was the most abundant in the charge distribution of peptide LB-ECGF-1H, but its MS2 spectrum was of such poor quality that it was not included for database searching. LB-ADIR peptides are from an ARF. ACC1+ Cys represents a special case in which the cysteine residue in the epitope can be modified by formation of an S–S bridge with free cysteines. This is relevant for both in vivo recognition and mass spectrometric interpretation
Sample preparation for determination of the EBV-LCL ligandome
Cell collection, preparation, and HLA elutions
Peripheral blood samples were obtained from healthy donors after approval by the Leiden University Medical Center Institutional Review Board and informed consent according to the Declaration of Helsinki. Mononuclear cells (MNC) were isolated by Ficoll-Isopaque separation and cryopreserved. Stable Epstein–Barr virus (EBV)-transformed B cell lines (EBV-LCL) were generated using standard procedures. EBV-LCL and HeLa cells were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM, BioWhittaker, Verviers, Belgium) supplemented with 10% bovine fetal serum (FBS, BioWhittaker).
Peptide isolation
Peptide isolation was performed with protein A beads (GE healthcare) covalently linked to the major histocompatibilty complex (MHC) class I mAb W6/32 (3 mg W6/32 on 1 ml of ProtA sepharose) using dimethyl pimelimidate according to the standard protocol (Stepniak et al. 2008).
The complex MHC-peptide pool was prefractionated on a C18 RP-HPLC system (2 mm × 15 cm; Reprosil-C18-AQ 3 um; Dr. Maisch GmbH, Ammerbuch, Germany), using a gradient 0–60% A to B. A: water, 5% Acetonitrile (ACN), 0.1% TFA, B: ACN, 0.1% TFA.
Liquid chromatography–mass spectrometry
Peptide fractions were reduced to near dryness and resuspended in 95/3/0.1 v/v/v water/acetonitrile/formic acid. These resuspended fractions were analyzed by on-line nano-HPLC mass spectrometry with a system described by Meiring et al (Meiring et al. 2002). Fractions were injected onto a precolumn (100 um × 15 mm; Reprosil-Pur C18-AQ 3 um, 5 um, Phenomenex) and eluted via an analytical nano-HPLC column (15 cm × 50 um; Reprosil-Pur C18-AQ 3 um). The gradient was run from 0% to 50% solvent B (10/90/0.1 v/v/v water/acetonitrile/formic acid) in 90 min. The nano-HPLC column was drawn to a tip of approximately 5 um and acted as the electrospray needle of the MS source.
The mass spectrometer was an LTQ-FT Ultra (Thermo, Bremen, Germany) and was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition. Full scan mass spectra were acquired in the FT-ICR with a resolution of 25,000 at a target value of 5,000,000. The two most intense ions were then isolated for accurate mass measurements by a selected ion monitoring scan in FT-ICR with a resolution of 50,000 at a target accumulation value of 50,000. The selected ions were then fragmented in the linear ion trap using collision-induced dissociation at a target value of 10,000. In a post analysis process, raw data were converted to peak lists using Bioworks Browser software, Version 3.1. For peptide identification, MS/MS data were submitted to the human IPI database using Mascot Version 2.2.04 (Matrix Science) with the following settings: 2 ppm and 0.8-Da deviation for precursor and fragment masses, respectively; no enzyme was specified. The Mascot output files were loaded into Scaffold (http://www.proteomesoftware.com) and exported to Excel as peptide reports and duplicates were removed.
Results
To investigate the value of our database, we studied two sets of samples. First, a test set comprising approximately 30% of all MiHA known today, as listed in Table 1, and second, a set of peptides eluted from HLA from an EBV-cell line.
Validation of HSPVdb with a test set of known MiHA
Our test set of known polymorphic peptides and allelic counterparts were synthesized and measured in standard on-line nanoHPLC/MS experiments, as in our normal proteomics workflow on HLA-ligands (Oliveira et al. 2010). Of all significantly occurring charge states, tandem spectra were recorded. Tandem mass spectra of varying quality are present in this dataset, reflecting a “real-world” situation, where the spectral quality depends on intrinsic peptide properties. A combined peak list was constructed from these spectra for searching the databases used in this work. This led to a set of 31 experimental tandem MS derived from 15 peptides (Table 2).
