

Abstract
Purpose of the review
Epigenetics investigates heritable changes in gene expression occurring without changes in DNA sequence. Several epigenetic mechanisms, including DNA methylation, histone modifications, and microRNA (miRNA) expression, can change genome function under exogenous influence. Here, we review current evidence indicating that epigenetic alterations mediate toxicity from environmental chemicals.
Recent findings
In-vitro, animal, and human investigations have identified several classes of environmental chemicals that modify epigenetic marks, including metals (cadmium, arsenic, nickel, chromium, methylmercury), peroxisome proliferators (trichloroethylene, dichloroacetic acid, trichloroacetic acid), air pollutants (particulate matter, black carbon, benzene), and endocrine-disrupting/reproductive toxicants (diethylstilbestrol, bisphenol A, persistent organic pollutants, dioxin). Most studies conducted so far have been centered on DNA methylation, whereas only a few investigations have studied environmental chemicals in relation to histone modications and miRNA.
Summary
For several exposures, it has been proved that chemicals can alter epigenetic marks and that the same or similar epigenetic alterations can be found in patients with the disease of concern or in diseased tissues. Future prospective investigations are needed to determine whether exposed subjects develop epigenetic alterations over time and, in turn, which such alterations increase the risk of disease. Also, further research is needed to determine whether environmental epigenetic changes are transmitted transgenerationally.
Keywords: Environment, Epigenetics, DNA Methylation, Histone modification, miRNA
Introduction
Identifying the effects of environmental exposures on human health is a major objective of life sciences and biomedical research. In environmental health, the recognition that exposures could produce DNA mutations represented a major landmark for risk assessment and prevention.[1] Consequently, chemical agents have been categorized according to their capability to alter the DNA sequence. Such information has been fundamental to determine environmental risks and shape current regulatory efforts for exposure reduction.[2] Recent evidence suggests that the molecular influence of the environment may extend well beyond the interaction with the DNA sequence.[3,4] Epigenetics is the study of heritable changes in gene expression that occur without changes in DNA sequence.[5] Epigenetic mechanisms are flexible genomic parameters that can change genome function under exogenous influence. Here, we review current evidence indicating that epigenetic mechanisms can mediate the toxicity of environmental chemicals.
• Overview of epigenetic mechanisms
The current field of epigenetics includes a number of mechanisms, including DNA methylation, histone modification, and microRNAs.[6,7] (Fig 1) DNA methylation is a covalent modification, heritable by somatic cells after cell division. 5-methyl-cytosine (5MeC) represents 2-5% of all cytosines in mammalian genomes and is found primarily on CpG dinucleotides.[8] DNA methylation is involved in regulating many cellular processes, including chromatin structure and remodeling, X-chromosome inactivation, genomic imprinting, chromosome stability, and gene transcription.[9,10] Generally, gene promoter hypermethylation is associated with decreased expression of the gene.[11] However, more than 90% of all genomic 5-methylcytosines are not directly related to gene function, as they lie on CpG dinucleotides located in transposable repetitive elements, also referred to as transposons.[12] Alu and LINE-1 (Long Interspersed Nuclear Element-1) are the most common and well-characterized transposable sequences and measurements of Alu and LINE-1 methylation have been used to estimate global genomic DNA methylation content.[12] Global hypomethylation, as well as hypomethylation of transposable repetitive elements, have been associated with reduced chromosomal stability and altered genome function.[13,14]
Figure 1. Potential mechanisms linking environmental exposures to epigenetic effects.
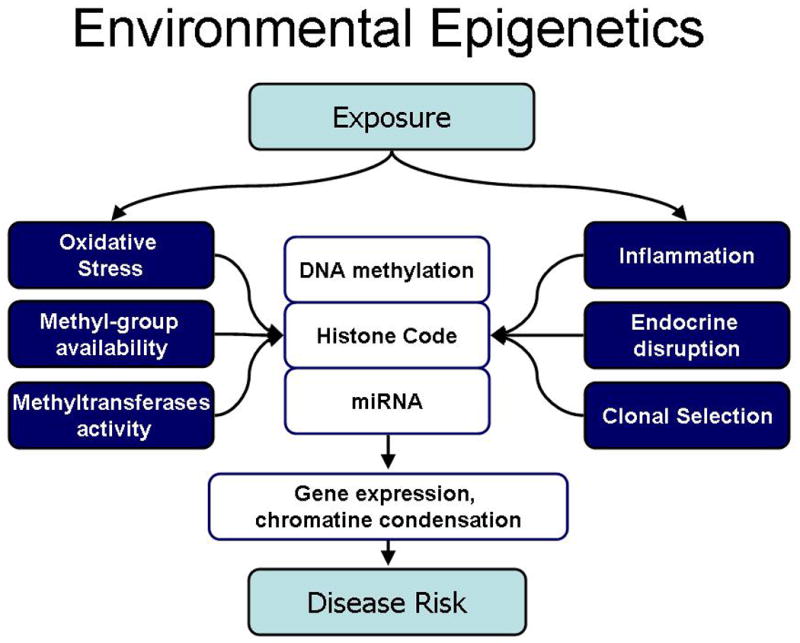
Environmental chemicals may modify multiple biological processes that affect epigenetic mechanisms, including DNA methylation, histone codes, and miRNA expression. These changes may, in turn, modify chromatin organization and condensation, gene expression, and affect disease risk.
Histones are globular proteins that undergo posttranslational modifications that alter their interaction with the DNA and other nuclear proteins.[15] H3 and H4 histones have long tails protruding from the nucleosome, which can be covalently modified by acetylation, methylation, ubiquitination, phosphorylation, sumoylation, citrullination, and ADP-ribosylation, and thus influence chromatin structure and gene expression (Fig 2).
Figure 2. Example of possible mechanisms of gene-regulation by histone modifications.
