

Abstract
We report a fluorescence-based assay for measuring the affinity of microtubule binding proteins for microtubules. The affinity of any fluorescently tagged protein for taxol-stabilized microtubules can be measured with this assay. We describe the assay and provide a detailed protocol. Using this assay we find that the affinity of the Dam1 complex for microtubules is decreased by the presence of free unpolymerized tubulin and is sensitive to the salt concentration in the binding buffer. These effects likely account for the previous differences in binding affinities reported.
Keywords: microtubule, microtubule binding protein, fluorescence assay
The biochemical analysis of microtubule-binding proteins has dramatically expanded in recent years as kinetochore proteins have been identified and produced in recombinant form. The kinetochore is the complex of proteins that connects chromosomes to the microtubules of the mitotic spindle. Kinetochores alter the dynamics of the spindle microtubules and maintain their attachment as the MTs polymerize and depolymerize under their grip. Models to account for this behavior have been developed but are difficult to test without knowing the affinity of the kinetochore proteins for MTs. Microtubule affinity has been measured by assays in which the amount of a MT binding protein co-sedimenting with MTs is quantified by SDS-PAGE and Coomassie blue staining or Western blots[1; 2; 3; 4; 5; 6]. These assays are unable to distinguish non-specific aggregation, from trapping by the microtubules, from true binding, any of which causes sedimentation of protein. We developed an assay that overcomes these difficulties and used it to measure the affinity of the Dam1 complex for microtubules[7]. The amount of GFP-labeled Dam1 complex bound to MTs was quantified by fluorescence microscopy. Using this assay, we measured a 30-fold higher affinity for microtubules than previously measured [6]. Here we characterize the assay and provide evidence of the increased sensitivity of our method over the current one widely used.
The assays were performed as described previously except as noted in the Figure legends [7]. A detailed protocol and video can be found at faculty.washington.edu/tdavis. Briefly, in a 250 μL assay, Dam1 complex (0–15 nM) was added to bovine brain taxol-stabilized MTs (2.5 nM, 1:41 Alexa-568-labeled tubulin/unlabeled tubulin) in BRB80 (80 mM PIPES buffer (pH 6.8), 1 mM EGTA, 1 mM MgCl2) containing 10 μM taxol. (Note, we started with PIPES in the acid form instead of K-PIPES to minimize the amount of K+ in the buffer.) The final buffer composition was 87.5% BRB80 and 12.5% Dam1 complex protein buffer (50 mM sodium phosphate buffer, pH 6.8, containing 350 mM NaCl). The mixture was incubated for 10 minutes at 21°C. The reaction was fixed by addition of 750 μL of 2% glutaraldehyde in BRB80 and incubated for 15 minutes at 21°C. A custom spacer (Ellard Instrumentation, Monroe, WA) was placed in a centrifuge tube (11 × 34 mm) and a polylysine coated coverslip (5 mm diameter) was placed on top of the spacer. A 1 mL cushion of 15% glycerol in Phosphate Buffered Saline (PBS) was added on top of the coverslip. An aliquot of the reaction mixture was layered onto the cushion. The MTs were pelleted onto the coverslip by centrifugation in a Beckman TLS55 rotor at 135,000 × g for 10 minutes at 21°C. The slides were imaged on a DeltaVision microscopy system (Applied Precision, Issaquah, WA). Ten sections (0.3 μm) were taken of 10 consecutive panels (512 × 512) for each slide using a TRITC filter set (555/28×, 617/73m) to detect Alexa-tubulin and then an FITC filter set (490/20×, 555/28m) to detect Dad1-GFP. (Dad1 is the component of the Dam1 complex labeled with GFP). The average pixel intensity for the second section of the FITC stack for each panel from a slide was averaged to give the observed fluorescence for the bound Dam1 complex. Background was measured on the slide in which MTs were incubated with no Dam1 complex. Values obtained on different days were corrected for lamp intensity using the photosensor value.
A standard curve was made to convert observed fluorescence to nM Dam1 complex bound as described [7]. Briefly, increasing concentrations of Dam1 complex (0 nM – 30 nM) were bound to saturating amounts of MTs (50 nM) such that all the Dam1 complex in the assay was bound to MTs. The MTs were pelleted and the fluorescence was measured as described above. An immunoblot of the supernatant detected no Dam1. A plot of fluorescence vs. Dam1 concentration fit a straight line with an R2 value of at least 0.99 (demonstrating that we were indeed at saturating conditions). Concentration of Dam1 bound to MTs (in nM) was calculated as the observed GFP fluorescence pelleting with MTs divided by the slope of that line. Concentration of free Dam1 complex was calculated as the concentration of total Dam1 complex added to the assay minus the concentration bound.
Our assay offers an important advantage over previously described assays because examination of the protein bound to microtubules by microscopy allowed for a clear distinction between binding and non-specific pelleting. Recombinant protein complexes are sensitive to buffering conditions, which must be optimized to prevent aggregation (Figure 1).
Figure 1.