For validation of our HSPVdb, we compared it to the MSIPI and PepHum databases that were specifically constructed to address the lack of peptide variation in common databases like IPI. A summary of the databases used in this study is shown in Table 3.
Table 3.
Overview of the databases used in this study, listing the number of entries and the number of amino acid residues present in each database. In addition, the presence of ARFs and the (type of) SNP information in the various databases is indicated. The number of residues of each database relative to the IPI database and the relative size of the HSPV subsets is given. The number of SNPs in MSIPI 3.67 is 170.242; the number of SNPs in HSPVdb (subsets 1 and 5) is 380.182
| Database | Number sequences | Number of residues | Size relative to IPI 3.69 | ARFs? | 0/1? | Unk? |
|---|---|---|---|---|---|---|
| IPI (HUMAN v3.69) | 87130 | 35200044 | 1.00 | – | – | |
| MSIPI (HUMAN v3.67) | 87040 | 42553286 | 1.21 | ✓ | ✓ | |
| PepHum | 75237 | 176019757 | 5.00 | ✓ | ✓ | ✓ |
| HSPV | 2634086 | 45422884 | 1.29 | ✓ | ✓ | ✓ |
| Rel. to set 5 | ||||||
| HSPV subset 1 | 423015 | 8344552 | 0.18 | ✓ | ✓ | |
| HSPV subset 2 | 377269 | 7440614 | 0.16 | ✓ | ||
| HSPV subset 3 | 106379 | 2108989 | 0.05 | |||
| HSPV subset 4 | 152125 | 3012927 | 0.07 | ✓ | ||
| HSPV subset 5 | 2634086 | 45422884 | 1.00 | ✓ | ✓ | ✓ |
| HSPV subset 6 | 2378073 | 41106669 | 0.90 | ✓ | ✓ | |
| HSPV subset 7 | 729721 | 12444311 | 0.27 | ✓ | ||
| HSPV subset 8 | 985734 | 16760526 | 0.37 | ✓ | ✓ |
The HSPVdb is similar to the size of the IPI and MSIPI databases, but it includes all SNP information in all forward and ARFs (MSIPI: 170.242 SNPs; HSPVdb: 380.182 SNPs). When leaving out the alternative reading frame information (i.e., HSPVdb subset 1, see Table 3), the size of our HSPVdb is reduced to only 25% of the size of IPI and MSIPI, which is of great importance when searching databases.
The test set containing the tandem mass spectra of known MiHA was searched against the IPI, MSIPI, PepHum, and our HSPVdb. Searches were performed using the Mascot search engine (Matrix science), with various settings for mass accuracy (1, 2, 5, 10, and 50 ppm) representing the mass accuracy of various MS and/or experimental set ups. The enzyme setting was “none”. It is important to note that in the elucidation of HLA-ligands, the peptide termini are unknown in contrast to the vast majority of cases in standard proteomics experiments, in which peptide matching against databases can be done with an additional and very stringent condition, namely an enzyme cleavage site (in most cases, trypsin). In the standard proteomics approach, the enzyme restriction has an enormous positive impact on specificity and search time. For the sequencing of T cell epitopes, enzyme restriction is not applicable. However, for binding to the presenting HLA molecule, HLA-ligands have to satisfy certain conditions imposed by the HLA molecule, the binding motif. This binding motif can be used as additional help to some extent to assess the value of the matched sequence by the search engine. In addition, netMHC, http://www.cbs.dtu.dk/services/NetMHC/, could be applied to some extent, but neither of the two can be directly applied in the database search as a fixed condition. The best proof of a correct peptide assignment, in spite of improvements in peptide matching algorithms, is still the comparison of the tandem spectrum of the proposed eluted epitope with its synthetic counterpart.
