

Abstract
Ever since it was shown that maintenance of muscle contraction required the presence of extracellular Ca2+, evidence has accumulated that Ca2+ plays a crucial role in excitation–contraction coupling. This culminated in the use of the photoprotein aequorin to demonstrate that [Ca2+]i increased after depolarization but before contraction in barnacle muscle. Green fluorescent protein was extracted from the same jellyfish as aequorin, so this work also has important historical links to the use of fluorescent proteins as markers in living cells. The subsequent development of cell-permeant Ca2+ indicators resulted in a dramatic increase in related research, revealing Ca2+ to be a ubiquitous cell signal. High-speed, confocal Ca2+ imaging has now revealed subcellular detail not previously apparent, with the identification of Ca2+ sparks. These act as building blocks for larger transients during excitation–contraction coupling in cardiac muscle, but their function in smooth muscle appears more diverse, with evidence suggesting both ‘excitatory’ and ‘inhibitory’ roles. Sparks can activate Ca2+-sensitive Cl− and K+ currents, which exert positive and negative feedback, respectively, on global Ca2+ signalling, through changes in membrane potential and activation of voltage-operated Ca2+ channels. Calcium imaging has also demonstrated that agonists that appear to evoke relatively tonic increases in average [Ca2+]i at the whole tissue level often stimulate much higher frequency phasic Ca2+ oscillations at the cellular level. These findings may require re-evaluation of some of our models of Ca2+ signalling to account for newly revealed cellular and subcellular detail. Future research in the field is likely to make increasing use of genetically coded Ca2+ indicators expressed in an organelle- or tissue-specific manner.
Live cell imaging: a terrible beauty
Imaging biological processes in living cells has had and will continue to have great impact on our scientific understanding of biological function. I believe it also has an amazing and largely untapped potential to stir the imagination of society at large by presenting the world of cell biology in an accessible and aesthetically stimulating way. Just as the amazing images from the Hubble telescope make cosmologists of us all, so too advances in imaging technology have provided us with pictures dramatic enough to engage and enthuse even those with the most limited interest in biology. The invisible has been made visible, revealing unsuspected beauty and endless movement. This is dramatically exemplified by the Ca2+ waves seen in Xenopus oocytes expressing muscarinic acetylcholine receptors (Fig. 1; Lechleiter et al. 1991). These spiral across the cell in endlessly evolving patterns, never repeating the same sequence twice but always obeying simple rules of propagation and annihilation. I will let this example stand for now not just as a great piece of science but also as a thing of wonder in its own right, easily appreciated by anyone who has ever stood delighted by a Catherine-wheel's fiery corkscrewing against the night sky.
Figure 1. Calcium ion spiral waves in Xenopus oocytes.

Three frames have been chosen from a movie of an oocyte to show how the wave patterns evolve over time. These spectacularly patterned signals were first described by Lechleiter et al. (1991). Reproduced with permission of Professor James Lechleiter PhD: http://www.uthscsa.edu/csb/faculty/lechleiter.asp.
What is so special about Ca2+ anyway?
Before looking at Ca2+ imaging itself, I want to provide a very brief and ridiculously selective overview of the evidence pointing to the importance of Ca2+ signalling. Although free Ca2+ inside the cytosol is a very small percentage of total body Ca2+, most of which is found in bone mineral, it is a crucial cell signal in many cell types. Some of the earliest evidence for this came from experiments carried out in the 1880s on frog heart muscle by Sydney Ringer. He wanted to develop a salt solution suitable for in vitro studies on isolated tissues. Early experiments were promising, but subsequent attempts to repeat the work gave much less satisfactory results. Some detective work led to the discovery (Ringer, 1883) ‘that the saline solution which I had used’ (i.e. in the earlier successful study) ‘had not been prepared with distilled water, but with pipe water supplied by the New River Water Company … . . It is obvious therefore that the effects I had obtained are due to some of the inorganic constituents of the pipe water.’ It is generally accepted that Ringer's technician was probably responsible for the initial oversight in using pipe water rather than distilled water, but the consequences were positive, since they demonstrated both that the heart could be kept beating and that this required some ingredient missing from saline prepared with distilled water (Miller, 2004). Ringer went on to determine which of the ‘inorganic constituents’ in pipe water was important and discovered that his artificial saline solution only supported consistent cardiac contractions when Ca2+ was present, suggesting that this was necessary for normal cell function (Fig. 2). By such serendipity knowledge is advanced.
Figure 2. An early demonstration of the importance of Ca2+ for muscle contractility.
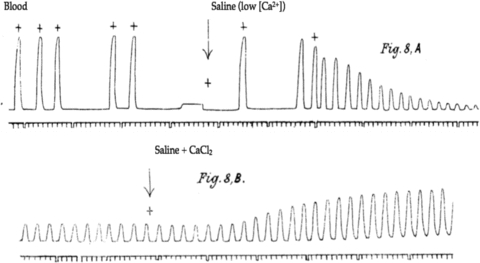
Changes in volume were recorded from an isolated frog ventricle perfused with blood or saline solutions (more detail on the technique and Ringer's work is presented by Miller, 2004). Full contractions were only seen when Ca2+ was present in the perfusate. Figure reproduced with permission from Ringer (1883).
Since these early observations, a large amount of evidence has accumulated suggesting that changes in intracellular [Ca2+] ([Ca2+]i) play a crucial role in the control of cell function, particularly in muscle. The sliding filament hypothesis of striated muscle contraction was established independently (by two different Huxleys) based on structural and functional experiments in the 1950s (see review, Huxley, 2000). By the early 1960s it was apparent that biochemical extracts of the contractile machinery could interact in the test tube to form a superprecipitate. This process required millimolar MgATP as an energy source but was also highly sensitive to [Ca2+], with measurable superprecipitation and ATP breakdown when [Ca2+] exceeded 10−7m (Weber & Winicur, 1961). This mimicking of muscle contraction in a test tube suggested that Ca2+ played some sort of role in controlling the contractile process. The development of techniques allowing muscle cells to be permeabilized, so that intracellular and extracellular [Ca2+] become equal, demonstrated a very similar relationship between [Ca2+]i and contractile force in permeabilized fibres and the Ca2+ dependence of superprecipitation, with activation of muscle contraction when [Ca2+]i exceeds 100 nm and maximal contraction above 1 μm (Fig. 3; Filo et al. 1965). Interestingly, the same relationship applied to both smooth and striated muscle, even though we now know there are major differences in the way contraction is regulated in these different muscle types (Fig. 3). We will return to this point later.
