

Abstract
In chorioamnionitis, intra-amniotic infections render the amniotic fluid an adverse environment for the fetus and increase the risk of fetal mortality and morbidity. It remains unclear how infection crosses the amniotic barrier, which is made up of tight junctions (TJs). In this study, we investigated whether amniotic TJs are disrupted in inflammatory conditions such as chorioamnionitis. Amniotic TJs were disrupted by single applications of interleukin (IL)-1β, IL-6, tumour necrosis factor-α(TNF-α), and prostaglandin E2. In organ-cultured amniotic membranes, these inflammatory mediators decreased the claudin-3 and claudin-4 levels at the apical junction at different times. Injecting IL-6 into the amniotic cavity concurrently induced the disruption of amniotic TJs by decreasing the claudin-3 and claudin-4 levels at the apical junction, and the dysfunction of the amniotic barrier; in contrast, injecting TNF-α weakened the amniotic barrier by inducing apoptosis of the amniotic epithelial cells, with no decrease in claudin-3 and claudin-4 at the apical junction. Furthermore, inflammation in the amniotic membrane, which was induced by the administration of lipopolysaccharide to pregnant mice, concurrently caused dysfunction of the amniotic barrier and disruption of TJs, involving the decrease of claudin-3 and claudin-4 levels at the apical junction and apoptosis in the amniotic epithelium. These results indicate that the adverse effects of the inflammatory mediators on amniotic TJs cause severe dysfunction of the amniotic barrier.
Introduction
Amniotic fluid contributes to providing an appropriate physiological environment for normal fetal development in mid-pregnancy (Menon et al. 2010). However, septic inflammation of the amniotic fluid impairs the growth of preterm fetuses (Murthy & Kennea, 2007) and causes other adverse developmental outcomes, such as chronic lung disease (Lee et al. 2009), periventricular leukomalacia (Rocha et al. 2006), oligohydramnios (Lee et al. 2010) and fetal death (Newton, 2005). Although intra-amniotic infection increases the risk of fetal mortality and morbidity, it remains unclear how infection spreads into the intra-amniotic cavity across the amniotic membrane.
The spread of microbes across epithelial tissue is blocked by TJs, which function as barriers (Moyer et al. 2008; Li et al. 2009; Bishop et al. 2010). TJs are the most apical junctional complexes and restrict the paracellular pathway of epithelial sheets (Tsukita et al. 2001). The tightness of these junctions is determined by claudins, which play a role in determining TJ structure at the apical junction (Furuse & Tsukita, 2006). In the amniotic epithelium, the TJs are composed of claudin-3 and claudin-4, and restrict paracellular transport between the placenta and amniotic cavity in mid-pregnancy (Kobayashi et al. 2009, 2010). Therefore, it is suggested that amniotic TJs block the spread of microbes in mid-pregnancy.
Some studies have reported inflammation-related disruption of TJs in the intestinal epithelium (Schulzke et al. 2009), corneal epithelium (Yi et al. 2000), epidermis (Kirschner et al. 2009) and blood–brain barrier (Stamatovic et al. 2008). In these cases, the disruption of TJs was caused by decrease in the levels of TJ components at the apical junctions or by apoptosis induced by inflammatory cytokines such as interleukin (IL)-1β (Al-Sadi & Ma, 2007), IL-6 (Tazuke et al. 2003), or tumour necrosis factor (TNF)-α (Schulzke et al. 2006). Prostaglandin E2 (PGE2) also induces the disassembly of the apical junctional complex of TJs in association with an increase in permeability (Tanaka et al. 2008). Interestingly, these inflammatory mediators also proliferate in the fetal membrane and amniotic fluid in the case of chorioamnionitis (Goldenberg et al. 2000; Saji et al. 2000; Christiaens et al. 2008).
In this study, we aimed to determine whether amniotic TJs are disrupted under inflammatory conditions. Inflammation was induced in the amniotic membranes of pregnant mice by administering lipopolysaccharide (LPS) in order to evaluate the effects of inflammation on amniotic TJs. The effects of IL-1β, IL-6, TNF-α and PGE2 on amniotic TJs were also investigated in organ-cultured amniotic membranes. Furthermore, IL-6 and TNF-α were injected into the amniotic cavity in vivo. Our results revealed that inflammatory mediators weaken the amniotic barrier by disrupting amniotic TJs.
Methods
Animals
Pregnant Institute of Cancer Research (ICR) mice were purchased from Japan SLC Inc. (Shizuoka, Japan). The day on which a vaginal plug was detected in the mice was designated as embryonic day (E)0. The pregnant mice were deeply anaesthetized with halothane and then decapitated. The amniotic membranes were quickly separated from other placental tissues and transferred to a solution containing 0.5 mm CaCl2 and 0.5 mm MgCl2-containing phosphate-buffered saline (mPBS), which was kept on ice. The dissected amniotic membranes were immediately used for the experiments. All the animal protocols used in the present study were approved by the Animal Resource Committee of the School of Medicine, Keio University, and comply with the policies and regulations of The Journal of Physiology (Drummond, 2009).