All output of the Mascot search engine was assessed manually, and a summary of the results for a 1-ppm mass accuracy is shown in Table 2, and a full report of the searches is given in Supplementary Table 1.
Table 2 shows a selection of the searches in the four databases with a 1-ppm mass measurement accuracy. For every individual tandem mass spectrum, the Mascot ion score is reported. The results from the database search were classified by the following criteria: (1) was the tandem mass spectrum correctly identified by the search engine (indicated by black and gray filling in the first column) for each database? and (2) was the identification score above (indicated by black filling in the second column for each database) or below the Mascot significance threshold (cut-off score)? Therefore, “the blacker the better”. The presence (“Pr”) of each peptide in the particular database is indicated by “Y” in the appropriate column. Supplementary Table 1 shows the results of all searches performed with the test set of 31 tandem mass spectra to the IPI 3.69, MSIPI 3.67, PepHum, and HSPVdb.
From Table 2, it is immediately clear that the IPI database is not useful for finding MiHA, since it lacks essential variation information.
The PepHum database, based on expressed sequence tags (ESTs) information, including ARFs, is relatively large, by which relevant information for finding our polymorphic epitopes is “diluted”, and consequently, a serious amount of “noise” is generated, increasing the chance of finding false positives. The consequence of this is reflected in the outcome of the database search for PepHum. The number of significantly scoring peptides is only 5 as compared to the 19 peptides identified by our HSPVdb, see also Fig. 1a. This low score is only partially rescued by the number of correctly assigned peptides with a score below the Mascot significance threshold. In addition, ESTs may be more prone to experimental sequencing errors, leading to occurrence of false SNPs.
Fig. 1.
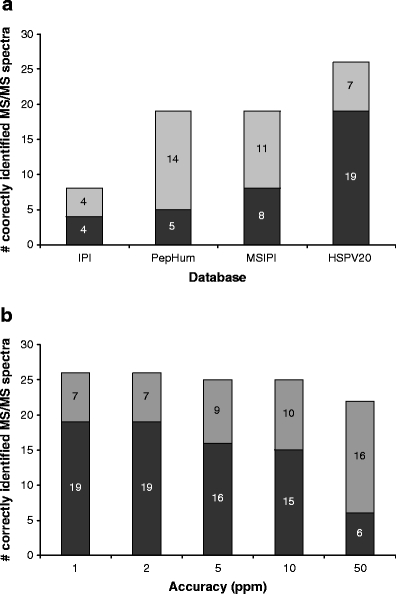
a Summary of the searches with 1-ppm accuracy against the IPI, MSIPI, PepHum, and HSPV databases. The color coding is as follows: black correct hit and above the MASCOT significance threshold; gray correct hit, but below the significance threshold. b Summary of the searches against HSPVdb with various mass measurement accuracies. b Summary of the searches with various mass accuracies, 1, 2, 5, 10, and 50-ppm accuracy against the HSPV database. The color coding is as above
The elegantly produced MSIPI does quite well, but also here, most correct peptide hits are below the statistical significance threshold score, which makes it hard to decide if a hit is true or a false positive in a “non-test set” setting. In addition, the MSIPI does not contain information from ARFs and UTRs.
For the HSPVdb, out of 31 MS/MS spectra, 19 are identified correctly above the Mascot significance threshold, while another 7 are also correctly identified, but below the significance threshold. Only three tandem mass spectra were wrongly assigned (false positives).
These wrong assignments are caused by the poor quality of the tandem mass spectra of these peptides, due to intrinsic peptide properties. To two tandem mass spectra, no match was assigned. These tandem mass spectra represent two peptides, “YIGEVLVSV”, which yields a bad mass spectrum and “RPHAIRRPLAL”, which is not present in the HSPVdb subset, because it is derived from a SNP not found in the dbSNP database. The HSPVdb, designed to reduce non-informative sequence information, outperforms the other databases.