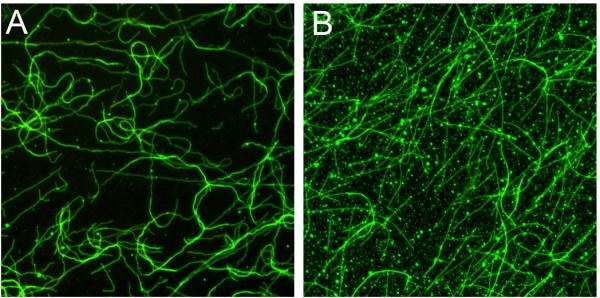
Visualization of microtubule binding can distinguish between non-specific aggregation and microtubule binding.
A. Dam1 complex bound to microtubules. Microtubule binding assay was performed as described except that the buffer conditions were BRB80 containing 33 mM NaCl, 3 mM HEPES, and 3 mM sodium phosphate. Green is Dam1 complex tagged with GFP. In this assay 10 nM Dam1 complex was incubated for 5 minutes with taxol-stabilized microtubules and then 2.5 nM unlabeled Ndc80 complex was added.
B. Aggregates of the Dam1 complex. The Dam1 complex can form aggregates in the binding assay. In this assay, the order of addition was reversed. 2.5 nM unlabeled Ndc80 complex was incubated with taxol-stabilized microtubules for 5 minutes, and then 10 nM Dam1 complex was added.
Fixation of the microtubules prior to imaging is required. We tested if fixation altered the results of the assay. In our normal protocol we dilute the binding assay into fixative and then separate the bound Dam1 complex from the unbound by sedimentation through a glycerol cushion. We reasoned that if the fixation process was driving the binding reaction then we would obtain different results if we fixed after separating the bound Dam1 complex from the unbound by sedimentation. Instead, we found that fixation of the bound Dam1 complex before sedimentation gave the same result as fixation after sedimentation (Figure 2A). Therefore, fixation was not changing the amount of Dam1 complex bound to microtubules. In fact we found that the results were generally insensitive to fixation conditions. Changing the concentration of glutaraldehyde from 0.5 to 9%, the duration of fixation from 2 to 20 minutes and the dilution by the fixative from 30% to 500% had little effect on the amount of binding detected (Figure 2 B, C, D). Binding is complete by 1 minute (Figure 2E). Figure 2F shows the binding of the Dam1 complex compared to a negative control (GFP). The negative control shows no binding. The binding curve for the Dam1 complex could be fit to the McGhee and VonHippel model for binding as described [7]. This fit gives a Kd of 6.5 nM with positive cooperativity of 2.3.
Figure 2.

Characterization of the microtubule binding assay.
The microtubule binding assays were performed and quantified as described in the text except as noted below.
A. Fixation prior to sedimentation gives the same result as fixation after sedimentation. After: Dam1 complex (2.5 or 5 nM) was incubated with taxol-stabilized microtubules (2.5 nM) for 10 minutes. The solution was not fixed. Instead, the bound Dam1 complex was separated from unbound by sedimentation through a 15% glycerol cushion made in BRB80 buffer. The supernatant and cushion were removed by aspiration and the coverslip was dipped in 2% glutaraldehyde before mounting on the slide. Before: Dam1 complex (2.5 or 5 nM) was incubated with taxol-stabilized microtubules for 10 minutes and then the solution was fixed before sedimentation through the glycerol cushion. The error bars represent standard error of the mean (SEM) for 4 replicates.
B. The effect of glutaraldehyde concentration on measuring MT binding
Dam1 complex (5 nM) was incubated with taxol-stabilized MTs (2.5 nM) for 10 minutes. An aliquot of glutaraldehyde (750 μl) was added to each assay tube (250 μl) and the samples were incubated for 15 min. The final concentrations of glutaraldehyde were as shown. The error bars represent the SEM for 4 replicates.
C. The effect of fixation time on measuring MT binding
Dam1 complex (5 nM) was added to taxol-stabilized MTs (3.2 nM) and incubated for 10 minutes. Samples were fixed for the given times. The error bars represent the SEM for 3 replicates.
D. The effect of fixation volume on measuring MT binding
Dam1 complex (5 nM) was added to taxol-stabilized MTs (2.5 nM) and incubated for 10 minutes. Different volumes of glutaraldehyde were added to each assay to yield the given ratio of volume of glutaraldehyde/volume of assay. The final concentration of glutaraldehyde was constant (1.5%). Error bars represent the SEM for 3–7 replicates.
E. Time course of Dam1 complex binding to MTs.
Dam1 complex (10 nM) was added to taxol-stabilized microtubules (2.5 nM). At the given times, glutaraldehyde was added to end the reaction. The error bars represent the SEM for 2–8 replicates.