Figure 3. Effect of changing [Ca2+] on contraction in permeabilized muscle.
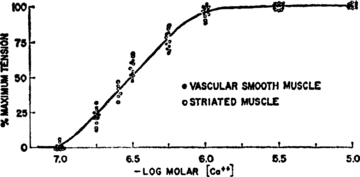
Permeabilized muscle fibre preparations were used, allowing intracellular [Ca2+] to be controlled simply by changing the extracellular [Ca2+]. The otherwise intact fibres developed active tension when [Ca2+] exceeded 100 nm, with maximal contraction above 1 μm. Interestingly, the relationships in striated and smooth muscles were almost identical. Reproduced with permission from Filo et al. (1965).
The evidence up to this point supported a model in which excitation of a striated muscle fibre by the propagation of an action potential across the cell membrane was coupled to mechanical contraction via the intracellular machinery through a Ca2+-dependent signalling process. However, this evidence was largely indirect and so the hunt was now on for some way to record changes in [Ca2+]i during electrically evoked muscle contraction in intact cells, so as to more closely reflect physiological events.
Measurements of [Ca2+]i in living cells
The relevant information was eventually derived from experiments using a Ca2+ indicator from a most unlikely source. Certain species of luminescent jellyfish manufacture the photoprotein aequorin, which emits a blue glow when it interacts with Ca2+. The protein was first isolated and purified from Aequoria aequoria by Osamu Shimomura, a biochemist with a keen interest in light-emitting molecules from biological sources (bioluminescence), while working in the laboratory of Frank Johnson at Princeton. They showed that the amount of light released could be used as a measure of the [Ca2+] in an aequorin-containing solution, opening the way for biological applications of the technique (Shimomura et al. 1963). Aequorin was used to demonstrate that Ca2+ signalling played a key role in excitation–contraction coupling within barnacle muscle (Ashley & Ridgway, 1968, 1970). This has large-diameter fibres, greatly facilitating intracellular aequorin injection. Contractions were stimulated by electrically depolarizing the fibres, and the resulting changes in membrane potential, Ca2+-dependent light emission by the aequorin and contractile force measured simultaneously (Fig. 4). This allowed both the amplitudes and the temporal sequencing of these events to be assessed. Membrane depolarization was followed by a rise in [Ca2+], which was, in turn, followed by muscle contraction. Since cause must precede effect, this ruled out the possibility that the [Ca2+]i increase was some sort of epiphenomenon or ‘side-effect’ of contraction itself, but was consistent with the hypothesis that excitation of the muscle fibre causes an increase in intracellular [Ca2+], which activates the formation of cross-bridges between myosin and actin. We now know that, in striated muscle, this results from the binding of Ca2+ to the troponin–tropomyosin complex on the thick myofilament, thereby relieving steric inhibition of cross-bridge formation between myosin heads and actin (Galinska-Rakoczy et al. 2008). Calcium also plays an important role in smooth muscle signalling using very different downstream mechanisms, with Ca2+–calmodulin activating myosin light chain kinase. The resulting phosphorylation upregulates myosin ATPase activity, releasing the chemical energy needed for cross-bridge cycling and cell shortening (Itoh et al. 1989).
Figure 4. A direct demonstration that Ca2+ is involved in excitation–contraction coupling.
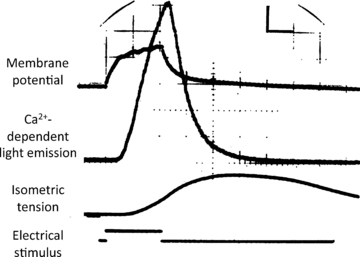
Barnacle muscle fibres were injected with aequorin, a protein which chemiluminesces when it binds Ca2+. The fibres were stimulated electrically, mimicking depolarization by an action potential, and changes in membrane potential, [Ca2+] (light generated by aequorin) and tension were measured simultaneously. Depolarization, a rise in [Ca2+]i and contraction occurred in that sequence, consistent with the hypothesis that Ca2+ signalling plays an important role in excitation–contraction coupling. Reproduced with permission from Ashley & Ridgway (1970).
This work established the involvement of intracellular Ca2+ in excitation–contraction coupling. Aequorin was difficult to use, however, since it had to be isolated and purified from jellyfish and then injected into each cell to be studied. The only alternative approach, which involved impaling cells with a Ca2+-selective microelectrode, was equally laborious and technically difficult, and could not always follow very rapid changes in [Ca2+]i (Lee, 1981). Better, more user-friendly Ca2+ indicators were clearly needed. This challenge was identified and taken on by Roger Tsien, first in the University of Cambridge and later in the University of California, Berkeley. He recognized that, just as physiologists had been obliged to design and manufacture the electronic equipment needed for the study of neuronal action potentials in a previous generation, so too there was a need to develop the new chemical tools needed for cell-signalling research. A range of fluorescent, Ca2+-sensitive dyes resulted from this work, along with a chemical strategy allowing them to be easily introduced into intact cells (Kresge et al. 2006). Acetoxymethyl esters are lipid soluble and cell permeant but are readily hydrolysed by intracellular enzymes, releasing the active dye in its anionic form. This remains trapped within the cell, and changes in fluorescence can then be used as a measure of [Ca2+]i. Based on its use in research papers, the most successful of these compounds was fura-2, a ratiometric Ca2+ indicator. These dyes circumvent movement artefacts and open the door to calibration of the signal in terms of absolute [Ca2+] (Grynkiewicz et al. 1985).