Next to the size of the database, relieving the accuracy condition from 1 to 50 ppm (Fig. 1b) has a detrimental effect on both the number of correctly assigned peptides above and below the Mascot significance threshold. This effect can even lead to a false-positive score, as illustrated by a high and significant Mascot score of 63 (!) for MS/MS/query #6 (in HSPVdb, 50 ppm), see supplementary Table 1a. This result emphasizes the value of high mass accuracy.
So far, the good performance in the MS/MS-based identification of T cell epitopes of HSPVdb can be attributed to the compact nature and the special focus on polymorphic peptides. A reduced database size directly translates to a lower noise level in the database search, which is especially important in high-throughput T cell epitope elucidation, where search space limiting constraints like an enzyme cleavage site cannot be used. Another parameter affecting search quality is mass accuracy, which is also proven to be a prominent factor in avoiding false positives.
To further improve the quality of our HSPVdb, we focused on the quality of the SNPs in dbSNP, since we noted that the reported frequency of a substantial number of SNPs in dbSNP is “0” or “1” or “unknown”. This made us decide to study a random set of 52 SNPs with no frequency reported in dbSNP. We developed a SNP assay to screen a random HLA-A*02-positive Dutch donor population using the KASPar assay (92 DNA samples) and a SNP array (192 DNA samples). In our test population, 46 out of the 52 SNPs (90%) were not polymorphic, having an allele frequency of 1 or 0 in the SNP assays. Two SNPs (4%) were very rare (allele frequencies of 0.97, and 0.99), and 4 SNPs (8%) had a reasonable distribution in our population (0.77; 0.70; 0.20; 0.13).
A large number of reported “SNPs” in dbSNP is apparently not polymorphic, thereby contaminating our proteomics approach and the chance of finding suitable patient/donor MiHA pairs. Therefore, since reduction of the search space greatly enhances the chance of finding true positives in database searches, we decided to test our HSPVdb after removal of either “unknowns” or “0” and “1”, or both. The results are shown in supplementary Table 1b. Subset 3, the leanest form of HSPVdb with both “0” and “1” and “unknown frequency” SNPs removed and without ARFs, is reduced to only one fourth of its original size. Therefore, the significance threshold is clearly lowered (from 28 to 22 for 1-ppm mass accuracy), increasing the chance of finding true positives. In particular, those derived from tandem mass spectra of relatively poor quality with accompanying intrinsic low Mascot scores. Only one true positive is lost, because its frequency is not reported in the dbSNP. Similarly, the other subsets (subsets 1–8) of HSPVdb have reduced significance thresholds (data not shown). The application of these various forms of the database can be adapted to the needs of the user.
So far, we have shown that the selective reduction of the database size by removal of both the non-polymorphic peptide stretches and the SNPs of limited value, leads to a comprehensive high quality database file dedicated to improving the elucidation of MiHA.
Database quality and inconsistencies
During this work, we discovered inconsistencies in the number of SNPs included in several RefSeq and MSIPI versions, see Fig. 2a and b.
Fig. 2.

Number of incorporated SNPs per release of RefSeq (a) and of MSIPI (b)
The number of reported human SNPs dropped by 50% going from RefSeq release 28 to release 30, and by more than 50% in MSIPI going from version 37 to version 38. We reported this in October 2008 to the respective database producers who acknowledged there were problems and improved their efforts. Recently, we encountered a problem with the SNPs reported by 1000genomes.org in dbSNP which is being solved. Therefore, we continued using version 3.32 (on our website the HSPVdb version based on either Refseq release 32 or release 40 can be chosen). We would like to warn users for the status of the RefSeq with respect to this. MSIPI, also being a secondary database, suffered from the same errors during several versions, but this has been repaired, starting from version 49, although a strong decrease can be seen in version 3.67 (Fig. 2b). In general, as a user of these databases, it is very hard to judge the value of the database, so caution should be taken: newer versions are not always better.