Materials
IL-1β, IL-6 and TNF-α were purchased from PeproTech (Rocky Hill, NJ, USA). Chemicals containing LPS from Escherichia coli 055:B5 and PGE2 were purchased from Sigma-Aldrich (St Louis, MO, USA). The following antibodies were used as primary antibodies for immunological studies: rabbit polyclonal antibodies against claudin-3 and claudin-4 (Invitrogen/Zymed Laboratories, San Francisco, CA, USA), and mouse monoclonal antibodies against occludin (Invitrogen/Zymed Laboratories). Secondary Alexa Fluor 488-conjugated goat anti-rabbit and Alexa Fluor 546-conjugated goat anti-mouse antibodies were purchased from Invitrogen (Carlsbad, CA, USA). Annexin V–fluorescein staining kit was purchased from Wako Pure Chemical Industries (Osaka, Japan).
Intraperitoneal LPS administration
LPS is soluble in PBS at a concentration of 0.5 mg ml−1. The LPS solution (50 μg) or vehicle was intraperitoneally administered to pregnant mice at E15.5. The amniotic membranes were extracted at 6 (n= 9), 9 (n= 8), or 12 h (n= 12) after administering LPS. Amniotic membranes obtained from pregnant mice that were administered phosphate buffered saline (PBS) were designated as controls (n= 9).
Measurement of transepithelial resistance and paracellular permeability
The amniotic membranes were allowed to float in mPBS and then placed on a cell culture insert for a 24-well plate (0.4 μm pore size; BD Biosciences, San Jose, CA, USA). For measuring transepithelial resistance (TER), the upper and lower chambers of the insert were filled with mPBS. The electrodes of a Millicel-ERS system (Millipore Co., Billerica, MA, USA) were placed in the upper and lower chambers, and the TER from the basolateral (placental side) to the apical side (fetal side) was measured. For measuring paracellular permeability, the upper chamber of the insert was filled with mPBS containing 0.1 mg ml−1 fluorescein sodium salt (molecular weight, 376; Sigma-Aldrich), and the lower chamber was filled with mPBS. To prevent tracer leakage, a silicon O-ring and a glass cylinder were placed on the amniotic membrane. The media from the lower compartment were collected 30 min after the addition of tracers, and the paracellular flux from the basolateral to the apical side was measured using a fluorometer (excitation, 492 nm; emission, 520 nm).
Immunostaining
The amniotic membrane was fixed with methanol for 10 min at −20°C and then in 1% paraformaldehyde in mPBS (pH 7.4) for 10 min at 4°C. The fixed amniotic membrane was permeabilized with 0.2% Triton X-100 in mPBS (pH 7.4) for 5 min and then incubated with mPBS containing 5% bovine serum albumin as a blocking solution to saturate the non-specific sites of antibody binding. The amniotic membrane was then sequentially treated as follows: the membrane was exposed to primary antibodies for 1.5 h at room temperature in a blocking solution, followed by washing and exposure to secondary antibodies for 1 h at room temperature in a blocking solution. The control amniotic membranes were treated in a similar manner, except that primary antibodies were not included in the treatment. Images of the stained sections were obtained using a fluorescence microscope (BX-51; Olympus, Tokyo, Japan) or a confocal laser scanning microscope (LSM 510; Zeiss, Oberkochen, Germany). The fluorescence intensities of claudin-3, claudin-4 and occludin were measured using the Plot Profile function of ImageJ (downloaded from the National Institutes of Health, USA; available at http://rsb.info.nih.gov/ij) on each of the immunostaining images. The claudins at the apical junction, but not in the lateral membrane, contribute to TJ function (Chiba et al. 2008). The position of the apical junction was determined by the localization of occludin – a representative TJ component that is mainly localized to the apical junction (Schneeberger & Lynch, 2004).
Preparation of Triton X-100-soluble and Triton X-100-insoluble protein fractions
The amniotic membrane was lysed in Triton X-100 buffer (1% Triton X-100, 100 mm NaCl, 10 mm Hepes at pH 7.4, 2 mm EDTA, and a protease inhibitor mixture) and then passed 30 times through a 21-gauge needle. The lysates were then centrifuged at 15,000 g for 30 min at 4°C. The supernatant was considered the Triton X-100-soluble fraction. The pellet was solubilized in Triton X-100 buffer containing 1% sodium dodecyl sulphate (SDS) by using an ultrasonic disintegrator and then centrifuged at 15,000 g for 5 min at 4°C. The supernatant was designated as the Triton X-100-insoluble fraction. Each fraction was diluted with an equal volume of sample buffer (100 mm Tris at pH 6.8, 100 mm dithiothreitol, 2% SDS, 0.2% bromophenol blue, and 20% glycerol), incubated for 10 min at 70°C, and then stored at −20°C as samples for Western blotting.
Western blotting analysis
The samples were electrophoresed using a 12% SDS-polyacrylamide gel and transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked for 1 h with PBS containing 3% non-fat dried milk and 0.05% Tween 20, and then incubated overnight at 4°C with primary antibodies diluted in PBS containing 5% bovine serum albumin. Subsequently, the membranes were washed in PBS containing 0.05% Tween 20 and incubated for 45 min at room temperature with the appropriate secondary horseradish peroxidase-conjugated antibodies diluted in PBS containing 3% non-fat dried milk and 0.05% Tween 20. An enhanced chemiluminescence detection system (GE Healthcare Bio-Sciences, Tokyo, Japan) was used to visualize specific immunoreactive proteins. The protein concentration in each sample was determined by the Bradford method. To evaluate the ratio of the Triton X-100-insoluble and Triton X-100-soluble fractions, the bands were quantified by densitometry (LAS-4000 mini; GE Healthcare).