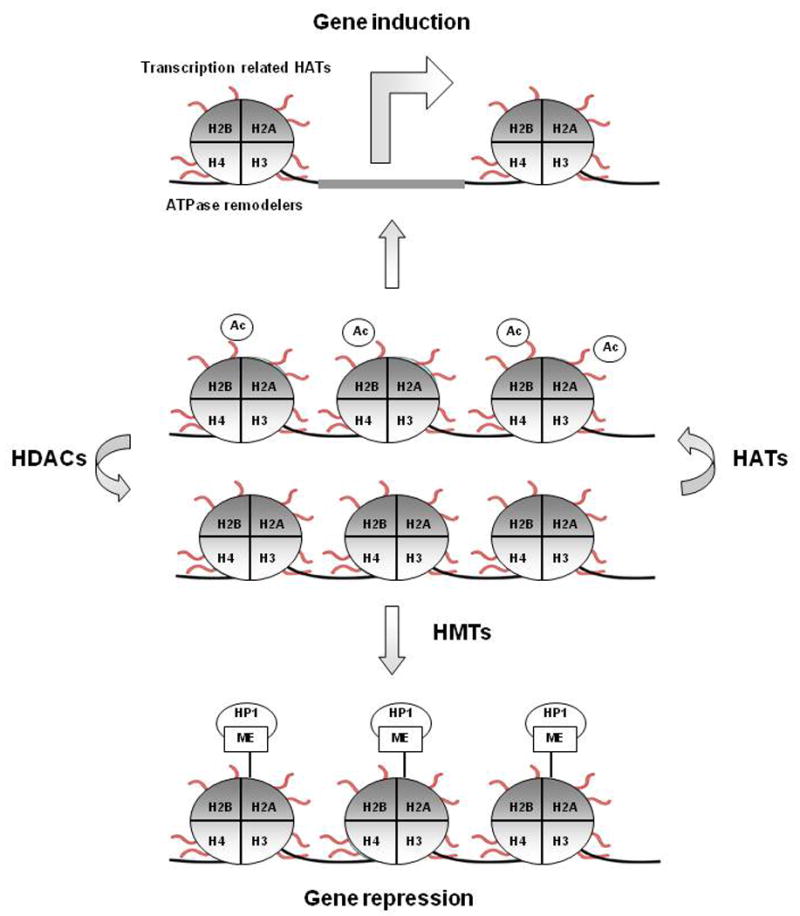
Modifications under histone methylases (HMTs), histone acetyltransferases (HATs) and histone deacetylases (HDACs) control alter gene expression. Modifications may generate a structure that contains bromo- and chromo-domains allowing the recruitment of ATP-dependent chromatin remodelling factors to open the chromatin. Effects of histone modifications on gene expression is dependent on the specific position of the chemical modification on the histone subunit.
microRNAs (miRNA) are single-stranded RNAs of ≈21–23 nucleotides in length that are transcribed from DNA but not translated into proteins (non-coding RNAs); Mature miRNAs are partially complementary to one or more messenger RNA (mRNA) molecules. miRNA main function is to down-regulate gene expression by interfering with mRNA functions (Fig 3).[16,17]
Figure 3. miRNA Processing and Activity.
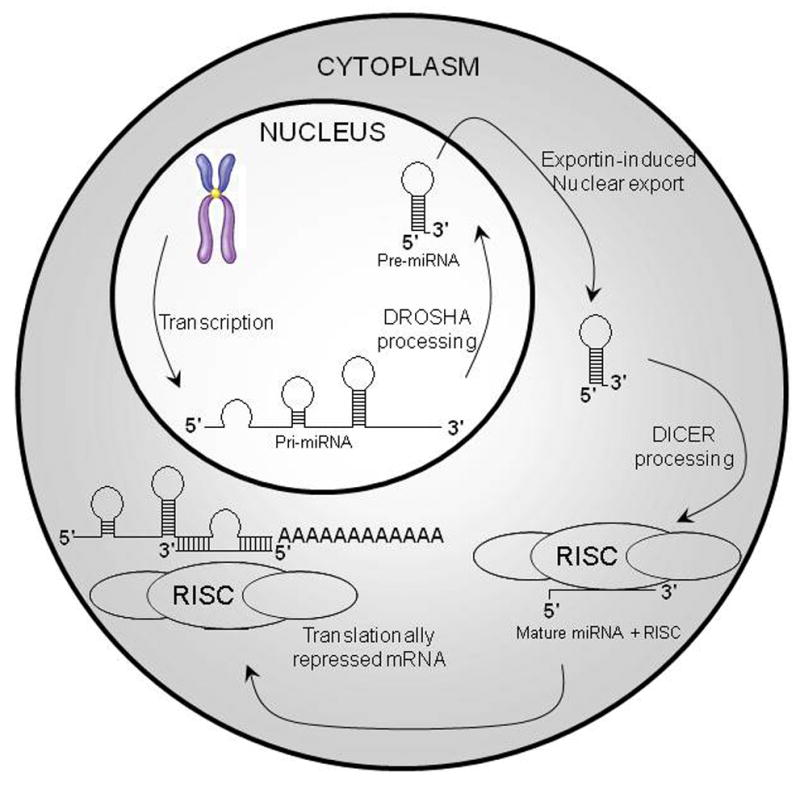
miRNAs are initially transcribed by RNA Polymerase II and expressed as a part of primary miRNAs (pri-miRNAs). The miRNA portion of the pri-miRNA transcript likely forms a hairpin with signals for dsRNA-specific nuclease cleavage. The dsRNA-specific ribonuclease Drosha digests the pri-miRNA in the nucleus to release hairpin that is called pre-miRNA (approximately 70 nt). Exportin-5 exports pre-miRNAs from the nucleus to the cytoplasm, where Dicer cleaves the pre-miRNA approximately 19 bp from the Drosha cut giving a mature miRNA. Each mature miRNA is complementary to a part of one or more messenger RNAs (mRNAs). The annealing of the miRNA to the mRNA(s) inhibits protein translation.
• Epigenetics and the environment
Some environmental factors have been linked to aberrant changes in epigenetic pathways both in experimental and epidemiological studies. In addition, epigenetic mechanisms may mediate specific mechanisms of toxicity and responses to certain chemicals. Whereas mechanisms of action of some of these agents are understood, for others the mode of action remains to be completely elucidated.[18] Because these epigenetic changes are small, potentially cumulative, and they may develop over time, it may be difficult to establish the cause-effect relationships among environmental factors, epigenetic changes and diseases. Below, we review the effects of individual environmental toxicants on epigenetic mechanisms, as derived from in-vitro, animal, or human studies, which we summarized in Table 1 (effects on DNA methylation), Table 2 (effects on histones modifications), and Table 3 (effects on miRNA).