Application of HSPVdb to finding potential MiHA presented in HLA on EBV-cells
To investigate the effects of application of our database to a representative HLA-ligand elution experiment, we eluted peptides from an EBV-LCL cell line (EBV-JY). After lysis, affinity purification was performed with BB7.2 antibody for HLA-A2, followed by separation of HLA and peptides. Subsequently, the complex peptide pool was analyzed by on-line nanoHPLC-tandem MS. The tandem mass spectra were matched against several databases for comparison, in particular, MSIPI and various subsets of our HSPV database.
Here, MSIPI is compared to the smallest subset of our HSPV database without ARFs (subset 3) and with ARFs included (subset 7), the advantages of which have been illustrated for the test set described above. These trimmed subsets do not include SNPs of which the frequencies in dbSNP are reported to be 0/1 or unknown. By searching against the smaller compact database containing all relevant SNPs, intermediate scoring peptides appear in the database search that would otherwise fall below the significance threshold when matching tandem mass spectra against larger databases.
This is illustrated by the number of intermediate scoring peptides, i.e., those peptides that score below the Mascot significance threshold when matching against MSIPI, and are, therefore, peptides not found otherwise. An additional 130 peptides were found for subset 3 and an additional 400 for subset 7. These extra peptides need to be checked for false positives (peptides with tandem mass spectra that match better with non-SNP containing peptides), and for the presence of a SNP. The extra peptides found can, e.g., be evaluated by application of netMHC. This approach, starting from our small experimental elution experiment, yielded eight peptides from subset 7 (including ARFs), and five peptides from subset 3 with a netMHC score below 50 (i.e., a stringent condition for strong binding). These peptides, shown in Table 4, are currently evaluated as potential MiHA.
Table 4.
Exclusive peptides with selected info from the HSPVdb. Peptides are either in frame (y) or in an ARF (n). The position of a SNP is indicated in the column SNP. In addition, the heterozygosity and NetMHC score is given
| Peptide | mRNA | Gene | Protein | rel2cds | In frame | dbSNP | SNP | Het | NetMHC |
|---|---|---|---|---|---|---|---|---|---|
| FLIPKTLVGV | NM_017700 | FLJ20184 | NP_060170.1 | downstream | y | rs2121558 | FLIPKTLVG[E/V] | 0.47 | 9 |
| SLSDLIYAL | NM_001080837 | SEBOX | NP_001074306.2 | inside | y | rs9910163 | SLSDLIYA[L/S] | 0.13 | 7 |
| GLWEQENHL | NM_024713 | C15orf29 | NP_078989.1 | inside | y | rs34998154 | GLW[E/K]QENHL | 0.05 | 41 |
| FIVTVIHTI | NM_024607 | PPP1R3B | NP_078883.2 | downstream | n | rs330915 | FIVTVIHT[I/F] | 0.49 | 30 |
| FLSEHPNVTL | NM_145298 | APOBEC3F | NP_660341.2 | inside | y | rs17000697 | FL[A/S]EHPNVTL | 0.28 | 19 |
| FLNQRSIML | NM_030956 | TLR10 | NP_112218.2 | upstream | n | rs9998678 | FLNQ[R/W]SIML | 0.05 | 29 |
| LLQSLVSI | NM_198889 | ANKRD17 | NP_942592.1 | inside | n | rs6855349 | LLQS[S/L]VSI | 0.46 | 46 |
| TLLDPNEKYLL | NM_016243 | CYB5R1 | NP_057327.2 | inside | y | rs2232842 | TLLDP[N/S]EKYLL | 0.31 | 31 |
All peptides found only by searching against the dedicated HSPV database increase the chance of finding relevant MiHA. The excellent annotation of the SNPs reported in our HSPV database enables the user to directly jump to the relevant information about the polymorphism, a feature that was largely lacking so far.
The HSPV database described here is an integral part of a complete peptidomics pipeline for finding therapeutically useful MiHA, a strategy that is currently under development.
Availability and web interface
A flat file with the content of the HSPV database can be requested by sending an email to hspv@bioinformatics.nl. A simple interactive query interface is available at: http://srs.bioinformatics.nl/hspv/.
