

Abstract
Mahogunin Ring Finger-1 (Mgrn1) null mutant mice have a pleiotropic phenotype that includes the absence of yellow hair pigment, abnormal head shape, reduced viability and adult-onset spongiform neurodegeneration. Mgrn1 encodes a highly conserved E3 ubiquitin ligase with 4 different isoforms which are differentially expressed and predicted to localize to different subcellular compartments. To test whether loss of specific isoforms causes different aspects of the mutant phenotype, we generated transgenes for each isoform and bred them onto the null mutant background. Mice expressing only isoform I or III appeared completely normal. Isoform II rescued or partially rescued the mutant phenotypes, while isoform IV had little or no effect. Our data show that different Mgrn1 isoforms are not functionally equivalent in vivo and that the presence of only isoform I or III is sufficient for normal development, pigmentation and neuronal integrity.
Keywords: Mahogunin Ring Finger-1, isoforms, spongiform neurodegeneration, pigmentation, craniofacial patterning, transgenesis
INTRODUCTION
The E3 ubiquitin ligase MGRN1 is likely to have multiple targets with diverse and important physiologic functions since Mgrn1 null mutant mice have a pleiotropic phenotype that includes hyperpigmentation (Miller et al., 1997), abnormal craniofacial patterning, reduced embryonic viability due to mispatterning of the left-right body axis (Cota et al., 2006), curly hair and whiskers, and progressive adult-onset spongiform neurodegeneration associated with mitochondrial dysfunction and elevated oxidative stress (He et al., 2003b; Sun et al., 2007). Heterozygotes for the Mgrn1md-nc (null) allele have no discernable phenotypes. Hypomorphic mutants that express Mgrn1 at reduced levels have mild pigmentation phenotypes, where their dorsal hairs are dark but hairs on their belly and flank are agouti-banded (He et al., 2003b; Miller et al., 1997). Mice homozygous for the Mgrn1md-2J allele, which results from an intracisternal A particle (IAP) insertion in exon 13, also have reduced viability associated with left-right patterning defects but do not develop spongiform neurodegeneration (Bagher et al., 2006; Cota et al., 2006). It is unlikely that Mgrn1md-2J mice express any full-length, wild-type Mgrn1, given the nature of their mutation. These observations suggest that spongiform neurodegeneration is only manifested in mice completely lacking MGRN1, while pigment-type switching is more sensitive to MGRN1 levels. It is unclear whether embryonic patterning defects reflect more severe reductions in MGRN1 levels or the importance of MGRN1 sequences downstream of the region encoded by exon 13.
The only reported ubiquitination target of MGRN1 is tumor susceptibility gene 101 (TSG101), a component of the endocytic trafficking machinery (Kim et al., 2007). MGRN1 multimonoubiquitinates TSG101 and appears to regulate its activity, but it is not known whether loss of TSG101 ubiquitination directly contributes to any of the defects observed in Mgrn1 null mutant mice. The pigmentation phenotype of Mgrn1 mutants implicates MGRN1 in the agouti-melanocortin receptor pathway, which mediates the normal switch in pigment synthesis between eumelanin (black/brown pigment) and pheomelanin (yellow/red pigment) during the mid-point of the hair growth cycle (He et al., 2003a). Activation of MC1R results in eumelanin production. Pheomelanin is produced when MC1R is inactive, either due to presence of agouti signaling protein (which acts as an inverse agonist of MC1R) or homozygosity for a loss-of-function mutation in Mc1r itself (reviewed by Wolff, 2003). As Mgrn1 acts genetically upstream of Mc1r but downstream of agouti transcription, it is possible that MGRN1 ubiquitinates the MC1R or proteins involved in its down-regulation and/or desensitization, but its target(s) in this pathway have not yet been identified.
Alternative splicing produces four Mgrn1 isoforms (Figure 1 and Bagher et al., 2006). Inclusion of exon 12 incorporates an additional 22 amino acids in isoforms II and IV, while alternative splicing of exon 17 results in the last 13 amino acids of isoforms I and II being different from the last 37 amino acids of isoform III and IV. These Mgrn1 isoforms have tissue specific expression patterns (Bagher et al., 2006). In wild-type adult skin, Mgrn1 isoforms II and IV were most abundant, while isoform III was expressed at low levels and isoform I was not detectable. In the brain, isoform I was strongly expressed, isoform II and III were present at lower levels, and isoform IV was detected at very low levels. These differences in expression suggest that different MGRN1 isoforms have different physiologic functions and that loss of specific isoforms may underlie specific aspects of the Mgrn1 null mutant phenotype.