Organ culture of amniotic membranes
The amniotic membranes were removed on E15.5, immersed in Dulbecco's modified Eagle's medium (DMEM)/F-12 (GIBCO-BRL, Grand Island, NY) containing 0.4% agarose, and chilled on ice for gelification. The amniotic membranes were now immobilized in agarose gel and were immersed in DMEM/F-12 containing 10% fetal bovine serum (FBS), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. IL-1β, IL-6 and TNF-α were added to the culture medium at a final concentration of 10 ng ml−1, and PGE2 was added at a final concentration of 1 μm. The amniotic membranes were cultured in the presence of inflammatory mediators for 1, 6 and 12 h at 37°C. The amniotic membranes cultured in the absence of inflammatory mediators are considered as 0 h.
IL-6 and TNF-α injection into the amniotic cavity
IL-6 and TNF-α were found to be soluble in PBS at a concentration of 2 μg ml−1. On E15.5, the pregnant mice were deeply anaesthetized with Somnopentyl (pentobarbital sodium, 50 mg kg−1), and their uterine horns were gently pulled. We injected 5 μl of IL-6 (n= 10) or TNF-α (n= 9) solution through the exposed uterine wall into each amniotic cavity by using a 33-gauge needle. The uterine horn was returned to the abdominal cavity, and the abdominal muscle wall and skin were closed. The amniotic membranes were extracted 14 h after injection. Specimens from pregnant mice injected with PBS were treated in the same manner as the specimens obtained from the mice from the vehicle group (n= 9).
Detection of apoptotic cells
Apoptosis was detected using the Annexin V–fluorescein kit, according to the manufacturer's instructions. In brief, the amniotic membrane was washed with a binding buffer and then immersed in the binding buffer containing Annexin V–fluorescein and propidium iodide for 10 min in the dark at room temperature. The excess unbound Annexin V–fluorescein was removed by washing with the binding buffer. The explants were fixed in cold methanol and then in 1% paraformaldehyde in mPBS for immunostaining.
Statistical analysis
Data are expressed as the mean [standard deviation (s.d.)]. The statistical significance of the differences between mean values was verified using Student's t test for densitometric analysis of Western blotting, and using the Mann-Whitney U test for TER and paracellular permeability. Statistical significance is indicated by asterisks. All the experiments were performed a minimum of 4 times to ensure reproducibility.
Results
Weakening of the amniotic membrane barrier on administering LPS
LPS administration induced a time-dependent decrease in the TER of the amniotic membranes (Fig. 1). A clear effect was detected 9 h after administration, and the TER 12 h after administration was less than half of that in the controls. The amount of fluorescein that passed through the amniotic membrane with LPS administration (n= 12, 11.3 [9.33]) before 12 h was significantly greater than the amount that passed through the control amniotic membranes injected with the vehicle (n= 9, 1.63 [2.39]). These data indicate that the amniotic barrier was weakened by LPS.
Figure 1. Lipopolysaccharide (LPS) administration-induced dysfunction of the amniotic barrier.
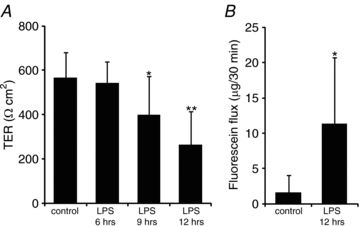
LPS was intraperitoneally administered to pregnant mice on embryonic day (E)15.5. The amniotic membranes were extracted 6, 9, or 12 h after administration of LPS in order to measure the transepithelial resistance (TER) (A) and fluorescein flux (B) through the amniotic membrane. The controls are the amniotic membranes injected with the vehicle. Data are represented as the mean and standard deviation (s.d.) (n= 8–12). *P < 0.05 versus control; **P < 0.005 versus control.
Decrease in the claudin-3 and claudin-4 levels at the apical junction on administering LPS
In the amniotic membranes in which LPS had not been administered, claudin-3 was mainly localized at the region identified as the apical junction by using occludin (Fig. 2A). The plot profiles of the fluorescence intensity of claudin-3 showed sharp peaks at the same position as the peaks for occludin. Similar localization patterns for claudin-3 and occludin were observed in normal amniotic membranes (Kobayashi et al. 2009). However, in the amniotic membranes injected with LPS, the linear localization of claudin-3 along the apical junction became indistinct and discontinuous (Fig. 2B). The claudin-3 peak in the plot profile became indistinct, and the fluorescence intensity of the lateral membrane increased. LPS slightly decreased the localization of occludin at the apical junction and induced the loss of the linear localization of claudin-4 along the apical junction; this localization was visualized as a distinct line in the controls (Fig. 3A). The plot profiles clearly showed the decrease in claudin-4 at the apical junction and the increase in claudin-4 in the lateral membrane (Fig. 3B).
Figure 2. LPS administration-induced subcellular localization changes in claudin-3.
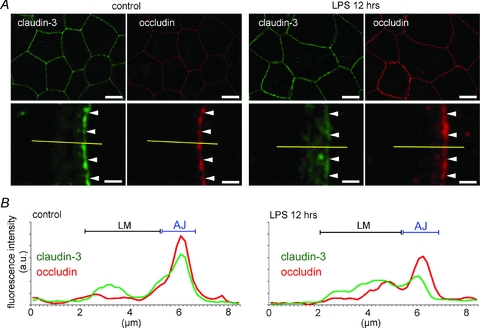
The amniotic membranes were extracted 12 h after administering LPS, and whole-mount immunostaining was performed. A, immunostaining images for claudin-3 (green) and occludin (red) show a vertical view of the amniotic membranes. In the magnified images, the arrowheads show the position of the apical junction of amniotic epithelial cells. B, plot profiles show the representative distribution of the fluorescence intensities of claudin-3 and occludin along the yellow lines in A. The positions of the apical junction (AJ) and lateral membrane (LM) were determined from the plot profiles of occludin. Scale bar: Upper row, 20 μm; lower row, 2 μm.