Table 1. Effects of Environmental Chemicals on DNA Methylation.
| Exposure | +/-* | Genes | Type | Tissue | Ref. |
|---|---|---|---|---|---|
| Arsenic | ↓ | Global | Rat | Liver | Zhao, 1997 [30] |
| ↑ | P53 | In-vitro | A549 Cells | Mass and Wang, 1997 [34] | |
| ↑↓ | Multiple genes | In-vitro | Human Kidney Cells | Zhong and Mass, 2001 [33] | |
| ↑ | p16, p53 | Human | PBL | Chanda, 2006 [31] | |
| ↑ | Global | Human | PBL | Pilsner, 2007 and 2008 [38,39] | |
| ↑ | p16 | Human | PBL | Zhang, 2007 [85] | |
| Cadmium | ↓ | Global | In-vitro | Rat Liver Cells | Takiguchi, 2003 [29] |
| Nickel | ↑ | ATF-1, HIF-1, Rb | In-vitro | G12 Cell Line | Lee, 1995 [40] |
| ↑ | P16 | Mouse | Histiocytomas | Govindarajan, 2002 [46] | |
| Chromium | ↑ | P16 | Human | Lung | Kondo, 2006 [48] |
| Methylmercury | ↑ | BDNF | Mouse | Hippocampus | Onishchenko, 2008 [50] |
| TCE, DCA, TCA | ↑ | c-jun, c-myc | Mouse | Liver | Tao, 1999 [51] |
| Air pollution | ↓ | Global (ALU, LINE1) | Human | Buffy Coat | Tarantini, 2008 [57] |
| ↓ | iNOS | ||||
| Benzene | ↓ | Global (ALU, LINE1) | Human | Blood | Bollati, 2007 [63] |
| ↑ | P15 | ||||
| ↓ | MAGE | ||||
| Vinclozolin | ↑ | gene-specific | Ray | Testis | Anway, 2005 [83] |
| DES | ↓ | global | Mouse | Uterus | Li, 1997 [86] |
| BPA | ↓ | Agouti gene, CabpIAP | Mouse | Embryo | Dolinoy, 2007 [72] |
| POPs | ↓ | ALU, LINE | Human | Blood | Rusiecki, 2008 [73] |
Increase (+) or decrease (-) in DNA methylation
Table 2. Effects of Environmental Chemicals on Histones.
| Exposure | +/- | Modification | Type | Tissue | Ref. |
|---|---|---|---|---|---|
| ↓ | acetylation | ||||
| ↑ | H3K9 dimethylation | In-vitro | Liver, brain | Ke, 2006 [41] | |
| Nickel | ↑ | H2A and H2B monoubiquitination | |||
| ↓ | H4K12 acetylation | In-vitro | Yeast cells | Broday, 2000 [42] | |
| ↓ | H4K4 acetylation | In-vitro | Mammalian cells | ||
| ↑ | H3K9 mono- and dimethylation | In-vitro | G12 cell line | Chen, 2006 [43] | |
| ↓ | Acetylation of histone H2B | In-vitro | HAE and NRK cell lines | Golebiowski, 2005 [45] | |
| ↓↑ | H2B ubiquitination | In-vitro | HAEo- and HPL1D cell lines | Karaczyn, 2006 [44] | |
| Arsenic | ↑ | H3K9 dimethylation | |||
| ↓ | H3K27 trimethylation | In-vitro | A549 cell line | Zhou, 2008 [87] | |
| ↑ | H3K4 trimethylation |
Increase (+) or decrease (-) in histone modification
Table 3. Effects of Environmental Chemicals on microRNA.
| Exposure | +/- | Genes | Type | Tissue | Ref. |
|---|---|---|---|---|---|
| RDX | ↓ | tumor suppressing miRNAs | Mouse | Liver, brain | Zhang and Pan, 2008 [65] |
| ↑ | oncogenic miRNAs | ||||
| Arsenic | ↓↑ | Multiple miRNAs | In-vitro | Lymphoblastoid cells | Marsit, 2006 [18] |
Increase (+) or decrease (-) in microRNA expression
1. Metals
Several studies have established an association between DNA methylation and environmental metals, including nickel, cadmium, lead, and particularly arsenic.[19-21] Metal-induced oxidative stress may represent a unifying process to account for these findings across different metals.[22] Metals are known to increase production of reactive oxygen species (ROS) in a catalytic fashion via redox cycling.[23,24] Oxidative DNA damage can interfere with the ability of methyltransferases to interact with DNA,[25] thus resulting in a generalized altered methylation of cytosine residues at CpG sites.[26]
a. Cadmium
Cadmium is an established carcinogen that has very low mutagenicity.[27] Many possible mechanisms of cadmium carcinogenesis have been suggested and, among them, induction of ROS and alteration of DNA methylation seem to play a predominant biological role.[28] Takiguchi et al. showed that cadmium reduces genome methylation, inhibiting DNA methyltransferases in a noncompetitive manner. This finding is suggestive of interference in enzyme-DNA interaction, possibly through an interaction of cadmium with the methyltransferase DNA binding domain.[29] Cadmium can also inhibit DNA methylation in proto-oncogenes inducing oncogene expression and resulting in cell proliferation.[28,29]
b. Arsenic
Arsenic is an established carcinogen that lacks carcinogenicity in animal models. Inorganic arsenic is enzymatically methylated for detoxication, using up S-adenosyl-methionine (SAM) in the process. The observation that DNA methyltransferases also require SAM as their methyl donor suggested a role for DNA methylation in arsenic carcinogenesis and other arsenic-related effects. [30] In rat-liver epithelial cell lines treated with chronic low arsenic doses, Zhao et al. showed malignant transformation associated with depressed SAM levels, global DNA hypomethylation, and decreased DNA methyltrasferase activity.[30] Following this findings, several studies have shown that arsenic is associated with gene-specific hypermethylation [31-34], as well as global DNA hypomethylation [35-37]. An unexpected finding was recently reported in vivo, as a global dose-dependent hypermethylation of blood DNA was observed in Bangladeshi adults with chronic arsenic exposure.[38] This effect was modified by folate, suggesting that arsenic-induced increases in DNA methylation were dependent from methyl availability.[38] The same group, however, reported that lower blood DNA methylation was a risk factor for arsenic-induced skin lesions in a related Bangladeshi population.[39]
In a human study from India, significant DNA hypermethylation of p53 and p16 promoter regions was observed in blood DNA of subjects exposed to toxic level of arsenic compared to controls.[31] In this study, hypermethylation showed a dose-response relationship with arsenic measured in drinking water.