Figure 1.
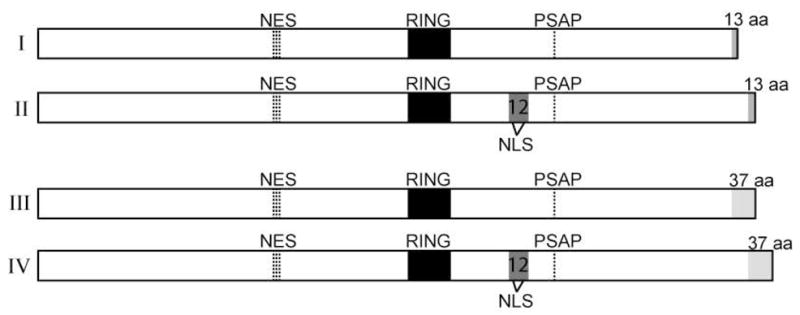
MGRN1 isoforms. Mgrn1 encodes a C3HC4 RING domain-containing E3 ubiquitin ligase (RING domain indicated by black box). Alternative splicing of exon 12 results in a 22 aa insertion carboxy-terminal to the RING domain in isoforms II and IV (indicated by gray box, labeled 12). This sequence contains a putative bipartite nuclear targeting sequence (NLS, sequence: KKSKSHPASLASKKPKR, at amino acids (aa) 359-375). All MGRN1 isoforms also contain a putative nuclear export signal (NES, sequence: DLDRGVFPVVIQ at aa 191-202) and a sequence (PSAP) that has been shown to mediate its association with tumor susceptibility gene 101 (TSG101). Alternative splicing of exon 17 results in the last 13 aa of isoforms I and II differing from the last 37 aa of isoforms III and IV (indicated by gray shading). Isoform numbers are indicated by roman numerals on the left. 70×27mm (300×300 DPI)
To test whether the four Mgrn1 isoforms have distinct functions in vivo, we generated transgenic mice that ubiquitously over-express each isoform and mated them onto the Mgrn1 null background (Mgrn1md-nc/md-nc). The presence of only isoform I or III was sufficient for normal development, pigmentation and neuronal integrity, while isoforms II and IV had limited or no effect on Mgrn1 null mutant phenotypes. One isoform I line completely rescued all phenotypes while the other showed only partial rescue of pigment-type switching and spongiform neurodegeneration, suggesting a position effect on expression of the transgene. These isoform I lines provide a system to assess the contribution of potential causative mechanisms to neurodegeneration; for example, they had equivalent levels of cytochrome c oxidase activity in mitochondria isolated from their brains, indicating that mitochondrial dysfunction is not likely to be the primary cause of spongiform neurodegeneration. Our data indicate that future studies to investigate the function of MGRN1 at the cellular level should focus on isoforms I and III, and that their common targets are important for normal development and neuronal integrity.
RESULTS
Generation of Mgrn1 transgenic mice
To determine the in vivo function of MGRN1 isoforms, transgenes were generated to express the full-length Mgrn1 cDNA of each isoform under control of the human β actin (ACTB) promoter. Based on previous studies (He et al., 2001; Walker et al., 2007), this transgene vector was expected to result in ubiquitous expression. Two independent transgenic founders were obtained for each isoform and mated to Mgrn1md-nc/+ and Mgrn1md-nc/md-nc mice to obtain transgenic animals heterozygous for the Mgrn1 null allele. These animals were then mated to Mgrn1 null homozygotes to obtain Mgrn1 null mutants segregating for the transgene. Gross external examination of non-mutant mice carrying each transgene did not reveal any obvious phenotypes due to Mgrn1 over-expression. As the isoform III A1 line appeared to rescue all the known Mgrn1 null mutant phenotypes, a full phenotypic analysis was not performed on C6 line mice.