As is often the case, the availability of new research tools that were relatively easily applied led to a massive explosion in Ca2+-signalling research, with many thousands of papers published in the last 25 years. It quickly became apparent that increases in [Ca2+]i were involved in the transduction of external stimuli into cellular responses in almost every cell type studied. Calcium signalling is genuinely ubiquitous, affecting a wide variety of molecular targets in many different types of cells. No one reference can really capture this diversity, but a simple PubMed search for the term ‘Ca-signalling’ generated over 7900 hits in June 2010. The importance of Ca2+ signalling is indicated by the very low basal [Ca2+]I found in most cells, which is maintained by a variety of Ca2+ pumps and exchangers. This allows relatively small concentration increases to be resolved by Ca2+-sensitive proteins. An analogy can be found in astronomy, in which viewing conditions improve as background ‘light pollution’ decreases, making faint objects easier to detect. At about 100 nm or so, resting [Ca2+]i is more than four orders of magnitude lower than the free extracellular [Ca2+]i in mammals, and at least 100 times less than that in the sarcoplasmic/endoplasmic reticulum, the major intracellular Ca2+ store (McCarron et al. 1992; Wray & Burdyga, 2010). Despite the large electrochemical gradients involved, however, Ca2+ does not simply flood into the cytosol, since it cannot readily cross lipid membranes. Calcium can only diffuse into the cell if appropriate ion channels are activated, providing a Ca2+-permeable access route. Many such channels have been identified in the cell membranes of both excitable and non-excitable tissues, and are activated by very diverse stimuli. Intracellular stores can release Ca2+, mainly via opening of ryanodine receptor (RyR)-gated or inositol 1,4,5-trisphosphate receptor (IP3R)-gated release channels. Some of these Ca2+ channels will be mentioned in what follows, but no attempt will be made to provide an overview of this vast field.
Bioluminescence and biofluorescence: a Nobel calling
Before going on to consider the new insights gained from Ca2+ imaging itself, it is worth mentioning the close conceptual and historical connections between the development of Ca2+ indicators and other live cell imaging techniques based on fluorescent protein markers. In Shimomura and Johnson's original work on Aequoria, they noticed that the jellyfish produced a second optically active protein, which generated green fluorescence when excited by the light emitted from the aequorin itself (Shimomura, 2009). At the time, this work was simply motivated by the researchers’ interest in the mechanisms responsible for light production in animals, and the discovery of green fluorescent protein (GFP) in 1962 (Shimomura et al. 1962) remained of apparently limited interest for over 30 years. However, the cloning of the GFP gene in 1992 led to its expression in the nematode in 1994, the first demonstration that it could be used as a marker of gene expression in living cells (Prasher et al. 1992; Chalfie et al. 1994). By introducing the DNA code for GFP linked to a gene of choice, the expression and trafficking of any specified protein could now be visualized in living cells. Roger Tsien applied his understanding of fluorescent chemistry, first stimulated by the need to design improved Ca2+ indicators, to develop a range of modified fluorescent proteins, allowing different gene products to be imaged simultaneously in the same organism or cell (Tsien, 2010). This work led to the rapid and diverse application of live cell imaging to address key questions in biology, a technical revolution for which Shimomura, Chalfie and Tsien were awarded the 2008 Nobel Prize for Chemistry (Chalfie, 2009; Shimomura, 2009; Zimmer, 2009; Tsien, 2010). Once again, new tools led to new knowledge. All of this came from Shimomura and Johnson's fascination with creatures that glow. At the time, neither they nor anyone else could have guessed what amazing scientific and technological implications their findings would have. They just wanted to know! Perhaps this stands as a reminder that we need to leave room for curiosity-driven enquiry, no matter how little obvious application the work might have, since nobody can foretell where the next crucial finding will come from. If we could, it wouldn't really be research.
Confocal technology, Ca2+ imaging and Ca2+ sparks
Getting back to the Ca2+ story, the step from Ca2+ measurement to Ca2+ imaging came with the development of new microscopy techniques. Recordings using Ca2+ indicators often involve a technique referred to as microfluorimetry, in which the overall (i.e. spatially summed) intensity of any emitted fluorescence is recorded. Although a microscope objective is used to collect the light, no fluorescent image is captured. There are several technical reasons for doing this, including increased sensitivity and speed of recording, important with rapidly changing signals. However, the development of confocal microscopy and ever more sensitive CCD-based cameras made true Ca2+ imaging a viable option, in which local changes in [Ca2+]i within individual cells can be visualized. Confocal microscopes combine a scanning laser to excite cell fluorescence with a pinhole to exclude out-of-focus light coming back into the imaging scanhead, allowing clear optical sections to be recorded from much thicker tissues. Initially, this technique was used to improve the resolution of images from fixed specimens stained with fluorescent dyes, and to generate three-dimensional tissue models by imaging different planes in the same sample and combining the data (Brakenhoff et al. 1989). It quickly became obvious, however, that this technology was also ideal for Ca2+ imaging using fluorescent indicators in living cells (Niggli & Lederer, 1990). Rather than simply recording the average signal emitted from the sample, as is the case in Ca2+ microfluorimetry, it was now possible to repeatedly image the Ca2+-dependent fluorescence in a given optical section of a cell or tissue, generating a ‘movie’ showing how Ca2+ changed over time in different cells and even in different parts of the same cell (Fig. 5). The only problem with these recordings was that they were too slow to faithfully reproduce the rapid changes in [Ca2+]i seen in muscle cells, which were the first focus of interest. This limitation could be largely overcome by adapting the technique so that the laser did not scan across the whole of the image plane but repeatedly scanned back and forth along a single line. Temporal resolution was greatly increased (from about 1 frame s−1 to about 500 lines s−1), but at the cost of one spatial dimension. This sacrifice was soon shown to be justified, however, as the resulting linescan images revealed subcellular Ca2+ events previously unsuspected in cardiac muscle cells. These brief, spatially localized increases in [Ca2+]i were dubbed ‘Ca2+ sparks’ and were shown to represent spontaneous release of Ca2+ from the sarcoplasmic reticulum via cardiac ryanodine receptors (Cheng et al. 1993). It should also be noted in passing that localized spontaneous Ca2+-release events mediated by clusters of IP3Rs have also been identified in a range of excitable and non-excitable tissues and given the name ‘Ca2+ puffs’ (Smith et al. 2009; Wray & Burdyga, 2010). These will not be considered further.
Figure 5. Application of confocal microscopy to Ca2+ imaging in living cells.
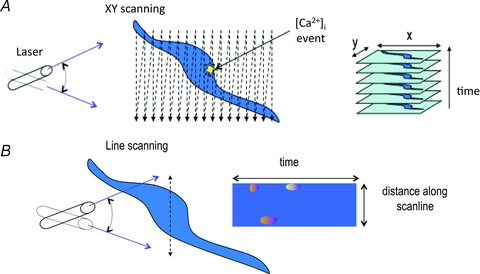
Cells are loaded with a fluorescent Ca2+ indicator (e.g. fluo-4) and excited with a scanning laser. A, in XY scanning mode a single image plane is scanned repeatedly, allowing any [Ca2+]i rises to be recorded and their location identified. B, line scanning greatly increases the speed of acquisition by imaging only along a single line rather than a full XY plane. The resulting images show how [Ca2+]i changes locally at each point on the line as time passes. This is particularly useful when imaging fast events, such as Ca2+ sparks.