This web interface allows the biologist to query the database for peptide sequences. It returns a list of RefSeq mRNA entries that contain a continuous reading frame encoding the query peptide, the start position of that reading frame, the position of the encoding nucleotide sequence with respect to any annotated CDS, and a description of the variations if the peptide contains any, see Fig. 3a. This is a great feature for the initial assessment of the quality and potential usefulness of the output of our database searches.
Fig. 3.
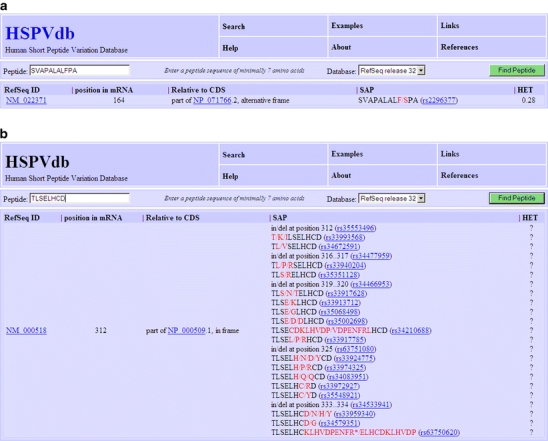
Screen shots show the output of a query for the peptides SVAPALALFPA (upper panel) and TLSELHCD (lower panel). It clearly illustrates the effect of the large number of annotated variations at the amino acid level
The richness of SNP information of our database is shown in Fig. 3b, for the peptide “TLSELHCD” displaying SAPs at every position in the peptide.
Conclusions
We have shown that selective reduction of the database size by removal of both the non-polymorphic peptide stretches and the non-polymorphic “SNPs” leads to a comprehensive high quality database file dedicated to improving the elucidation of MiHA.
Improvements in the quality and quantity of dbSNP entries, among others by the 1000 genomes project (http://www.1000genomes.org), if well controlled, will greatly enhance the use of our database by reporting useful frequencies and removal of spurious frequencies in the current dbSNP releases.
The website (http://srs.bioinformatics.nl/hspv/) provides easy access to relevant information about SNPs by its good annotation and hyperlinks incorporated in the HSPVdb.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Overview of the polymorphic peptides contained in the MGF-file which was used for all database searches. The charge of the precursors, chosen for the MS2 experiments, is included. ACC1+ Cys represents a special case in which the cysteine residue in the epitope can be modified by formation of an S–S bridge with free cysteines. This is relevant for both in vivo recognition and mass spectrometric interpretation. *Names according to: http://www.lumc.nl/dbminor. #Charge state 4+ was the most abundant in the charge distribution of peptide LB-ECGF-1 H, but its MS2 spectrum was of such poor quality that it was not included for database searching. LB-ADIR peptides are from an ARF (XLS 47 kb)
Overview of the polymorphic peptides contained in the MGF-file which was used for all database searches. The charge of the precursors, chosen for the MS2 experiments, is included. ACC1+ Cys represents a special case in which the cysteine residue in the epitope can be modified by formation of an S–S bridge with free cysteines. This is relevant for both in vivo recognition and mass spectrometric interpretation.*Names according to: http://www.lumc.nl/dbminor. #Charge state 4+ was the most abundant in the charge distribution of peptide LB-ECGF-1 H, but its MS2 spectrum was of such poor quality that it was not included for database searching. LB-ADIR peptides are from an ARF (XLS 42 kb)
Acknowledgments
The authors would like to thank David Kloet for the initial work on the project. Peter de Koning and Antoinette Teixeira are thanked for peptide synthesis. H.N. was supported by the BioAssist program of the Netherlands Bioinformatics Centre. This research was made possible by the financial assistance of the Landsteiner Foundation for Blood Transfusion Research. The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- ARF
alternative reading frame
- SNP
single nucleotide polymorphism
- UTR
untranslated region
- MiHA
minor histocompatibility antigen
- GvL
graft versus leukemia
- HSPVdb
Human Short Peptide Variation Database
- ACN
acetonitrile
- TFA
trifluoroacetic acid
Footnotes
Harm Nijveen and Michel G.D. Kester share the first authorship and Jack A.M. Leunissen and Peter van Veelen share the senior authorship.