Pigmentation
The effect of each transgene on the Mgrn1 null mutant pigmentation phenotype was assessed by external examination (Figure 2). All Mgrn1 null mutant mice without the transgene were black. All Mgrn1 null mutant mice carrying isoform I (line C3) transgenes were agouti and indistinguishable from wild-type mice (Figure 2a and data not shown). Mgrn1 null mutants in the isoform I transgenic line B7 were dark agouti (Figure 2a), similar to mice homozygous for the hypomorphic Mgrn1XC712 allele (Bagher et al., 2006). One isoform II line (A3) had no effect on the Mgrn1 null mutant pigmentation phenotype (Figure 2b). Mice heterozygous for the isoform II transgene in the other line (A1) produced a limited amount of yellow pigment on their hairs (Figure 2c), while transgene homozygotes were only slightly darker than wild-type mice (Figure 2d). Mgrn1 null mutant mice carrying an isoform III transgene (line A1 or C6) had agouti coats indistinguishable from wild-type mice (Figure 2e and data not shown), while those carrying an isoform IV transgene (line A2 or A3) were black and indistinguishable from null mutants lacking the transgene (Figure 2f and data not shown). Mgrn1 null mutant mice also have curly hair and whiskers, but since the penetrance of this phenotype can be influenced by modifier genes and the transgenic mice were on a mixed genetic background, it was not possible to assess this phenotype with certainty.
Figure 2.

Mgrn1 isoforms I, II and III alter the pigmentation phenotype of Mgrn1 null mutant mice. (a) While Mgrn1 null mutant mice have black rather than agouti hairs, mutants carrying isoform I transgene had dark agouti (line B7) or normal agouti (line C3) hairs. (b) Mgrn1 null mutant mice carrying the isoform II, line A3 transgene were indistinguishable from non-transgenic mutants. (c–d) The hairs of Mgrn1 null mutant mice heterozygous for the isoform II, line A1 transgene contained a very small amount of yellow pigment (c), while transgenic homozygotes had agouti-banded hairs and appeared only slightly darker than wild-type mice (d). (e) Mgrn1 null mutant mice carrying isoform III transgenes were indistinguishable from wild-type mice (line A1 shown; line C6 looked similar and is not shown). (f) Mgrn1 null mutant mice carrying isoform IV transgenes were indistinguishable from non-transgenic mutant mice (line A3 shown; line A2 looked similar and is not shown). 63×70mm (400×400 DPI)
Craniofacial patterning
We had previously noted that face shape of Mgrn1 null mutant mice is abnormal, with their eyes being more deep-set (Figure 3a) and their skull vault having a more domed appearance (Figure 3b) than wild-type mice. The dimensions of the calvarial plates of inbred Mgrn1 null mutant and wild-type (heterozygous) mice were measured after digesting tissues away with papain. Significant differences (p<0.05) were observed in the length, width and height of mutant skulls. The distance from the tip of the snout to the posterior end of the frontal plate was 4.0% shorter in Mgrn1 null mutants than in heterozygotes, while the distance from the tip of the snout to the posterior end of the parietal plate was 4.4% shorter in null mutant mice (Figure 3c–e and Table 1). The posterior ends of the frontal and parietal plates of Mgrn1 null mutants were 11.5% wider and 6.4% narrower, respectively, while the skull vault, at its highest point, was 10.9% higher in null mutants than in control (heterozygous) mice (Figure 3c–g and Table 1). These measurements did not differ significantly between male and female mice of the same genotype. The developmental aspects of this phenotype will be described in detail elsewhere.
Figure 3.

Abnormal craniofacial patterning in Mgrn1 null mutants. (a) The faces of Mgrn1 null mutant mice appear broader than those of wild-type mice and their eyes appear to be more deep-set. (b) From the side, the skull vault of Mgrn1 null mutant mice appears to be more domed (arrow) than that of wild-type mice. (c) Sketch of top-view of a mouse skull, showing parietal and frontal plates. The length measurements that differed significantly between wild-type and Mgrn1 null mutant mice are indicated. (d–e) Top-view of Mgrn1 null mutant (d) and wild-type (e) skulls. (f) Sketch of side-view of a mouse skull. The height of vault measurement that differed significantly between wild-type and Mgrn1 null mutant mice are indicated. (g) Side-view of Mgrn1 null mutant (left) and wild-type (right) skulls. Arrow indicates the area of increased vault height of the mutant skull. 63×38mm (400×400 DPI)
Table 1.