We will return to the function of Ca2+ sparks later, but first I want to consider smooth muscle, which will be the main focus for the rest of this discussion. Interestingly, evidence that something similar to Ca2+ sparks must occur in smooth muscle had been reported nearly a decade before they were imaged. Electrophysiological recordings, in which the membrane potential of a smooth muscle cell could be clamped at a specific voltage and transmembrane ion currents recorded, had revealed brief, relatively short-lived and outwardly directly K+ currents (Fig. 6A). These were given the descriptive title ‘spontaneous transient outward currents (STOCs)’ (Benham & Bolton, 1986). When caffeine was applied to release stored Ca2+ by activating RyRs, there was an initial burst of STOC activity, followed by relative quiescence. This response could be inhibited by introducing EGTA, a Ca2+ chelator, to the intracellular solution via the recording pipette, indicating that the current was activated by a rise in [Ca2+]i. Taken together, these findings suggested that STOCs reflected spontaneous Ca2+ release from the sarcoplasmic reticulum via RyRs, leading to the activation of Ca2+-sensitive K+ channels conducting the outward current (IKCa). Such channels (also known as large-conductance K+ channels, or BK channels) had already been identified in smooth muscle (Walsh & Singer, 1981; Benham et al. 1986). It was not until 1995, however, that this indirect evidence for localized spontaneous Ca2+ release in smooth muscle was confirmed by the direct visualization of Ca2+ sparks, again using confocal imaging of Ca2+ indicator fluorescence (Fig. 6B; Nelson et al. 1995). Both sparks and STOCs were inhibited by using ryanodine to block RyR activity and by agents that depleted the Ca2+ stores. Calcium imaging therefore allowed spontaneous Ca2+ release to be directly observed in smooth muscle cells, confirming a hypothesis originally postulated on the basis of indirect evidence.
Figure 6. Indirect and direct evidence for Ca2+ sparks in smooth muscle.
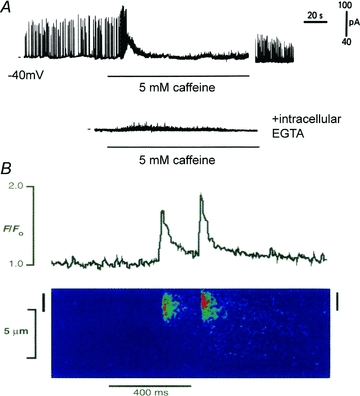
A, electrophysiological recordings from voltage-clamped smooth muscle reveals spontaneous outward transient currents (STOCs) carried by K+. Activation of RyRs with caffeine to release the sarcoplasmic reticulum Ca2+ store resulted in an initial burst of STOC activity and then quiescence. Inclusion of EGTA in the recording pipette to buffer changes in [Ca2+]i inhibited STOCs. This indirect but highly persuasive evidence suggests that there must be spontaneous Ca2+ release via RyRs, resulting in the activation of Ca2+-activated BK channels. Reproduced with permission from Benham & Bolton (1986). B, confocal linescan image of an arterial smooth muscle cell loaded with the indicator fluo-3. Normalized fluorescence (F/F0) is used as a measure of [Ca2+]i. Two spontaneous Ca2+ sparks have been recorded. Reproduced with permission from Nelson et al. (1995).
What do sparks do?
Physiology is driven by functional questions. Usually, this expresses itself in the desire to understand the mechanisms responsible for the function of a molecule, cell, organ or organism. In the case of Ca2+ sparks, however, we are presented with a cell mechanism whose function needs to be defined. In the case of cardiac muscle, it was immediately proposed that they might act as fundamental building blocks from which larger and global Ca2+ transients are constructed during the excitation–contraction process (Cheng et al. 1993). This view has been confirmed over the intervening years. Depolarization of cardiac myocytes activates voltage-operated (L-type) Ca2+ channels in the plasma membrane, leading to an influx down the electrochemical gradient from the extracellular fluid. This stimulates the opening of near-membrane RyRs, which are directly sensitive to the local [Ca2+]i, leading to an almost synchronous cell-wide Ca2+ rise and contraction (Cheng & Lederer, 2008). Individual sparks are not easily identified in confocal linescans of normal Ca2+ transients but become obvious when relatively small depolarizations are applied. Thus, in cardiac muscle at least, sparks act as excitatory events whose summation generates the intracellular signal responsible for contraction.
The story in smooth muscle, however, does not appear to be so straightforward. In the initial report of Ca2+ sparks in arterial smooth muscle, considerable evidence was provided for an inhibitory function. Interventions that inhibited sparks resulted in constriction of pressurized arteries, suggesting that sparks normally exert a dilatory influence (Nelson et al. 1995). It was proposed that this reflects negative feedback via spark activation of BK channels, resulting in outward, hyperpolarizing current (Fig. 7). Activation of voltage-operated Ca2+ channels is reduced in these circumstances, limiting Ca2+ influx. Localized Ca2+ release in the form of Ca2+ sparks could, therefore, favour a reduction in global [Ca2+]i, relaxing smooth muscle. Considerable evidence supports this model in many different smooth muscles, particularly in vascular and urinary myocytes (Perez et al. 2001; Heppner et al. 2003; Burdyga & Wray, 2005; McGahon et al. 2007).
Figure 7. Sparks activate Ca2+-sensitive membrane currents in smooth muscle.