References
- Bleakley M, Riddell SR. Molecules and mechanisms of the graft-versus-leukaemia effect. Nat Rev Cancer. 2004;4:371–380. doi: 10.1038/nrc1365. [DOI] [PubMed] [Google Scholar]
- Edwards NJ. Novel peptide identification from tandem mass spectra using ESTs and sequence database compression. Mol Syst Biol. 2007;3:102. doi: 10.1038/msb4100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenlohr LC, Huang L, Golovina TN. Rethinking peptide supply to MHC class I molecules. Nat Rev Immunol. 2007;7:403–410. doi: 10.1038/nri2077. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR., 3rd An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database . Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Engelhard VH. The contributions of mass spectrometry to understanding of immune recognition by T lymphocytes. Int J Mass Spectrom. 2007;259:32–39. doi: 10.1016/j.ijms.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzold T, Ulyanov A, Argos P. SRS: information retrieval system for molecular biology data banks. Methods Enzymol. 1996;266:114–128. doi: 10.1016/S0076-6879(96)66010-8. [DOI] [PubMed] [Google Scholar]
- Falkenburg JH, van de Corput L, Marijt EW, Willemze R. Minor histocompatibility antigens in human stem cell transplantation. Exp Hematol. 2003;31:743–751. doi: 10.1016/S0301-472X(03)00190-5. [DOI] [PubMed] [Google Scholar]
- Hambach L, Goulmy E. Immunotherapy of cancer through targeting of minor histocompatibility antigens. Curr Opin Immunol. 2005;17:202–210. doi: 10.1016/j.coi.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Hiemstra HS, Duinkerken G, Benckhuijsen WE, Amons R, de Vries RR, Roep BO, Drijfhout JW. The identification of CD4+ T cell epitopes with dedicated synthetic peptide libraries. Proc Natl Acad Sci USA. 1997;94:10313–10318. doi: 10.1073/pnas.94.19.10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen N, Stevanovic S. Contribution of mass spectrometry-based proteomics to immunology. Expert Rev Proteomics. 2006;3:653–664. doi: 10.1586/14789450.3.6.653. [DOI] [PubMed] [Google Scholar]
- Ho O, William R, Green WR. Alternative translational products and cryptic T cell epitopes: expecting the unexpected. J Immunol. 2006;177:8283–8289. doi: 10.4049/jimmunol.177.12.8283. [DOI] [PubMed] [Google Scholar]
- Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. The International Protein Index: an integrated database for proteomics experiments. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- Kessler JH, Melief CJ. Identification of T-cell epitopes for cancer immunotherapy. Leukemia. 2007;21:1859–1874. doi: 10.1038/sj.leu.2404787. [DOI] [PubMed] [Google Scholar]
- Marijt WA, Heemskerk MH, Kloosterboer FM, Goulmy E, Kester MG, van der Hoorn MA, van Luxemburg-Heys SA, Hoogeboom M, Mutis T, Drijfhout JW, van Rood JJ, Willemze R, Falkenburg JH. Hematopoiesis-restricted minor histocompatibility antigens HA-1- or HA-2-specificT cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci USA. 2003;100:2742–2747. doi: 10.1073/pnas.0530192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiring HD, van der Heeft E, ten Hove GJ, de Jong APJM. Nanoscale LC-MS(n): technical design and applications to peptide and protein analysis. J Sep Science. 2002;25:557–568. doi: 10.1002/1615-9314(20020601)25:9<557::AID-JSSC557>3.0.CO;2-F. [DOI] [Google Scholar]
- Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods. 2007;4:787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- Oliveira CC, van Veelen PA, Querido B, de Ru A, Sluijter M, Laban S, Drijfhout JW, van der Burg SH, Offringa R, van Hall T. The nonpolymorphic MHC Qa-1b mediates CD8+ T cell surveillance of antigen-processing defects. J Exp Med. 2010;207(1):207–221. doi: 10.1084/jem.20091429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisinger F, Martens L. Database on Demand—an online tool for the custom generation of FASTA-formatted sequence databases. Proteomics. 2009;9(18):4421–4424. doi: 10.1002/pmic.200900254. [DOI] [PubMed] [Google Scholar]