Figure 3. LPS administration-induced subcellular localization changes in claudin-4.
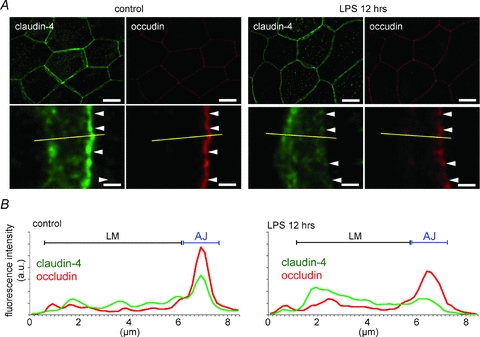
A, a vertical view of amniotic membranes that were extracted 12 h after administering LPS and immunostained for claudin-4 (green) and occludin (red). In the magnified images, the arrowheads show the position of the apical junction of amniotic epithelial cells. B, plot profiles show the representative distributions of the fluorescence intensities of claudin-3 and occludin along the yellow lines in A. Scale bar: Upper row, 20 μm; lower row, 2 μm.
To confirm the decrease in the claudin-3 and claudin-4 levels at the apical junction, immunoblotting experiments were performed to compare the detergent-soluble fractions (representative of claudins in the cytosol or lateral membranes) with the detergent-insoluble fractions (representative of claudins involved in the apical junctional complex of TJs) (Nishiyama et al. 2001). The amounts of claudin-3 and claudin-4 in the detergent-insoluble fractions gradually decreased in a time-dependent manner on LPS administration (Fig. 4). The significant decrease in claudin-3 in the detergent-insoluble fractions was observed after 12 h, and claudin-4 was also nearly undetectable after 12 h.
Figure 4. LPS administration-induced increase in the detergent solubility of claudin-3 and claudin-4.
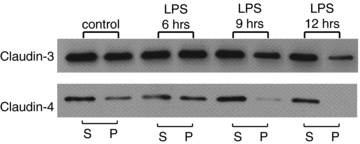
LPS was intraperitoneally administered to pregnant mice on E15.5, and the amniotic membranes were extracted after 6, 9, or 12 h. The detergent-soluble (S) and detergent-insoluble (P) fractions of the amniotic membrane were isolated, and Western blotting for claudin-3 and claudin-4 was performed.
Inflammatory mediators differentially affect the detergent-insoluble fractions of claudin-3 and claudin-4
The amniotic membranes were organ cultured in the presence of IL-1β, IL-6, TNF-α, or PGE2 (Fig. 5). IL-1β and TNF-α induced a clear decrease in the amounts of claudin-3 and claudin-4 in the detergent-insoluble fractions after 1 h of treatment. The same result was obtained on 12 h treatment with IL-6 and on 1- and 6 h treatments with PGE2. Treatment with TNF-α for 12 h induced an increase in the amounts of claudin-3 and claudin-4 in the detergent-insoluble fractions.
Figure 5. Effects of inflammatory mediators on the detergent solubility of claudin-3 and claudin-4 in vitro.
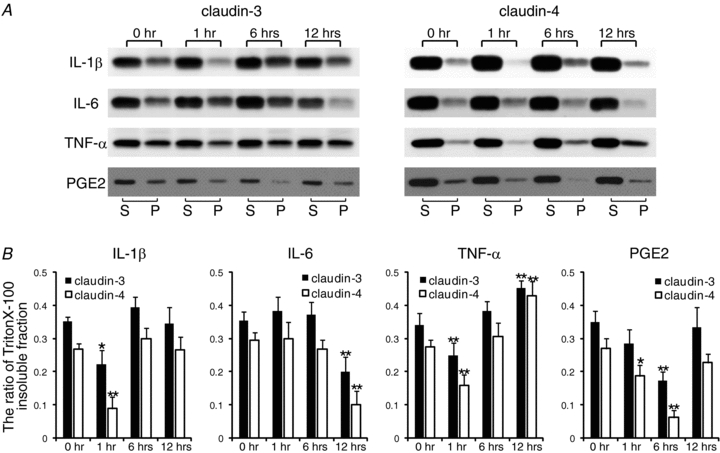
Amniotic membranes obtained from pregnant mice on E15.5 were immobilized using agarose gel and were organ cultured. A, the amniotic membranes were exposed to interleukin (IL)-1β, IL-6, tumour necrosis factor-α(TNF-α), and prostaglandin E2 (PGE2) for 1, 6, or 12 h. The detergent-soluble (S) and detergent-insoluble (P) fractions of the amniotic membrane were isolated, and Western blotting for claudin-3 and claudin-4 was performed. B, the ratio of insoluble fractions was densitometrically analysed and was calculated using the formula P/(S + P). Data are represented as means and s.d. (n= 4). *P < 0.05 versus 0 h; **P < 0.005 versus 0 h.