F. Non-specific binding was not detected
Dam1 complex (filled circles) or GFP (open circles) (0.5–15 nM) were assayed. The data for the Dam1 complex are reproduced for comparison from [7]. They are plotted as a Hill plot [9] and the dotted line shows the fit to the McGhee and VonHippel model as described [7]. For the Dam1 complex the error bars represent the SEM for 8–11 replicates. The assays for binding GFP were done in quadruplicate. The SEM for the GFP data were smaller than the size of the symbols and are not shown.
G. Dam1 complex binding to MTs is decreased by free tubulin.
Dam1 complex binding to MTs was measured in the presence of the given quantities of unpolymerized tubulin (in nM). Unpolymerized tubulin in BRB80 containing 1 mM GTP was diluted 1/100 in BRB80 containing 10 μM taxol before addition to the assay. The error bars represent the SEM for 2–5 replicates.
H. Dam1 complex binding to MTs is sensitive to salt.
Dam1 complex (7.5 nM) was added to taxol-stabilized microtubules (2.5 nM) in BRB80 buffer containing the given concentration of NaCl. (Note that BRB80 is 120 mM K+). Error bars represent SEM for 3 replicates.
Because 80–90% of the tubulin polymerizes, a small amount of free tubulin is present in each assay. We tested whether free tubulin competed with the binding of Dam1 complex to polymerized tubulin. We measured binding in the presence of increasing concentrations of unpolymerized tubulin (Figure 2G). The free tubulin decreased binding when present at 500 nM, but did not have an effect at lower concentrations of free tubulin. Therefore, the low amount of free tubulin in our standard assay (0.25 – 0.3 nM) is not competing with polymerized tubulin for the Dam1 complex. However, assays that measure binding at high concentrations of taxol-stabilized microtubules (e.g. 4 μM total tubulin with about 0.4 μM free) might be affected by the presence of the unpolymerized tubulin [6; 8]. This may partially explain the difference between the Kd we measured [7] and the Kd measured previously by Westermann and coworkers (200 nM) [6].
Binding decreases with the addition of salt, and is nearly undetectable when 150 mM NaCl is present in the binding buffer in addition to the 120 mM K+ in BRB80 (Figure 2H). Because the binding of Dam1 complex to microtubules is sensitive to the salt concentration of the binding buffer, careful reporting of the buffer conditions for each binding assay is crucial for comparison of binding constants measured in different labs.
In vitro microtubule binding techniques provide key insights into the binding affinities of individual protein complexes, uncovering potential functions of individual kinetochore complexes. In our fluorescence microtubule binding assay we rigorously tested several parameters to ensure the careful measurement of dissociation constants, providing visualization as well as a greater sensitivity in measuring binding. The detailed description of our protocol provides a useable resource in revealing both qualitative and quantitative variations in binding affinities that might be missed in a more standard pelleting assay.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ciferri C, Pasqualato S, Screpanti E, Varetti G, Santaguida S, Dos Reis G, Maiolica A, Polka J, De Luca JG, De Wulf P, Salek M, Rappsilber J, Moores CA, Salmon ED, Musacchio A. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell. 2008;133:427–39. doi: 10.1016/j.cell.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006;127:983–97. doi: 10.1016/j.cell.2006.09.039. [DOI] [PubMed] [Google Scholar]
- [3].Hofmann C, Cheeseman IM, Goode BL, McDonald KL, Barnes G, Drubin DG. Saccharomyces cerevisiae Duo1p and Dam1p, novel proteins involved in mitotic spindle function. J Cell Biol. 1998;143:1029–40. doi: 10.1083/jcb.143.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cheeseman IM, Brew C, Wolyniak M, Desai A, Anderson S, Muster N, Yates JR, Huffaker TC, Drubin DG, Barnes G. Implication of a novel multiprotein Dam1p complex in outer kinetochore function. J Cell Biol. 2001;155:1137–45. doi: 10.1083/jcb.200109063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wei RR, Al-Bassam J, Harrison SC. The Ndc80/HEC1 complex is a contact point for kinetochore-microtubule attachment. Nat Struct Mol Biol. 2007;14:54–9. doi: 10.1038/nsmb1186. [DOI] [PubMed] [Google Scholar]
- [6].Westermann S, Avila-Sakar A, Wang HW, Niederstrasser H, Wong J, Drubin DG, Nogales E, Barnes G. Formation of a dynamic kinetochore- microtubule interface through assembly of the Dam1 ring complex. Mol Cell. 2005;17:277–90. doi: 10.1016/j.molcel.2004.12.019. [DOI] [PubMed] [Google Scholar]
- [7].Gestaut DR, Graczyk B, Cooper J, Widlund PO, Zelter A, Wordeman L, Asbury CL, Davis TN. Phosphoregulation and depolymerization-driven movement of the Dam1 complex do not require ring formation. Nat Cell Biol. 2008;10:407–14. doi: 10.1038/ncb1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lampert F, Hornung P, Westermann S. The Dam1 complex confers microtubule plus end-tracking activity to the Ndc80 kinetochore complex. J Cell Biol. 2010;189:641–9. doi: 10.1083/jcb.200912021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hill A. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. 1910;40:iv–vii. [Google Scholar]