Arsenic toxicity has been recently related to changes in miRNA expression. Marsit et al. showed alterations in miRNA profiles of human lymphoblastoid cells grown under sodium arsenite treatment.[18] Interestingly, Arsenic altered expression of specific miRNAs that were involved in one-carbon metabolism.[18]
c. Nickel
The mechanisms underlying nickel health-related effects, including carcinogenicity and cardiorespiratory disease, are still largely unknown. It has been proposed that nickel may replace magnesium in DNA interactions, enhance chromatin condensation, and trigger de novo DNA methylation.[40] In chinese hamster G12 cells transfected with E.Coli gtp gene, Lee et al. demonstrated nickel-induced hypermethylation leading to the inactivation of the expression of the transfected gene.[40]
Several studies have shown that nickel affects histone modifications. Exposure to soluble NiCl2 has been shown to reduce histone acetylation, increase demethylation of H3K9, and increase monoubiquitination of H2A and H2B in-vitro.[41] Broday et al. studied nickel effects, at nontoxic levels, on yeast and mammalian cells and found a decrease in histone H4 acetylation, affecting only lysine 12 in mammalian cells and all of the four H4 lysines in yeasts.[42]
Nickel ion exposure has been shown to increase global H3K9 mono- and dimethylation, both of which have been associated with increased DNA methylation and long-term gene silencing. Nickel ions also interfere with the removal of histone methylation in vivo and directly decrease the activity of a Fe(II)-2-oxoglutarate-dependent histone H2K9 demethylase in nuclear extract in vitro.[43] In human lung cells exposed to soluble nickel compounds, three major changes in histone modifications have been observed: i) loss of acetylation of H2A, H2B, H3 and H4; ii) increased H3K9 dimethylation; iii) increased ubiquitinylation of H2A and H2B.[41-46]
It has been proposed that the binding of Ni2+ is able to promote a secondary structure with organized side-chain orientation on the N-terminal tail of histone H4. Acetylation of lysine 12 and 16 in yeast exposed to nickel was more robustly affected than lysine 5 and 8. Nickel binding to histidine 18 in histone H4 may be accountable for this effect, acting as an anchoring binding site for metal ions.[47]
d. Chromium
Kondo et al.[48] investigated p16 methylation using a methylation-specific PCR method in 30 lung cancer cases associated with chromate exposure and 38 non-chromate lung cancers. A variety of genetic changes in lung cancers from chromate-exposed subjects is known, but the epigenetic effects of chromium are still poorly understood. Kondo et al showed that chromate exposure influenced p16 hypermethylation measured in lung cancer tissues, compared to tissues from non-chromate lung cancer.[48] Chromium has been shown to reduce in-vitro H3 phosphorilation and trimethylation, as well as various acetylation marks in H3 and H4.[49]
e. Methylmercury
Methylmercury is an environmental contaminant and a potential neurotoxic agent that may be present at high levels in sea-food. Perinatal exposure to methylmercury causes persistent changes in learning and motivational behavior in mice. Developmental exposure to low levels of methylmercury induces epigenetic suppression of BDNF (Brain Derived Neurotrophic Factor) gene expression in the hippocampus and predisposes mice to depression.[50]
2. Trichloroethylene (TCE), dichloroacetic acid (DCA), and trichloroacetic acid (TCA)
Trichloroethylene (TCE), dichloroacetic acid (DCA), and trichloroacetic acid (TCA) are environmental contaminants that are peroxisome proliferators and carcinogenic in mouse liver. Decreased methylation in the promoter regions of the c-jun and c-myc genes and increased levels of their mRNAs and proteins were found in livers of mice exposed to TCE, DCA, and TCA. Methionine supplement prevented both the decreased methylation and the increased levels of the mRNAs and proteins of the two protooncogenes.[51] This work supported the hypothesis that these carcinogens may act by depleting the availability of SAM, whereas methionine would prevent DNA hypomethylation by maintaining adequate SAM levels.[51]
3. Air pollution
Exposure to air pollution, particularly to particulate matter (PM), has been associated with increased morbidity and mortality from cardiorespiratory disease, as well as with lung cancer risk.[52-56] In a human study, we recently investigated the effects of particulate matter (PM) exposure on global (estimated through Alu and LINE-1 repeated elements) and gene-specific methylation in workers of a steel plant with well-characterized exposure to PM with aerodynamic diameters <10 μm (PM10). iNOS (inducible Nitric Oxide Synthase) promoter methylation was significantly lower in post-exposure blood samples compared to baseline.[57] Long-term exposure to PM10 was negatively associated with methylation in both Alu and LINE-1. In a recent investigation, we showed that exposure to black carbon (BC), a marker of traffic particles, was also associated with decreased DNA methylation in LINE-1, measured in 1,097 blood DNA samples from the Normative Aging Study (NAS), a repeated measure investigation of elderly men in the Boston area. As global DNA hypomethylation has been found in patients with cancer [58] or cardiovascular disease,[59] such changes may reproduce epigenetic processes related to disease development and represent mechanisms by which particulate air pollution affects human health.[60]
In an animal study, Yauk et al. showed that sperm DNA of mice exposed to steel plant air was hypermethylated compared to control animals and this change persisted following removal from the environmental exposure.[61] This finding calls for further research to determine whether air pollutants produce DNA methylation changes that are transmitted transgenerationally.
4. Benzene
In a recent investigation, we investigated whether DNA methylation changes are induced by low-benzene exposure in peripheral blood DNA of gasoline station attendants and traffic police officers. High-level exposure to benzene has been associated with increased risk of acute myelogenous leukemia (AML), [62] which is characterized by aberrant global hypomethylation and gene-specific hypermethylation/hypomethylation. In our study, airborne benzene exposure was associated with a significant reduction in global methylation measured in LINE-1 and Alu. Airborne benzene was also associated with hypermethylation in p15 and hypomethylation of the MAGE-1 cancer-antigen gene.[63] This findings show that low-level benzene exposure may induce altered DNA methylation reproducing the aberrant epigenetic patterns found in malignant cells.
5. Hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX)
RDX is a common environmental pollutant resulting from military and civil activities that has been associated with neurotoxicity, immunotoxicity and increased risk of cancer.[64] Zhang and Pan evaluated the role of RDX in modifying miRNA expression in mouse liver and brain, as measured by miRNA expression microarrays.[65] Several miRNAs were found to be differentially expressed in exposed mice, with specific miRNA expression profiles in gene pathways related to cancer, toxicant-metabolizing enzymes, and neurotoxicity.[65]
6. Endocrine-disrupting Chemicals and Reproductive Toxicants
Developing organisms are extremely sensitive to perturbation by endocrine-disrupting chemicals with ormone-like activity. In mammal germ cells and preimplantation embryos, DNA methylation undergoes two distinct cycles of demethylation/remethylation in which methylation patterns are reprogrammed genome wide and cells with a broad developmental potential are generated.[10] Available evidence from animal models indicate that exposure to xenobiotics during critical periods of mammalian development may induce persistent and heritable changes of epigenetic states.
a. Diethylstilbestrol (DES)
Diethylstilbestrol (DES) is a developmental stage-specific non-genotoxic carcinogen that was used in the past to prevent miscarriages in pregnant women. [66] In mice models, pre- and neonatal DES exposure causes a wide range of gene expression changes. Exogenous estrogen treatment results in persistent expression of certain genes, including lactoferrin, epidermal growth factor, and protooncogenes such as c-fos, c-jun and c-myc.[67-70] Xie et al. have provided data supporting an indirect relationship between exogenous estrogen and methylation changes, showing that estrogen inhibited catechol-O-methyltransferase (COMT) gene transcription.[71] COMT is an ubiquitous enzyme catalyzing the transfer of the methyl groups from SAM to one of the hydroxyl groups of catechols in presence of Mg2+. Inhibition of this enzyme results in inhibition of the methylation process.[71]
b. Bisphenol A (BPA)
BPA is a chemical with estrogenic properties that is present in many commonly used products including food and beverage containers, baby bottles, and dental composites. Dolinoy et al [72] used a rat exposure model to show that maternal exposure to BPA modified methylation of the metastable loci Avy and CapbIAP. Interestingly, this effect on DNA methylation and the associated change in coat color of the exposed animals were prevented by maternal dietary supplementation with a source of methyl group such as folic acid or with the phytoestrogen genistein. [72]
c. Persistent Organic Pollutants (POPs)
Rusiecki et al. evaluated the relationship between plasma POP concentrations and blood global DNA methylation, estimated in Alu repeated elements, in 70 Greenlandic Inuit, a population presenting some of the highest reported levels of POPs worldwide. In this work, a significant inverse linear relationships was found for DDT, DDE, β-BHC, oxychlordane, α-chlordane, mirex, several PCBs, and sum of all POPs. [73]
d. Dioxin
Dioxin toxicity is mediated by the Aryl-hydrocarbon Receptor (AhR) (reviewed in [74-76]) and appears to require altered transcription of target genes.[75] Moffat et al. hypothesized that miRNAs might be responsible for this mRNA downregulation in dioxin/AhR-related pathways but, using two different miRNA array platforms and Real time PCR, they just found a few changes of small entity in miRNA levels, thus indicating a limited role of miRNA in dioxin toxicity.[77]
• Potential roles of environmental epigenetic effects in determining trans-generational risks and fetal origins of diseases
In animal studies, several chemicals including alloxan,[78] cyclophosphamide,[79] orthoaminoasotoluol,[80], benzopyrene,[81], diethylstilbestrol (DES),[82] and vinclozolin[83] have been reported to induce transgenerational phenotypic effects. Transgenerational transmission of chemically-induced epigenetic changes have been suggested as a potential mechanisms for these effects. Anway et al.[83] showed that gestational exposure of female rats to the endocrine disruptor vinclozolin at the time of gonadal sex determination caused a variety of abnormalities in the offspring that were then transmitted down the male line for at least three generations. The high incidence of the defects (approximately 90% of all males in all generations) and the absence of abnormalities when passed down the female line suggested gametic epigenetic inheritance. In this study, altered DNA methylation in two candidate genes was seen in sperm from vinclozolin-exposed males, and these abnormal methylation patterns were inherited. These results indicate that exposure of germ cells, possibly at a specific developmental stage, is necessary to produce heritable epigenetic changes. In addition, epigenetic mechanisms may underlie the effects of in utero and early life exposures on adult health, as in-utero/early-life exposures to epigenetically-active chemicals may produce health effects later in life even independently of environmental risk factors in adults.[84] As reported in the sections above, most of the studies on epigenetic effects of environmental chemicals have shown changes in DNA methylation, histone modifications or microRNA in somatic cells of adult individuals. Whether epigenetic changes observed in somatic cells are correlated with germline epigenetic changes is uncertain. Environmentally-induced epigenetic somatic alterations may be sufficient to cause anomalies in biological functions, but these changes are not heritable per se and may not be associated to any transgenerational risk.
Conclusions
In the last few years, several investigations have examined the relation between exposure to environmental chemicals and epigenetics, and identified several toxicants that modify epigenetic marks. Most of the studies conducted so far have been centered on DNA methylation, whereas only a few recent investigations have studied the effects of environmental chemicals on histone modications and miRNA. Epigenetics holds substantial potential for developing biological markers to predict which exposures would put exposed subjects at risk and which individuals will be more susceptible to develop disease. In human studies, this will require the use of laboratory methods with enhanced precision and sensitivity, so that epigenetic changes can be detected as early as possible and well ahead of disease diagnosis. It is worth noting that several human studies have investigated the effects of environmental exposures on tissues, such as blood, that are easy to obtain, but that, as effects of environmental chemicals may be tissue or even cell specific, do not necessarily represent epigenetic patterns in the target tissues. For several exposures, however, it has been proved that chemicals can alter epigenetic marks and that the same or similar epigenetic alterations can be found in patients with the disease of concern or in diseased tissues. At the current stage, the missing link is to determine whether environmentally-induced epigenetic alterations are part of the causative pathways that leads to the disease development. Future prospective investigations are needed to determine whether exposed subjects develop epigenetic alterations over time and, in turn, whether such alterations increase the risk of disease.