Effects of IL-6 and TNF-α injection into the amniotic cavity on the claudin-3 and claudin-4 levels at the apical junction
No differences were observed between the control amniotic membranes and the amniotic membranes injected with the vehicle, with respect to the localization patterns and plot profiles of claudin-3, claudin-4 and occludin (Figs 2 and 3). In the IL-6-injected amniotic membranes, claudin-3 and claudin-4 were mainly observed in the lateral membrane, and their linear localization patterns along the apical junction became discontinuous and indistinct (Fig. 6A). In the plot profiles of claudin-3 and claudin-4, the peak at the apical junction disappeared, and the fluorescence intensity increased in the lateral membrane (Fig. 6B). In the IL-6-injected amniotic membranes, the localization patterns of occludin along the apical junction also became discontinuous. However, TNF-α injection did not induce an increase in the claudin-3, claudin-4 and occludin levels in the lateral membrane, and claudin-3 and claudin-4 mainly localized at the apical junctions, similar to occludin (Fig. 7A). Similar to the occludin peaks, the claudin-3 and claudin-4 peaks at the apical junction were sharp (Fig. 7B). The effects of IL-1β and PGE2 could not be examined because the injected IL-1β caused fetal death within 12 h, and PGE2 induced premature birth a few hours after injection.
Figure 6. IL-6 administration-induced decrease in the claudin-3 and claudin-4 levels at the apical junction.
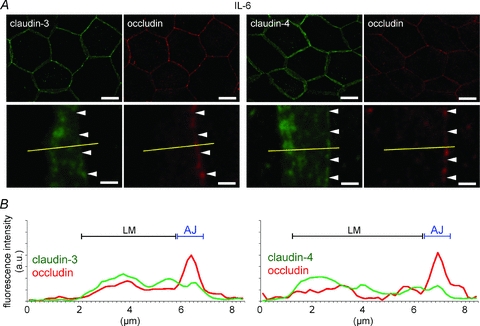
A, IL-6 was injected into the amniotic cavities on E15.5. The amniotic membranes were extracted 12 h after injection and immunostained for claudin-3 (green), claudin-4 (green) and occludin (red). The magnified images show the position of the apical junction of amniotic epithelial cells (arrowhead). B, the plot profiles show the representative distributions of the fluorescence intensities of claudin-3, claudin-4 and occludin along the yellow lines in A. Scale bar: Upper row, 20 μm; lower row, 2 μm.
Figure 7. Effects of TNF-α on the subcellular localization of claudin-3 and claudin-4 in vivo.
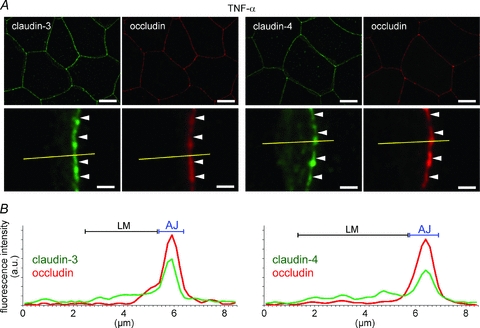
A, TNF-α was injected into the amniotic cavities on E15.5 The amniotic membranes were extracted 12 h after injection and immunostained for claudin-3 (green), claudin-4 (green), and occludin (red). The magnified images show the position of the apical junction of amniotic epithelial cells (arrowhead). B, the plot profiles show the representative distributions of the fluorescence intensities of claudin-3, claudin-4 and occludin along the yellow lines in A. Scale bar: Upper row, 20 μm; lower row, 2 μm.
The amounts of claudin-3 and claudin-4 in the detergent-insoluble fractions significantly decreased on injecting IL-6, whereas TNF-α injection induced only a slight decrease in the amounts of claudin-3 and claudin-4 in the detergent-insoluble fractions (Fig. 8A). Densitometric analysis revealed that there were significant differences between treatment with IL-6 and the vehicle with respect to the amounts of claudin-3 and claudin-4 in the detergent-insoluble fractions; however, no significant differences were observed between treatment with TNF-α and the vehicle in this regard (Fig. 8B). These results indicate that IL-6 induced the translocation of claudin-3 and claudin-4 from the apical junction to the lateral membrane.
Figure 8. Effects of IL-6 and TNF-α on the detergent solubility of claudin-3 and claudin-4 in vivo.
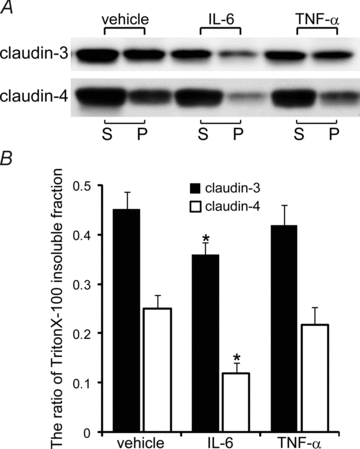
A, IL-6 and TNF-α were injected into the amniotic cavities on E15.5, and the amniotic membranes were extracted after 12 h. The detergent-soluble (S) and detergent-insoluble (P) fractions of the amniotic membranes were isolated, and Western blotting of claudin-3 and claudin-4 was performed. B, the ratio of the insoluble fractions was densitometrically analysed and was calculated using the formula P/(S + P). Data are represented as means and s.d. (n= 4). *P < 0.001 versus vehicle.