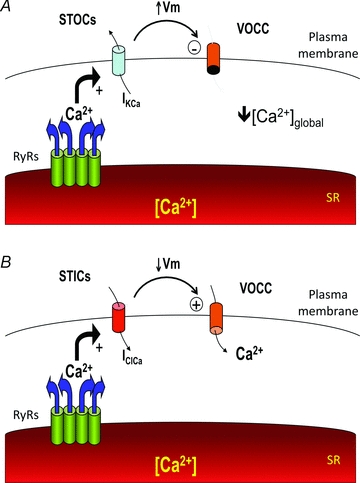
A, activation of Ca2+-activated K+ channels by Ca2+ sparks results in spontaneous transient outward currents (STOCs), hyperpolarizing the plasma membrane (increased membrane potential; Vm). This reduces activation of voltage-operated Ca2+ channels (VOCCs), decreasing Ca2+ influx and global [Ca2+]. This can act as a negative feedback mechanism, whereby localized Ca2+ release from the sarcoplasmic reticulum (SR) lowers mean cytosolic [Ca2+], restricting further store loading. B, spark-dependent activation of Ca2+-activated Cl− channels causes spontaneous transient inward currents (STICs), with membrane depolarization and Ca2+ channel activation. This results in positive feedback, with Ca2+ release from the SR promoting further Ca2+ influx.
This is conceptually quite different from their proposed role in cardiac muscle, where sparks are used to construct global Ca2+ signals. Evidence is now accumulating, however, to suggest that Ca2+ sparks may also play an excitatory role in some smooth muscles. We have used confocal linescan imaging to investigate Ca2+ signalling in the smooth muscle of retinal arterioles, which regulate blood flow to the inner retina. These myocytes generate both brief localized Ca2+ sparks and more prolonged Ca2+ oscillations (Fig. 8). Rather than reducing global [Ca2+]i, sparks often appear to summate, generating oscillations, which sometimes result in obvious contraction of the relevant myocyte (Curtis et al. 2004). In more recent experiments using faster two-dimensional Ca2+ imaging, Ca2+ sparks again appear to initiate cell-wide Ca2+ oscillations and contraction, again suggesting an excitatory rather than an inhibitory role (Tumelty et al. 2007). Pharmacological interventions that reduce spark frequency also reduce spontaneous oscillations, and constrictor agonists increase both sparks and oscillations (Tumelty et al. 2007; Jeffries et al. 2010). Similar observations have been made by other groups, with intracellular sites that demonstrate high levels of spontaneous spark activity (termed ‘frequent discharge sites’) acting as points of initiation for cell-wide spontaneous Ca2+ waves and evoked Ca2+ transients. This suggests that, as in cardiac muscle, sparks in smooth muscle can also act as ‘elementary building blocks’, from which excitatory global Ca2+ signals are constructed (Bolton et al. 2004). It should also be noted that, although sparks can exert negative feedback by activating outward IKCa, the presence of Ca2+-activated Cl– channels in the plasma membrane also opens up the possibility of positive feedback following a rise in [Ca2+]i (Fig. 7; Large & Wang, 1996; Jackson, 2000; McGahon et al. 2009). In smooth muscle, Cl− currents are inwardly directed at normal resting potentials (outward movement of negatively charged Cl− ions equates to inward conventional current). The resulting spontaneous transient inward currents (STICs) tend to depolarize the plasma membrane, increasing the steady-state activation of voltage-operated Ca2+ channels and so raising cell [Ca2+]i. Calcium sparks have been shown to generate STICs in a number of different smooth muscle types (Bao et al. 2008). Indeed, depending on the conditions, a single spark can activate a STOC followed by a STIC. The resulting biphasic current has been rather philosophically dubbed a STOIC (ZhuGe et al. 1998). The functional consequences of such complex signals are only just beginning to be explored (ZhuGe et al. 2010).
Figure 8. Calcium sparks and oscillations recorded from retinal arteriole myocytes in situ.
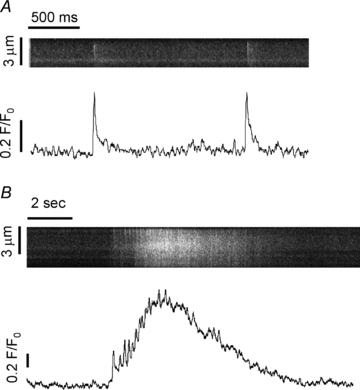
Arterioles were loaded with the Ca2+ indicator fluo-4, and scanned transversely across their diameter along a single scanline 500 s−1 using a confocal laser microscope (see Fig. 9B). Normalized fluorescence (F/F0) is a measure of local [Ca2+]. A, two spontaneous Ca2+ sparks, with a rapid rising phase and slower decay. The entire event typically lasts <150 ms. B, a more prolonged Ca2+ oscillation lasting several seconds. These are sometimes associated with myocyte contraction. Spark activity is clearly seen near the upper cell margin, and sparks appear to summate during the rising phase of the oscillation. For more detail see Curtis et al. (2004) and Tumelty et al. (2007). Image courtesy of Dr Tim Curtis.
What you see depends on how you look at things: agonist stimulation and Ca2+ oscillations
Another aspect of Ca2+ signalling in smooth muscle of considerable interest relates to its role in control of contraction by neurotransmitters and endocrine or paracrine messenger molecules. We have been interested in how endothelin-1 (Et-1), a highly potent endothelium-derived vasoconstrictor, regulates Ca2+ signals in retinal arterioles. Recording the average smooth muscle [Ca2+]i using microfluorimetry results in a relatively simple response, with an initial Ca2+ transient lasting approximately 100 s, which then falls back towards the control level but remains elevated above baseline (Fig. 9A). This triggers a sustained vessel constriction (Curtis et al. 2007). The intuitive model of stimulus–contraction coupling suggested by this result is one in which muscle contraction tracks the increase in [Ca2+]i, during the rising phase at least, consistent with the data from skinned muscle showing that tension is a function of the steady-state [Ca2+]i (Fig. 3). Calcium imaging reveals a very different picture, however, in which endothelin-1 stimulates phasic Ca2+ oscillations with a much shorter period (<5 s) than the transient seen at the whole vessel level (Fig. 9B; Curtis et al. 2007). Presumably, the asynchronous nature of these oscillations accounts for the much slower and relatively tonic increase in [Ca2+]i recorded using microfluorimetry. Importantly, no individual cell experiences the temporal profile of [Ca2+]i changes recorded in the spatially averaged recording. Any realistic model of cell signalling in response to Et-1 must, therefore, be able to account for the more complex pattern revealed by high-speed imaging. Similar oscillatory responses to constricting stimuli, first reported in vascular tissue in 1994 (Iino et al. 1994), have subsequently been described in a variety of other smooth muscles. Studies on pulmonary arteriolar and bronchiolar smooth muscle, for example, have demonstrated that increasing concentrations of agonist lead to an increase in the frequency of Ca2+ oscillations, and that the resulting contractions can be plotted as a function of frequency rather than ‘mean’[Ca2+]i (Fig. 10; Perez & Sanderson, 2005a,b;). Although high agonist concentrations lead to persistent elevation of global [Ca2+]i in most cells, it is reasonable to argue that Ca2+ imaging has revealed a major ‘digital’, ‘frequency-modulated’ aspect to smooth muscle Ca2+ signalling previously unsuspected from global measurements of average [Ca2+]i (Sanderson et al. 2010). Calcium ion spikes and oscillations had been demonstrated in a range of cell types, both excitable and non-excitable, prior to the widespread use of Ca2+ imaging (Thorn et al. 1993; Friel, 1995), but the ability to observe Ca2+ changes within individual myocytes embedded in their parent tissue using confocal techniques has demonstrated their importance in smooth muscle in a way not previously appreciated from Ca2+ microfluorimetry records. It remains to be seen what functional benefit results from the use of complexly patterned Ca2+ signals rather than simple variations in steady-state [Ca2+]i, although oscillations may allow for intermittent activation of Ca2+-dependent processes with a relatively high Ca2+ threshold while reducing the risk of cell damage (Berridge et al. 2000). The temporal patterning of Ca2+ signals may also contribute to signal targeting within the cell just as much as their spatial distribution. This may even play a role in the regulation of protein expression in smooth muscle by Ca2+ signals, a process referred to as ‘excitation–transcription coupling’ (Wamhoff et al. 2006).