References
- 1.Wogan GN. Molecular epidemiology in cancer risk assessment and prevention: recent progress and avenues for future research. Environ Health Perspect. 1992;98:167–178. doi: 10.1289/ehp.9298167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weisburger JH, Williams GM. The distinct health risk analyses required for genotoxic carcinogens and promoting agents. Environ Health Perspect. 1983;50:233–245. doi: 10.1289/ehp.8350233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reamon-Buettner SM, Mutschler V, Borlak J. The next innovation cycle in toxicogenomics: environmental epigenetics. Mutat Res. 2008;659:158–165. doi: 10.1016/j.mrrev.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286:481–486. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- 6.Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61:24R–29R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- 7.Allis CD, Jenuwein T, Reinberg D. Epigenetics edn. Cold Spring Harbor Laboratory Press; 2007. [Google Scholar]
- 8.Millar D, Holliday R, Grigg G. Five not four: History and significance of the fifth base. In: Beck S, Olek A, editors. The Epigenome, Molecular Hide and Seek. Wiley-VCH Verlag GmbH Co. KGaA; 2003. pp. 3–20. [Google Scholar]
- 9.Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- 10.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 11.Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108:439–451. doi: 10.1016/s0092-8674(02)00655-4. [DOI] [PubMed] [Google Scholar]
- 12.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulz WA. L1 retrotransposons in human cancers. J Biomed Biotechnol. 2006;2006:83672. doi: 10.1155/JBB/2006/83672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 15.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Jackson RJ, Standart N. How do microRNAs regulate gene expression? Sci STKE. 2007;2007:re1. doi: 10.1126/stke.3672007re1. [DOI] [PubMed] [Google Scholar]
- 17.Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–126. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 18.Marsit CJ, Eddy K, Kelsey KT. MicroRNA responses to cellular stress. Cancer Res. 2006;66:10843–10848. doi: 10.1158/0008-5472.CAN-06-1894. [DOI] [PubMed] [Google Scholar]
- 19.McVeigh GE, Allen PB, Morgan DR, Hanratty CG, Silke B. Nitric oxide modulation of blood vessel tone identified by arterial waveform analysis. Clin Sci (Lond) 2001;100:387–393. [PubMed] [Google Scholar]
- 20.Dolinoy DC, Weidman JR, Jirtle RL. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod Toxicol. 2007;23:297–307. doi: 10.1016/j.reprotox.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Bleich S, Lenz B, Ziegenbein M, Beutler S, Frieling H, Kornhuber J, Bonsch D. Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcohol Clin Exp Res. 2006;30:587–591. doi: 10.1111/j.1530-0277.2006.00068.x. [DOI] [PubMed] [Google Scholar]
- 22.Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 23.Fowler BA, Whittaker MH, Lipsky M, Wang G, Chen XQ. Oxidative stress induced by lead, cadmium and arsenic mixtures: 30-day, 90-day, and 180-day drinking water studies in rats: an overview. Biometals. 2004;17:567–568. doi: 10.1023/b:biom.0000045740.52182.9d. [DOI] [PubMed] [Google Scholar]
- 24.Shen XM, Dryhurst G. Iron- and manganese-catalyzed autoxidation of dopamine in the presence of L-cysteine: possible insights into iron- and manganese-mediated dopaminergic neurotoxicity. Chem Res Toxicol. 1998;11:824–837. doi: 10.1021/tx980036t. [DOI] [PubMed] [Google Scholar]
- 25.Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32:4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turk PW, Laayoun A, Smith SS, Weitzman SA. DNA adduct 8-hydroxyl-2′-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis. 1995;16:1253–1255. doi: 10.1093/carcin/16.5.1253. [DOI] [PubMed] [Google Scholar]
- 27.Filipic M, Hei TK. Mutagenicity of cadmium in mammalian cells: implication of oxidative DNA damage. Mutat Res. 2004;546:81–91. doi: 10.1016/j.mrfmmm.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Huang D, Zhang Y, Qi Y, Chen C, Ji W. Global DNA hypomethylation, rather than reactive oxygen species (ROS), a potential facilitator of cadmium-stimulated K562 cell proliferation. Toxicol Lett. 2008;179:43–47. doi: 10.1016/j.toxlet.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res. 2003;286:355–365. doi: 10.1016/s0014-4827(03)00062-4. [DOI] [PubMed] [Google Scholar]
- 30.Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc Natl Acad Sci U S A. 1997;94:10907–10912. doi: 10.1073/pnas.94.20.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chanda S, Dasgupta UB, Guhamazumder D, Gupta M, Chaudhuri U, Lahiri S, Das S, Ghosh N, Chatterjee D. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol Sci. 2006;89:431–437. doi: 10.1093/toxsci/kfj030. [DOI] [PubMed] [Google Scholar]
- 32.Chen H, Liu J, Zhao CQ, Diwan BA, Merrick BA, Waalkes MP. Association of c-myc overexpression and hyperproliferation with arsenite-induced malignant transformation. Toxicol Appl Pharmacol. 2001;175:260–268. doi: 10.1006/taap.2001.9253. [DOI] [PubMed] [Google Scholar]
- 33.Zhong CX, Mass MJ. Both hypomethylation and hypermethylation of DNA associated with arsenite exposure in cultures of human cells identified by methylation-sensitive arbitrarily-primed PCR. Toxicol Lett. 2001;122:223–234. doi: 10.1016/s0378-4274(01)00365-4. [DOI] [PubMed] [Google Scholar]
- 34.Mass MJ, Wang L. Arsenic alters cytosine methylation patterns of the promoter of the tumor suppressor gene p53 in human lung cells: a model for a mechanism of carcinogenesis. Mutat Res. 1997;386:263–277. doi: 10.1016/s1383-5742(97)00008-2. [DOI] [PubMed] [Google Scholar]
- 35.Reichard JF, Schnekenburger M, Puga A. Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem Biophys Res Commun. 2007;352:188–192. doi: 10.1016/j.bbrc.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sciandrello G, Caradonna F, Mauro M, Barbata G. Arsenic-induced DNA hypomethylation affects chromosomal instability in mammalian cells. Carcinogenesis. 2004;25:413–417. doi: 10.1093/carcin/bgh029. [DOI] [PubMed] [Google Scholar]