- Salimi N, Fleri W, Peters B, Sette A. Design and utilization of epitope-based databases and predictive tools. Immunogenetics. 2010;62(4):185–196. doi: 10.1007/s00251-010-0435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schandorff S, Olsen JV, Bunkenborg J, Blagoev B, Zhang Y, Andersen JS, Mann M. A mass spectrometry-friendly database for cSNP identification. Nat Methods. 2007;4:465–466. doi: 10.1038/nmeth0607-465. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spierings E, Hendriks M, Absi L, Canossi A, Chhaya S, Crowley J, Dolstra H, Eliaou JF, Ellis T, Enczmann J, Fasano ME, Gervais T, Gorodezky C, Kircher B, Laurin D, Leffell MS, Loiseau P, Malkki M, Markiewicz M, Martinetti M, Maruya E, Mehra N, Oguz F, Oudshoorn M, Pereira N, Rani R, Sergeant R, Thomson J, Tran TH, Turpeinen H, Yang KL, Zunec R, Carrington M, de Knijff P, Goulmy E. Phenotype frequencies of autosomal minor histocompatibility antigens display significant differences among populations. PLoS Genet. 2007;3:e103. doi: 10.1371/journal.pgen.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniak D, Wiesner M, de Ru AH, Moustakas AK, Drijfhout JW, Papadopoulos GK, van Veelen PA, Koning F. Large-scale characterization of natural ligands explains the unique gluten-binding properties of HLA-DQ2. J Immunol. 2008;180:3268–3278. doi: 10.4049/jimmunol.180.5.3268. [DOI] [PubMed] [Google Scholar]
- Storb R. Allogeneic hematopoietic stem cell transplantation—yesterday, today, and tomorrow. Exp Hematol. 2003;31:1–10. doi: 10.1016/S0301-472X(02)01020-2. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium The Universal Protein Resource (UniProt) Nucleic Acids Res. 2008;36:D190–D195. doi: 10.1093/nar/gkm895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bergen CA, Kester MG, Jedema I, Heemskerk MH, van Luxemburg-Heijs SA, Kloosterboer FM, Marijt WA, de Ru AH, Schaafsma MR, Willemze R, van Veelen PA, Falkenburg JH. Multiple myeloma-reactive T cells recognize an activation-induced minor histocompatibility antigen encoded by the ATP-dependent interferon-responsive (ADIR) gene. Blood. 2007;109:4089–4096. doi: 10.1182/blood-2006-08-043935. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overview of the polymorphic peptides contained in the MGF-file which was used for all database searches. The charge of the precursors, chosen for the MS2 experiments, is included. ACC1+ Cys represents a special case in which the cysteine residue in the epitope can be modified by formation of an S–S bridge with free cysteines. This is relevant for both in vivo recognition and mass spectrometric interpretation. *Names according to: http://www.lumc.nl/dbminor. #Charge state 4+ was the most abundant in the charge distribution of peptide LB-ECGF-1 H, but its MS2 spectrum was of such poor quality that it was not included for database searching. LB-ADIR peptides are from an ARF (XLS 47 kb)
Overview of the polymorphic peptides contained in the MGF-file which was used for all database searches. The charge of the precursors, chosen for the MS2 experiments, is included. ACC1+ Cys represents a special case in which the cysteine residue in the epitope can be modified by formation of an S–S bridge with free cysteines. This is relevant for both in vivo recognition and mass spectrometric interpretation.*Names according to: http://www.lumc.nl/dbminor. #Charge state 4+ was the most abundant in the charge distribution of peptide LB-ECGF-1 H, but its MS2 spectrum was of such poor quality that it was not included for database searching. LB-ADIR peptides are from an ARF (XLS 42 kb)