Figure 9. Calcium imaging reveals cell behaviour not apparent in ‘average’ recordings.
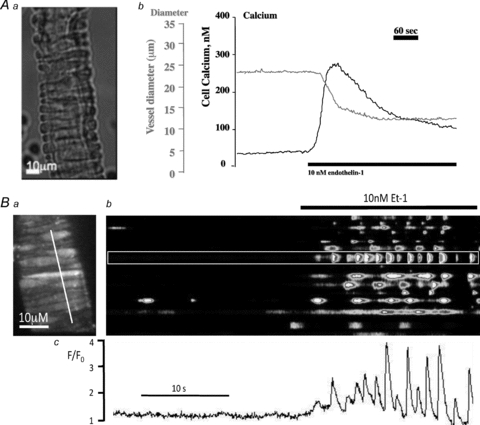
A, a retinal arteriole (Aa) was loaded with fura-2 AM and changes in mean [Ca2+]i and vessel diameter recorded (Ab). This technique records changes in smooth muscle but not endothelial [Ca2+] (Scholfield & Curtis, 2000). Addition of endothelin-1 (Et-1; 10 nm) leads to an increase in [Ca2+]i, followed by constriction. There is an initial transient Ca2+ peak, followed by a sustained rise but at a lower [Ca2+]i. Images courtesy of Drs C. N. Scholfield and T. M. Curtis. B, confocal Ca2+ imaging of retinal arteriole myocytes in situ within the vessel wall (Ba). Fluo-4-loaded cells have been imaged in a plane near the bottom of the organ bath, revealing an array of adjacent myocytes. These were then scanned along the indicated line to generate a linescan (Bb). Addition of endothelin stimulates asynchronous Ca2+ oscillations in adjacent cells. The temporal pattern of the cell response (Bc) does not mirror that of the global signal seen in A (note the faster time base in B).
Figure 10. Contraction of bronchiolar smooth muscle is a function of Ca2+ oscillation frequency.
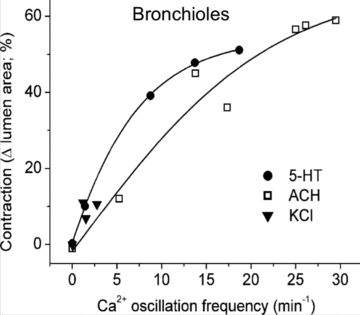
The results were obtained by using Ca2+ imaging in pulmonary slices and applying different concentrations of 5-hydroxytryptamine (5-HT) or acetylcholine (ACH), or by using K+ to depolarize the smooth muscle (KCl). Reproduced with permission from Perez & Sanderson (2005a). Qualitatively similar results were obtained for pulmonary arterioles (Perez & Sanderson, 2005b).
Conclusion
The application of Ca2+-imaging technology has radically altered the way we have to think about signalling at the cellular level. The complex and highly dynamic responses evoked in smooth muscle by agonists, for example, were not obvious in microfluorimetry recordings of average [Ca2+], and suggest that frequency modulation of phasic Ca2+ waves and oscillations plays an important role in determining the contractile response. Subcellular detail has also been revealed, with brief, spatially localized Ca2+-release events. These Ca2+ sparks can contribute directly to the elevation of cell [Ca2+] and contraction, as they do in cardiac excitation–contraction coupling and in some smooth muscles, or indirectly through activation of Ca2+-sensitive membrane conductances, altering membrane potential and thus regulating Ca2+ influx via changes in activation of voltage-operated channels. As is usual in science, the ability to look at Ca2+ signals in more detail has generated as many new questions as answers, and the challenge of integrating the relevant detail into functional models at the cell and tissue level will be considerable. Future developments will probably include increased use of genetically coded Ca2+ indicators, in which proteins with Ca2+-sensitive fluorescent or luminescent properties are expressed in specific organelles or tissues (Alvarez & Montero, 2002; Kotlikoff, 2007). This reflects the same basic strategy that underpinned the initial use of aequorin, but with molecules genetically engineered to increase the range of possible applications and target indicator expression to specific cell types or organelles (Eglen & Reisine, 2008).
Acknowledgments
The retinal arteriole research presented here has all been carried out in close collaboration with Tim Curtis and Norman Scholfield in the Centre for Vision and Vascular Science, Queen's University of Belfast.