- 37.Chen H, Li S, Liu J, Diwan BA, Barrett JC, Waalkes MP. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: implications for arsenic hepatocarcinogenesis. Carcinogenesis. 2004;25:1779–1786. doi: 10.1093/carcin/bgh161. [DOI] [PubMed] [Google Scholar]
- 38.Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. Genomic methylation of peripheral blood leukocyte DNA: influences of arsenic and folate in Bangladeshi adults. Am J Clin Nutr. 2007;86:1179–1186. doi: 10.1093/ajcn/86.4.1179. [DOI] [PubMed] [Google Scholar]
- 39.Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. Folate deficiency, hyperhomocysteinemia, low urinary creatinine and hypomethylation of leukocyte DNA are risk factors for Arsenic-induced skin lesions. Environ Health Perspect. 2008 doi: 10.1289/ehp.11872. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee YW, Klein CB, Kargacin B, Salnikow K, Kitahara J, Dowjat K, Zhitkovich A, Christie NT, Costa M. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: a new model for epigenetic carcinogens. Mol Cell Biol. 1995;15:2547–2557. doi: 10.1128/mcb.15.5.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ke Q, Davidson T, Chen H, Kluz T, Costa M. Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis. 2006;27:1481–1488. doi: 10.1093/carcin/bgl004. [DOI] [PubMed] [Google Scholar]
- 42.Broday L, Peng W, Kuo MH, Salnikow K, Zoroddu M, Costa M. Nickel compounds are novel inhibitors of histone H4 acetylation. Cancer Res. 2000;60:238–241. [PubMed] [Google Scholar]
- 43.Chen H, Ke Q, Kluz T, Yan Y, Costa M. Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Mol Cell Biol. 2006;26:3728–3737. doi: 10.1128/MCB.26.10.3728-3737.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karaczyn AA, Golebiowski F, Kasprzak KS. Ni(II) affects ubiquitination of core histones H2B and H2A. Exp Cell Res. 2006;312:3252–3259. doi: 10.1016/j.yexcr.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 45.Golebiowski F, Kasprzak KS. Inhibition of core histones acetylation by carcinogenic nickel(II) Mol Cell Biochem. 2005;279:133–139. doi: 10.1007/s11010-005-8285-1. [DOI] [PubMed] [Google Scholar]
- 46.Govindarajan B, Klafter R, Miller MS, Mansur C, Mizesko M, Bai X, LaMontagne K, Jr, Arbiser JL. Reactive oxygen-induced carcinogenesis causes hypermethylation of p16(Ink4a) and activation of MAP kinase. Mol Med. 2002;8:1–8. [PMC free article] [PubMed] [Google Scholar]
- 47.Zoroddu MA, Schinocca L, Kowalik-Jankowska T, Kozlowski H, Salnikow K, Costa M. Molecular mechanisms in nickel carcinogenesis: modeling Ni(II) binding site in histone H4. Environ Health Perspect. 2002;110 5:719–723. doi: 10.1289/ehp.02110s5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kondo K, Takahashi Y, Hirose Y, Nagao T, Tsuyuguchi M, Hashimoto M, Ochiai A, Monden Y, Tangoku A. The reduced expression and aberrant methylation of p16(INK4a) in chromate workers with lung cancer. Lung Cancer. 2006;53:295–302. doi: 10.1016/j.lungcan.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 49.Schnekenburger M, Talaska G, Puga A. Chromium cross-links histone deacetylase 1-DNA methyltransferase 1 complexes to chromatin, inhibiting histone-remodeling marks critical for transcriptional activation. Mol Cell Biol. 2007;27:7089–7101. doi: 10.1128/MCB.00838-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Onishchenko N, Karpova N, Sabri F, Castren E, Ceccatelli S. Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. J Neurochem. 2008;106:1378–1387. doi: 10.1111/j.1471-4159.2008.05484.x. [DOI] [PubMed] [Google Scholar]
- 51.Tao L, Ge R, Xie M, Kramer PM, Pereira MA. Effect of trichloroethylene on DNA methylation and expression of early-intermediate protooncogenes in the liver of B6C3F1 mice. J Biochem Mol Toxicol. 1999;13:231–237. doi: 10.1002/(sici)1099-0461(1999)13:5<231::aid-jbt2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 52.Brook RD, Franklin B, Cascio W, Hong Y, Howard G, Lipsett M, Luepker R, Mittleman M, Samet J, Smith SC, Jr, et al. Air pollution and cardiovascular disease: a statement for healthcare professionals from the Expert Panel on Population and Prevention Science of the American Heart Association. Circulation. 2004;109:2655–2671. doi: 10.1161/01.CIR.0000128587.30041.C8. [DOI] [PubMed] [Google Scholar]
- 53.Peters A. Particulate matter and heart disease: evidence from epidemiological studies. Toxicol Appl Pharmacol. 2005;207:477–482. doi: 10.1016/j.taap.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 54.Samet JM, Dominici F, Curriero FC, Coursac I, Zeger SL. Fine particulate air pollution and mortality in 20 U.S. cities, 1987-1994. N Engl J Med. 2000;343:1742–1749. doi: 10.1056/NEJM200012143432401. [DOI] [PubMed] [Google Scholar]
- 55.Vineis P, Husgafvel-Pursiainen K. Air pollution and cancer: biomarker studies in human populations. Carcinogenesis. 2005;26:1846–1855. doi: 10.1093/carcin/bgi216. [DOI] [PubMed] [Google Scholar]
- 56.Baccarelli A, Martinelli I, Zanobetti A, Grillo P, Hou LF, Bertazzi PA, Mannucci PM, Schwartz J. Exposure to particulate air pollution and risk of deep vein thrombosis. Arch Intern Med. 2008;168:920–927. doi: 10.1001/archinte.168.9.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, Cantone L, Rizzo G, Hou L, Schwartz J, et al. Effects of Particulate Matter on Genomic DNA Methylation Content and iNOS Promoter Methylation. Environ Health Perspect in press. 2008 doi: 10.1289/ehp.11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–5413. doi: 10.1038/sj.onc.1205651. [DOI] [PubMed] [Google Scholar]
- 59.Castro R, Rivera I, Struys EA, Jansen EE, Ravasco P, Camilo ME, Blom HJ, Jakobs C, Tavares de Almeida I. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin Chem. 2003;49:1292–1296. doi: 10.1373/49.8.1292. [DOI] [PubMed] [Google Scholar]
- 60.Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua A, Suh H, Zanobetti A, Sparrow D, Vokonas P, Schwartz J. Genomic DNA Methylation, Cardiovascular Disease, and Short-Term Exposure to Traffic Air Pollution. 2008 ISEE-ISEA Joint Conference; Pasadena, Cal ifornia USA. 2008. [Google Scholar]