TNF-α injected into the amniotic cavity induced apoptosis of amniotic epithelial cells
TNF-α is a representative ligand for death receptors, and the binding of TNF-α to death receptors induces apoptosis (Wilson et al. 2009). By using Annexin V–fluorescein labelling, we determined whether the apoptosis was induced because of TNF-α injection into the amniotic cavity. Apoptotic cells labelled with annexin V–fluorescein were unevenly distributed in the amniotic epithelia that were injected with TNF-α or LPS (Fig. 9). Apoptotic cells were not detected in the amniotic membranes that were injected with the vehicle or IL-6.
Figure 9. Effects of IL-6 and TNF-α on the apoptosis of amniotic epithelial cells in vivo.
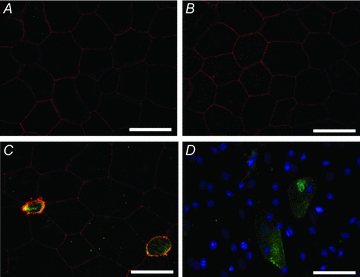
The vehicle (A), IL-6 (B), or TNF-α (C) was injected into the amniotic cavity on E15.5, and the amniotic membranes were extracted after 12 h. The fluorescence images show double staining with Annexin V–fluorescein (green) and anti-occludin antibodies(red). TNF-α induced apoptosis of amniotic epithelial cells. D, apoptosis of the amniotic epithelial cells was also observed in the amniotic membranes injected with LPS (green: Annexin V–fluorescein, blue: DAPI). Scale bars: 20 μm.
The injected IL-6 and TNF-α reduced the barrier function of the amniotic membrane
The injected IL-6 and TNF-α both induced a decrease in the TER of the amniotic membranes (Fig. 10A). TNF-α had a greater effect on amniotic TER than did IL-6; the effect of IL-6 was two-thirds that of the injected vehicle. The amount of fluorescein that passed through the amniotic membranes injected with IL-6 (n= 9, 4.45 [2.28]) and TNF-α (n= 8, 7.68 [5.95]) was significantly greater than the amount that passed through control amniotic membranes injected with the vehicle (n= 9, 1.96 [2.23]) (Fig. 10B).
Figure 10. IL-6-induced and TNF-α-induced dysfunction of the amniotic barrier.
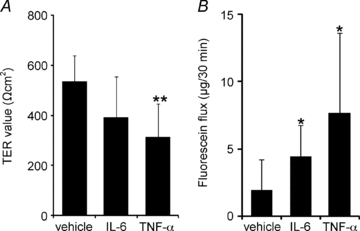
IL-6, TNF-α, or the vehicle was injected into the amniotic cavities on E15.5, and the amniotic membranes were extracted after 12 h to measure TER (A) and the fluorescein flux (B) through the amniotic membrane. Data are represented as means and s.d. (n= 8–9). *P < 0.05 versus vehicle; **P < 0.005 versus vehicle.
Discussion
Chorioamnionitis increases the risk of mortality and morbidity in newborn infants because of infection of the amniotic fluid (Romero et al. 1992; Menon et al. 2010). However, the mechanism by which infection crosses the amniotic barrier and spreads into the amniotic fluid remains unclear. In this study, we have shown that the amniotic barrier is weakened under inflammatory conditions induced by injected LPS. A single application of IL-6 weakened the amniotic barrier concurrently with the decrease in the claudin-3 and claudin-4 levels at the apical junction, and a single application of TNF-α weakened the amniotic barrier concurrently with apoptosis of the amniotic epithelial cells. The claudins at the apical junction, but not those in the lateral membrane, contribute to the barrier function of TJs, and the decrease in claudin levels at the apical junction weakens the TJ barrier (Krause et al. 2008; Tanaka et al. 2008). Apoptosis causes dysfunction of the epithelial TJs by increasing paracellular permeability (Flynn & Buret, 2008; Schwerk et al. 2010). The fluorescein permeability of the amniotic membrane via the paracellular pathway depends on amniotic TJs, and the apoptosis of amniotic epithelial cells increases the permeability of the membrane (Kobayashi et al. 2009). In this study, LPS, IL-6 and TNF-α increased the fluorescein permeability of the amniotic epithelium from the basolateral to the apical side. This suggests that the amniotic barrier weakens under inflammatory conditions because of a decrease in the claudin-3 and claudin-4 levels at the apical junction, and apoptosis of amniotic epithelial cells. Furthermore, the disruption of TJs, which restrict the paracellular pathway, facilitates microbial invasion of the epithelial and endothelial tissues (Moyer et al. 2008; Li et al. 2009; Bishop et al. 2010). Therefore, it is thought that the disruption of TJs by inflammatory mediators may facilitate intra-amniotic infection via the paracellular pathway.
The levels of several inflammatory mediators, including IL-1β, IL-6, TNF-α and PGE2, increase in the amniotic fluid under inflammatory conditions (Goldenberg et al. 2000; Menon et al. 2009). Each inflammatory mediator activates distinct signalling pathways which are reported to regulate TJs, for example the nuclear factor-κB, protein kinase A, protein kinase C, myosin light chain kinase, and apoptosis pathways (Desai et al. 2002; Schulzke et al. 2006; Shen et al. 2006; Al-Sadi & Ma, 2007; Kawedia et al. 2008; Koizumi et al. 2008; Wang et al. 2010). In fact, in this study, the amniotic TJs exhibited different responses to IL-1β, IL-6, TNF-α and PGE2. These inflammatory mediators mutually activate each other's production (Saji et al. 2000; Christiaens et al. 2008). Therefore, IL-1β, IL-6, TNF-α and PGE2 may induce the disruption of TJs in distinctly different ways. Interestingly, the levels of these inflammatory mediators spontaneously increase in the amniotic fluid in late pregnancy (Saji et al. 2000; Christiaens et al. 2008). We have previously reported that amniotic TJs are gradually disrupted in late pregnancy (Kobayashi et al. 2009, 2010). This disruption in late pregnancy may be caused by increase in the levels of these inflammatory mediators.