References
- Alvarez J, Montero M. Measuring [Ca2+] in the endoplasmic reticulum with aequorin. Cell Calcium. 2002;32:251–260. doi: 10.1016/s0143416002001860. [DOI] [PubMed] [Google Scholar]
- Ashley CC, Ridgway EB. Simultaneous recording of membrane potential, calcium transient and tension in single muscle fibers. Nature. 1968;219:1168–1169. doi: 10.1038/2191168a0. [DOI] [PubMed] [Google Scholar]
- Ashley CC, Ridgway EB. On the relationships between membrane potential, calcium transient and tension in single barnacle muscle fibres. J Physiol. 1970;209:105–130. doi: 10.1113/jphysiol.1970.sp009158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao R, Lifshitz LM, Tuft RA, Bellve K, Fogarty KE, ZhuGe R. A close association of RyRs with highly dense clusters of Ca2+-activated Cl– channels underlies the activation of STICs by Ca2+ sparks in mouse airway smooth muscle. J Gen Physiol. 2008;132:145–160. doi: 10.1085/jgp.200709933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham CD, Bolton TB. Spontaneous transient outward currents in single visceral and vascular smooth muscle cells of the rabbit. J Physiol. 1986;381:385–406. doi: 10.1113/jphysiol.1986.sp016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham CD, Bolton TB, Lang RJ, Takewaki T. Calcium-activated potassium channels in single smooth muscle cells of rabbit jejunum and guinea-pig mesenteric artery. J Physiol. 1986;371:45–67. doi: 10.1113/jphysiol.1986.sp015961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Bolton TB, Gordienko DV, Povstyan OV, Harhun MI, Pucovsky V. Smooth muscle cells and interstitial cells of blood vessels. Cell Calcium. 2004;35:643–657. doi: 10.1016/j.ceca.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Brakenhoff GJ, van Spronsen EA, Van Der Voort HT, Nanninga N. Three-dimensional confocal fluorescence microscopy. Methods Cell Biol. 1989;30:379–398. doi: 10.1016/s0091-679x(08)60987-5. [DOI] [PubMed] [Google Scholar]
- Burdyga T, Wray S. Action potential refractory period in ureter smooth muscle is set by Ca sparks and BK channels. Nature. 2005;436:559–562. doi: 10.1038/nature03834. [DOI] [PubMed] [Google Scholar]
- Chalfie M. GFP: lighting up life. Proc Natl Acad Sci U S A. 2009;106:10073–10080. doi: 10.1073/pnas.0904061106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Curtis TM, Scholfield C, McGeown DJ. Calcium signaling in ocular arterioles. Crit Rev Eukaryot Gene Expr. 2007;17:1–12. doi: 10.1615/critreveukargeneexpr.v17.i1.10. [DOI] [PubMed] [Google Scholar]
- Curtis TM, Tumelty J, Dawicki J, Scholfield CN, McGeown JG. Identification and spatiotemporal characterization of spontaneous Ca2+ sparks and global Ca2+ oscillations in retinal arteriolar smooth muscle cells. Invest Ophthalmol Vis Sci. 2004;45:4409–4414. doi: 10.1167/iovs.04-0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eglen RM, Reisine T. Photoproteins: important new tools in drug discovery. Assay Drug Dev Technol. 2008;6:659–671. doi: 10.1089/adt.2008.160. [DOI] [PubMed] [Google Scholar]
- Filo RS, Bohr DF, Ruegg JC. Glycerinated skeletal and smooth muscle: calcium and magnesium dependence. Science. 1965;147:1581–1583. doi: 10.1126/science.147.3665.1581. [DOI] [PubMed] [Google Scholar]
- Friel DD. Calcium oscillations in neurons. Ciba Found Symp. 1995;188:210–223. doi: 10.1002/9780470514696.ch12. discussion 223–234. [DOI] [PubMed] [Google Scholar]
- Galinska-Rakoczy A, Engel P, Xu C, Jung H, Craig R, Tobacman LS, Lehman W. Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J Mol Biol. 2008;379:929–935. doi: 10.1016/j.jmb.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Heppner TJ, Herrera GM, Bonev AD, Hill-Eubanks D, Nelson MT. Ca2+ sparks and KCa channels: novel mechanisms to relax urinary bladder smooth muscle. Adv Exp Med Biol. 2003;539:347–357. doi: 10.1007/978-1-4419-8889-8_26. [DOI] [PubMed] [Google Scholar]
- Huxley AF. Cross-bridge action: present views, prospects, and unknowns. J Biomech. 2000;33:1189–1195. doi: 10.1016/s0021-9290(00)00060-9. [DOI] [PubMed] [Google Scholar]
- Iino M, Kasai H, Yamazawa T. Visualization of neural control of intracellular Ca2+ concentration in single vascular smooth muscle cells in situ. EMBO J. 1994;13:5026–5031. doi: 10.1002/j.1460-2075.1994.tb06831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Ikebe M, Kargacin GJ, Hartshorne DJ, Kemp BE, Fay FS. Effects of modulators of myosin light-chain kinase activity in single smooth muscle cells. Nature. 1989;338:164–167. doi: 10.1038/338164a0. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Ion channels and vascular tone. Hypertension. 2000;35:173–178. doi: 10.1161/01.hyp.35.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries O, McGahon MK, Bankhead P, Lozano MM, Scholfield CN, Curtis TM, McGeown JG. cAMP/PKA-dependent increases in Ca sparks, oscillations and SR Ca stores in retinal arteriolar myocytes after exposure to vasopressin. Invest Ophthalmol Vis Sci. 2010;51:1591–1598. doi: 10.1167/iovs.09-4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlikoff MI. Genetically encoded Ca2+ indicators: using genetics and molecular design to understand complex physiology. J Physiol. 2007;578:55–67. doi: 10.1113/jphysiol.2006.120212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kresge N, Simoni RD, Hill RL. The chemistry of fluorescent indicators: the work of Roger Y. Tsien. J Biol Chem. 2006;281:e29–e31. [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl– conductance in smooth muscle. Am J Physiol Cell Physiol. 1996;271:C435–C454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Lechleiter J, Girard S, Peralta E, Clapham D. Spiral calcium wave propagation and annihilation in Xenopus laevis oocytes. Science. 1991;252:123–126. doi: 10.1126/science.2011747. [DOI] [PubMed] [Google Scholar]