- 61.Yauk C, Polyzos A, Rowan-Carroll A, Somers CM, Godschalk RW, Van Schooten FJ, Berndt ML, Pogribny IP, Koturbash I, Williams A, et al. Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location. Proc Natl Acad Sci U S A. 2008;105:605–610. doi: 10.1073/pnas.0705896105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Proceedings of the International Symposium on Recent Advances in Benzene Toxicity, Munich, Germany, 9-12 October 2004. Chem Biol Interact. 2005;153-154:1–270. [PubMed] [Google Scholar]
- 63.Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- 64.Jenkins TF, Hewitt AD, Grant CL, Thiboutot S, Ampleman G, Walsh ME, Ranney TA, Ramsey CA, Palazzo AJ, Pennington JC. Identity and distribution of residues of energetic compounds at army live-fire training ranges. Chemosphere. 2006;63:1280–1290. doi: 10.1016/j.chemosphere.2005.09.066. [DOI] [PubMed] [Google Scholar]
- 65.Zhang B, Pan X. RDX Induces Aberrant Expression of MicroRNAs in Mouse Brain and Liver. Environ Health Perspect in press. 2008 doi: 10.1289/ehp.11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. 1971;284:878–881. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- 67.Nelson KG, Sakai Y, Eitzman B, Steed T, McLachlan J. Exposure to diethylstilbestrol during a critical developmental period of the mouse reproductive tract leads to persistent induction of two estrogen-regulated genes. Cell Growth Differ. 1994;5:595–606. [PubMed] [Google Scholar]
- 68.Kamiya K, Sato T, Nishimura N, Goto Y, Kano K, Iguchi T. Expression of estrogen receptor and proto-oncogene messenger ribonucleic acids in reproductive tracts of neonatally diethylstilbestrol-exposed female mice with or without post-puberal estrogen administration. Exp Clin Endocrinol Diabetes. 1996;104:111–122. doi: 10.1055/s-0029-1211432. [DOI] [PubMed] [Google Scholar]
- 69.Yamashita S, Takayanagi A, Shimizu N. Effects of neonatal diethylstilbestrol exposure on c-fos and c-jun protooncogene expression in the mouse uterus. Histol Histopathol. 2001;16:131–140. doi: 10.14670/HH-16.131. [DOI] [PubMed] [Google Scholar]
- 70.Falck L, Forsberg JG. Immunohistochemical studies on the expression and estrogen dependency of EGF and its receptor and C-fos proto-oncogene in the uterus and vagina of normal and neonatally estrogen-treated mice. Anat Rec. 1996;245:459–471. doi: 10.1002/(SICI)1097-0185(199607)245:3<459::AID-AR2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 71.Xie S, Wang Z, Okano M, Nogami M, Li Y, He WW, Okumura K, Li E. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene. 1999;236:87–95. doi: 10.1016/s0378-1119(99)00252-8. [DOI] [PubMed] [Google Scholar]
- 72.Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC. Global DNA Hypomethylation Is Associated with High Serum-Persistent Organic Pollutants in Greenlandic Inuit. Environ Health Perspect. 2008;116:1547–1552. doi: 10.1289/ehp.11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Okey AB. An aryl hydrocarbon receptor odyssey to the shores of toxicology: the Deichmann Lecture, International Congress of Toxicology-XI. Toxicol Sci. 2007;98:5–38. doi: 10.1093/toxsci/kfm096. [DOI] [PubMed] [Google Scholar]
- 75.Bunger MK, Moran SM, Glover E, Thomae TL, Lahvis GP, Lin BC, Bradfield CA. Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J Biol Chem. 2003;278:17767–17774. doi: 10.1074/jbc.M209594200. [DOI] [PubMed] [Google Scholar]
- 76.Baccarelli A, Pesatori AC, Masten SA, Patterson DG, Jr, Needham LL, Mocarelli P, Caporaso NE, Consonni D, Grassman JA, Bertazzi PA, et al. Aryl-hydrocarbon receptor-dependent pathway and toxic effects of TCDD in humans: a population-based study in Seveso, Italy. Toxicol Lett. 2004;149:287–293. doi: 10.1016/j.toxlet.2003.12.062. [DOI] [PubMed] [Google Scholar]
- 77.Moffat ID, Boutros PC, Celius T, Linden J, Pohjanvirta R, Okey AB. microRNAs in adult rodent liver are refractory to dioxin treatment. Toxicol Sci. 2007;99:470–487. doi: 10.1093/toxsci/kfm189. [DOI] [PubMed] [Google Scholar]
- 78.Spergel G, Levy LJ, Goldner MG. Glucose intolerance in the progeny of rats treated with single subdiabetogenic dose of alloxan. Metabolism. 1971;20:401–413. doi: 10.1016/0026-0495(71)90102-8. [DOI] [PubMed] [Google Scholar]
- 79.Hales BF, Crosman K, Robaire B. Increased postimplantation loss and malformations among the F2 progeny of male rats chronically treated with cyclophosphamide. Teratology. 1992;45:671–678. doi: 10.1002/tera.1420450612. [DOI] [PubMed] [Google Scholar]
- 80.Popova NV. Transgenerational effect of orthoaminoasotoluol in mice. Cancer Lett. 1989;46:203–206. doi: 10.1016/0304-3835(89)90131-6. [DOI] [PubMed] [Google Scholar]
- 81.Csaba G, Inczefi-Gonda A. Transgenerational effect of a single neonatal benzpyrene treatment on the glucocorticoid receptor of the rat thymus. Hum Exp Toxicol. 1998;17:88–92. doi: 10.1177/096032719801700203. [DOI] [PubMed] [Google Scholar]
- 82.Newbold RR, Padilla-Banks E, Jefferson WN. Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology. 2006;147:S11–17. doi: 10.1210/en.2005-1164. [DOI] [PubMed] [Google Scholar]
- 83.Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in Utero and Early-Life Conditions on Adult Healt and Disease. New Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang AH, Bin HH, Pan XL, Xi XG. Analysis of p16 gene mutation, deletion and methylation in patients with arseniasis produced by indoor unventilated-stove coal usage in Guizhou, China. J Toxicol Environ Health A. 2007;70:970–975. doi: 10.1080/15287390701290808. [DOI] [PubMed] [Google Scholar]
- 86.Li S, Hursting SD, Davis BJ, McLachlan JA, Barrett JC. Environmental exposure, DNA methylation, and gene regulation: lessons from diethylstilbesterol-induced cancers. Ann N Y Acad Sci. 2003;983:161–169. doi: 10.1111/j.1749-6632.2003.tb05971.x. [DOI] [PubMed] [Google Scholar]
- 87.Zhou X, Sun H, Ellen TP, Chen H, Costa M. Arsenite alters global histone H3 methylation. Carcinogenesis. 2008;29:1831–1836. doi: 10.1093/carcin/bgn063. [DOI] [PMC free article] [PubMed] [Google Scholar]