In this study, we have shown that the amniotic membrane barrier weakened under inflammatory conditions. The weakening of the amniotic barrier was associated with the disruption of TJs. The inflammatory mediators – IL-1β, IL-6, TNF-α and PGE2 – affected amniotic TJs in distinctly different ways. The levels of these inflammatory mediators increase under inflammatory conditions such as chorioamnionitis (Goldenberg et al. 2000; Menon et al. 2009). However, it remains unclear whether the reduced barrier function of the amniotic membrane actually facilitates the invasion of microbes into the amniotic cavity via the paracellular pathway; therefore, further studies on the effects of inflammation on amniotic TJs are required.
Acknowledgments
This work was supported by a Grant-in-Aid for Young Scientists (20790177) from the Japan Society of Promotion of Science; and a Grant-in-Aid for Specially Promoted Research and the Global Center-of-Excellence Program for Human Metabolomic Systems Biology from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Glossary
Abbreviations
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- PGE2
prostaglandin E2
- TER
transepithelial resistance
- TJ
tight junction
- TNF-α
tumour necrosis factor-α
Author contributions
K.K.: design of the study, analysis and interpretation of data, and drafting and revision of the manuscript. H.M.: analysis and interpretation of data, and revision of the manuscript. M.Y.: concept and design of the study, interpretation of data, and revision of the manuscript. All authors approved the final version of the manuscript. All the experiments were performed at the Department of Pharmacology, School of Medicine, Keio University, Japan.
References
- Al-Sadi RM, Ma TY. IL-1β causes an increase in intestinal epithelial tight junction permeability. J Immunol. 2007;178:4641–4649. doi: 10.4049/jimmunol.178.7.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop BL, Lodolce JP, Kolodziej LE, Boone DL, Tang WJ. The role of anthrolysin O in gut epithelial barrier disruption during Bacillus anthracis infection. Biochem Biophys Res Commun. 2010;394:254–259. doi: 10.1016/j.bbrc.2010.02.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba H, Osanai M, Murata M, Kojima T, Sawada N. Transmembrane proteins of tight junctions. Biochim Biophys Acta. 2008;1778:588–600. doi: 10.1016/j.bbamem.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Christiaens I, Zaragoza DB, Guilbert L, Robertson SA, Mitchell BF, Olson DM. Inflammatory processes in preterm and term parturition. J Reprod Immunol. 2008;79:50–57. doi: 10.1016/j.jri.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Desai TR, Leeper NJ, Hynes KL, Gewertz BL. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J Surg Res. 2002;104:118–123. doi: 10.1006/jsre.2002.6415. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn AN, Buret AG. Caspases-3, -8, and -9 are required for induction of epithelial cell apoptosis by enteropathogenic E. coli but are dispensable for increased paracellular permeability. Microb Pathog. 2008;44:311–319. doi: 10.1016/j.micpath.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Furuse M, Tsukita S. Claudins in occluding junctions of humans and flies. Trends Cell Biol. 2006;16:181–188. doi: 10.1016/j.tcb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N Engl J Med. 2000;342:1500–1507. doi: 10.1056/NEJM200005183422007. [DOI] [PubMed] [Google Scholar]
- Kawedia JD, Jiang M, Kulkarni A, Waechter HE, Matlin KS, Pauletti GM, Menon AG. The protein kinase A pathway contributes to Hg2+-induced alterations in phosphorylation and subcellular distribution of occludin associated with increased tight junction permeability of salivary epithelial cell monolayers. J Pharmacol Exp Ther. 2008;326:829–837. doi: 10.1124/jpet.107.135798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner N, Poetzl C, von den Driesch P, Wladykowski E, Moll I, Behne MJ, Brandner JM. Alteration of tight junction proteins is an early event in psoriasis: putative involvement of proinflammatory cytokines. Am J Pathol. 2009;175:1095–1106. doi: 10.2353/ajpath.2009.080973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Inai T, Shibata Y, Yasui M. Dynamic changes in amniotic tight junctions during pregnancy. Placenta. 2009;30:840–847. doi: 10.1016/j.placenta.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Kadohira I, Tanaka M, Yoshimura Y, Ikeda K, Yasui M. Expression and distribution of tight junction proteins in human amnion during late pregnancy. Placenta. 2010;31:158–162. doi: 10.1016/j.placenta.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Koizumi J, Kojima T, Ogasawara N, Kamekura R, Kurose M, Go M, Harimaya A, Murata M, Osanai M, Chiba H, Himi T, Sawada N. Protein kinase C enhances tight junction barrier function of human nasal epithelial cells in primary culture by transcriptional regulation. Mol Pharmacol. 2008;74:432–442. doi: 10.1124/mol.107.043711. [DOI] [PubMed] [Google Scholar]
- Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta. 2008;1778:631–645. doi: 10.1016/j.bbamem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Lee J, Oh KJ, Yang HJ, Park JS, Romero R, Yoon BH. The importance of intra-amniotic inflammation in the subsequent development of atypical chronic lung disease. J Matern Fetal Neonatal Med. 2009;22:917–923. doi: 10.1080/14767050902994705. [DOI] [PubMed] [Google Scholar]
- Lee SE, Romero R, Lee SM, Yoon BH. Amniotic fluid volume in intra-amniotic inflammation with and without culture-proven amniotic fluid infection in preterm premature rupture of membranes. J Perinat Med. 2010;38:39–44. doi: 10.1515/JPM.2009.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhang Q, Wang C, Liu X, Li N, Li J. Disruption of tight junctions during polymicrobial sepsis in vivo. J Pathol. 2009;218:210–221. doi: 10.1002/path.2525. [DOI] [PubMed] [Google Scholar]
- Menon R, Peltier MR, Eckardt J, Fortunato SJ. Diversity in cytokine response to bacteria associated with preterm birth by fetal membranes. Am J Obstet Gynecol. 2009;201:306. doi: 10.1016/j.ajog.2009.06.027. e301–306. [DOI] [PubMed] [Google Scholar]
- Menon R, Taylor RN, Fortunato SJ. Chorioamnionitis: a complex pathophysiologic syndrome. Placenta. 2010;31:113–120. doi: 10.1016/j.placenta.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Moyer AL, Ramadan RT, Thurman J, Burroughs A, Callegan MC. Bacillus cereus induces permeability of an in vitro blood-retina barrier. Infect Immun. 2008;76:1358–1367. doi: 10.1128/IAI.01330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy V, Kennea NL. Antenatal infection/inflammation and fetal tissue injury. Best Pract Res Clin Obstet Gynaecol. 2007;21:479–489. doi: 10.1016/j.bpobgyn.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Newton ER. Preterm labor, preterm premature rupture of membranes, and chorioamnionitis. Clin Perinatol. 2005;32:571–600. doi: 10.1016/j.clp.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Nishiyama R, Sakaguchi T, Kinugasa T, Gu X, MacDermott RP, Podolsky DK, Reinecker HC. Interleukin-2 receptor β subunit-dependent and -independent regulation of intestinal epithelial tight junctions. J Biol Chem. 2001;276:35571–35580. doi: 10.1074/jbc.M106013200. [DOI] [PubMed] [Google Scholar]
- Rocha G, Proenca E, Quintas C, Rodrigues T, Guimaraes H. Chorioamnionitis and neonatal morbidity. Acta Med Port. 2006;19:207–212. [PubMed] [Google Scholar]
- Romero R, Salafia CM, Athanassiadis AP, Hanaoka S, Mazor M, Sepulveda W, Bracken MB. The relationship between acute inflammatory lesions of the preterm placenta and amniotic fluid microbiology. Am J Obstet Gynecol. 1992;166:1382–1388. doi: 10.1016/0002-9378(92)91609-e. [DOI] [PubMed] [Google Scholar]
- Saji F, Samejima Y, Kamiura S, Sawai K, Shimoya K, Kimura T. Cytokine production in chorioamnionitis. J Reprod Immunol. 2000;47:185–196. doi: 10.1016/s0165-0378(00)00064-4. [DOI] [PubMed] [Google Scholar]
- Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- Schulzke JD, Bojarski C, Zeissig S, Heller F, Gitter AH, Fromm M. Disrupted barrier function through epithelial cell apoptosis. Ann N Y Acad Sci. 2006;1072:288–299. doi: 10.1196/annals.1326.027. [DOI] [PubMed] [Google Scholar]
- Schulzke JD, Ploeger S, Amasheh M, Fromm A, Zeissig S, Troeger H, Richter J, Bojarski C, Schumann M, Fromm M. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci. 2009;1165:294–300. doi: 10.1111/j.1749-6632.2009.04062.x. [DOI] [PubMed] [Google Scholar]
- Schwerk C, Rybarczyk K, Essmann F, Seibt A, Molleken ML, Zeni P, Schroten H, Tenenbaum T. TNFα induces choroid plexus epithelial cell barrier alterations by apoptotic and nonapoptotic mechanisms. J Biomed Biotechnol. 2010;2010:307231. doi: 10.1155/2010/307231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE, Turner JR. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci. 2006;119:2095–2106. doi: 10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol. 2008;6:179–192. doi: 10.2174/157015908785777210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka MN, Diaz BL, de Souza W, Morgado-Diaz JA. Prostaglandin E2-EP1 and EP2 receptor signaling promotes apical junctional complex disassembly of Caco-2 human colorectal cancer cells. BMC Cell Biol. 2008;9:63. doi: 10.1186/1471-2121-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazuke Y, Drongowski RA, Teitelbaum DH, Coran AG. Interleukin-6 changes tight junction permeability and intracellular phospholipid content in a human enterocyte cell culture model. Pediatr Surg Int. 2003;19:321–325. doi: 10.1007/s00383-003-1003-8. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–293. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- Wang P, Zhu F, Lee NH, Konstantopoulos K. Shear-induced interleukin-6 synthesis in chondrocytes: the roles of E prostanoid (EP)2 and EP3 in cAMP/protein kinase A- and PI3-K/Akt-dependent NF-κB activation. J Biol Chem. 2010;285:24793–20804. doi: 10.1074/jbc.M110.110320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- Yi X, Wang Y, Yu FS. Corneal epithelial tight junctions and their response to lipopolysaccharide challenge. Invest Ophthalmol Vis Sci. 2000;41:4093–4100. [PubMed] [Google Scholar]