- Lee CO. Ionic activities in cardiac muscle cells and application of ion-selective microelectrodes. Am J Physiol Heart Circ Physiol. 1981;241:H459–H478. doi: 10.1152/ajpheart.1981.241.4.H459. [DOI] [PubMed] [Google Scholar]
- McCarron JG, McGeown JG, Reardon S, Ikebe M, Fay FS, Walsh JV., Jr Calcium-dependent enhancement of calcium current in smooth muscle by calmodulin-dependent protein kinase II. Nature. 1992;357:74–77. doi: 10.1038/357074a0. [DOI] [PubMed] [Google Scholar]
- McGahon MK, Dash DP, Arora A, Wall N, Dawicki J, Simpson DA, Scholfield CN, McGeown JG, Curtis TM. Diabetes downregulates large-conductance Ca2+-activated potassium β1 channel subunit in retinal arteriolar smooth muscle. Circ Res. 2007;100:703–711. doi: 10.1161/01.RES.0000260182.36481.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGahon MK, Needham MA, Scholfield CN, McGeown JG, Curtis TM. Ca2+-activated Cl– current in retinal arteriolar smooth muscle. Invest Ophthalmol Vis Sci. 2009;50:364–371. doi: 10.1167/iovs.08-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DJ. Sydney Ringer; physiological saline, calcium and the contraction of the heart. J Physiol. 2004;555:585–587. doi: 10.1113/jphysiol.2004.060731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Niggli E, Lederer WJ. Real-time confocal microscopy and calcium measurements in heart muscle cells: towards the development of a fluorescence microscope with high temporal and spatial resolution. Cell Calcium. 1990;11:121–130. doi: 10.1016/0143-4160(90)90065-3. [DOI] [PubMed] [Google Scholar]
- Perez GJ, Bonev AD, Nelson MT. Micromolar Ca2+ from sparks activates Ca2+-sensitive K+ channels in rat cerebral artery smooth muscle. Am J Physiol Cell Physiol. 2001;281:C1769–C1775. doi: 10.1152/ajpcell.2001.281.6.C1769. [DOI] [PubMed] [Google Scholar]
- Perez JF, Sanderson MJ. The frequency of calcium oscillations induced by 5-HT, ACH, and KCl determine the contraction of smooth muscle cells of intrapulmonary bronchioles. J Gen Physiol. 2005a;125:535–553. doi: 10.1085/jgp.200409216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JF, Sanderson MJ. The contraction of smooth muscle cells of intrapulmonary arterioles is determined by the frequency of Ca2+ oscillations induced by 5-HT and KCl. J Gen Physiol. 2005b;125:555–567. doi: 10.1085/jgp.200409217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 1992;111:229–233. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- Ringer S. A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J Physiol. 1883;4:29–42.3. doi: 10.1113/jphysiol.1883.sp000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson MJ, Bai Y, Perez-Zoghbi J. Ca2+ oscillations regulate contraction of intrapulmonary smooth muscle cells. Adv Exp Med Biol. 2010;661:77–96. doi: 10.1007/978-1-60761-500-2_5. [DOI] [PubMed] [Google Scholar]
- Scholfield CN, Curtis TM. Heterogeneity in cytosolic calcium regulation among different microvascular smooth muscle cells of the rat retina. Microvasc Res. 2000;59:233–242. doi: 10.1006/mvre.1999.2227. [DOI] [PubMed] [Google Scholar]
- Shimomura O. Discovery of green fluorescent protein (GFP) (Nobel Lecture) Angew Chem Int Ed Engl. 2009;48:5590–5602. doi: 10.1002/anie.200902240. [DOI] [PubMed] [Google Scholar]
- Shimomura O, Johnson FH, Saiga Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol. 1962;59:223–239. doi: 10.1002/jcp.1030590302. [DOI] [PubMed] [Google Scholar]
- Shimomura O, Johnson FH, Saiga Y. Microdetermination of calcium by aequorin luminescence. Science. 1963;140:1339–1340. doi: 10.1126/science.140.3573.1339. [DOI] [PubMed] [Google Scholar]
- Smith IF, Wiltgen SM, Shuai J, Parker I. Ca2+ puffs originate from preestablished stable clusters of inositol trisphosphate receptors. Sci Signal. 2009;2:ra77. doi: 10.1126/scisignal.2000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn P, Lawrie AM, Smith PM, Gallacher DV, Petersen OH. Ca2+ oscillations in pancreatic acinar cells: spatiotemporal relationships and functional implications. Cell Calcium. 1993;14:746–757. doi: 10.1016/0143-4160(93)90100-k. [DOI] [PubMed] [Google Scholar]
- Tsien RY. Nobel lecture: constructing and exploiting the fluorescent protein paintbox. Integr Biol (Camb) 2010;2:77–93. doi: 10.1039/b926500g. [DOI] [PubMed] [Google Scholar]
- Tumelty J, Scholfield N, Stewart M, Curtis T, McGeown G. Ca2+-sparks constitute elementary building blocks for global Ca2+-signals in myocytes of retinal arterioles. Cell Calcium. 2007;41:451–466. doi: 10.1016/j.ceca.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JV, Jr, Singer JJ. Voltage clamp of single freshly dissociated smooth muscle cells: current-voltage relationships for three currents. Pflugers Arch. 1981;390:207–210. doi: 10.1007/BF00590209. [DOI] [PubMed] [Google Scholar]
- Wamhoff BR, Bowles DK, Owens GK. Excitation–transcription coupling in arterial smooth muscle. Circ Res. 2006;98:868–878. doi: 10.1161/01.RES.0000216596.73005.3c. [DOI] [PubMed] [Google Scholar]
- Weber A, Winicur S. The role of calcium in the superprecipitation of actomyosin. J Biol Chem. 1961;236:3198–3202. [PubMed] [Google Scholar]
- Wray S, Burdyga T. Sarcoplasmic reticulum function in smooth muscle. Physiol Rev. 2010;90:113–178. doi: 10.1152/physrev.00018.2008. [DOI] [PubMed] [Google Scholar]
- ZhuGe R, Bao R, Fogarty KE, Lifshitz LM. Ca2+ sparks act as potent regulators of excitation-contraction coupling in airway smooth muscle. J Biol Chem. 2010;285:2203–2210. doi: 10.1074/jbc.M109.067546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZhuGe R, Sims SM, Tuft RA, Fogarty KE, Walsh JV., Jr Ca2+ sparks activate K+ and Cl– channels, resulting in spontaneous transient currents in guinea-pig tracheal myocytes. J Physiol. 1998;513:711–718. doi: 10.1111/j.1469-7793.1998.711ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer M. GFP: from jellyfish to the Nobel prize and beyond. Chem Soc Rev. 2009;38:2823–2832. doi: 10.1039/b904023d. [DOI] [PubMed] [Google Scholar]