Abnormal head shape of Mgrn1 null mutant mice.
| Measurement (SD)1 |
|||
|---|---|---|---|
| Skull element2 | Mgrn1md-nc/+ | Mgrn1md-nc/md-nc | % difference3 |
| Length: snout-frontal | 14.16 (0.48) | 13.59 (0.52) | −4.0 (p<0.0001) |
| n=21 | n=37 | ||
| Length: snout-parietal | 17.86 (0.29) | 17.08 (0.67) | −4.4 (p<0.0001) |
| n=20 | n=34 | ||
| Width of frontal | 5.98 (0.63) | 6.67 (0.28) | 11.5 (p<0.0001) |
| n=26 | n=42 | ||
| Width of parietal | 9.77 (0.13) | 9.14 (0.22) | −6.4 (p<0.0001) |
| n=17 | n=34 | ||
| Height of vault | 6.88 (0.14) | 7.63 (0.18) | 10.9 (p<0.0001) |
| n=20 | n=34 | ||
In millimeters, measured with digital calipers; SD: standard deviation.
Sample sizes (n) are variable as some skull structures were damaged during preparation/handing and could not be measured.
Elements measured as indicated in Figure 3c,f.
Mgrn1 null mutants smaller than wild-type when value is negative, larger when value is positive.
In order to assess the potential role of each Mgrn1 isoform in skull development, we measured the skull plates of Mgrn1 null mutant mice carrying each transgene and compared them to non-transgenic null mutants of the same mixed genetic background. We also compared their measurements to wild-type (Mgrn1md-nc/+) mice to determine whether structures that differed significantly from those of null mutant mice were partially rescued or fully restored to normal by any given transgene. Isoforms I and II were capable of rescuing all aspects of the craniofacial patterning phenotype: the length and width of the frontal and parietal bones and height of the vault were the same as wild-type mice and significantly different from non-transgenic null mutants (Table 2). For isoform III, all craniofacial measurements of Mgrn1 null mutant mice carrying the transgene differed significantly from those of non-transgenic null mutant mice and most did not differ significantly from wild-type measurements (Table 2); the one exception was the width of the parietal bone, which was intermediate to that of wild-type and null mutant mice, indicating partial rescue of this phenotype. A similar pattern was observed for both lines of mice carrying the isoform IV transgene (Table 2), where their craniofacial measurements were all significantly different from those of non-transgenic Mgrn1 null mutants and either the same as wild-type mice (width of parietal plate and both skull length measurements) or intermediate to those of wild-type and mutant mice (width of frontal plate and height of skull vault). It is not possible at this time to determine whether the failure of isoforms III and IV to completely rescue some aspects this phenotype is due to differences in their amino acid sequence (relative to isoforms I and II) or in the timing, pattern or levels of the expression of these transgenes during development.
Table 2.
Effect of transgenic over-expression of individual Mgrn1 isoforms on head shape.
| Measurement in mm (standard deviation)1 |
||||||||
|---|---|---|---|---|---|---|---|---|
| Skull element2 |
Non-transgenic Mgrn1md-nc/md-nc |
Mgrn1md-nc/md-nc; Tg(Mgrn1I)B7 |
Mgrn1md-nc/md-nc; Tg(Mgrn1I)C3 |
Mgrn1md-nc/md-nc; Tg(Mgrn1II)A1 |
Mgrn1md-nc/md-nc; Tg(Mgrn1II)A3 |
Mgrn1md-nc/md-nc; Tg(Mgrn1III)A1 |
Mgrn1md-nc/md-nc; Tg(Mgrn1IV)A2 |
Mgrn1md-nc/md-nc; Tg(Mgrn1IV)A3 |
| Length:snout-frontal | 13.82 (0.72) | 14.17* (0.94) | 14.53§ (0.46) | 14.34* (0.58) | 14.45§ (0.58) | 14.27* (1.1) | 14.37* (0.81) | 14.22* (0.74) |
| n=75 | n=31 | n=38 | n=31 | n=46 | n=28 | n=15 | n=23 | |
| Length: snout-parietal | 17.01 (0.76) | 17.75* (0.95) | 18.42§ (0.41) | 18.07* (0.62) | 18.23§ (0.72) | 17.85* (0.91) | 17.56* (0.95) | 17.67* (0.85) |
| n=75 | n=31 | n=38 | n=28 | n=44 | n=25 | n=15 | n=23 | |
| Width of frontal | 6.68 (0.25) | 6.02* (0.28) | 5.95* (0.19) | 5.93* (0.35) | 5.87* (0.34) | 6.19* (0.38) | 6.42† (0.30) | 6.37† (0.29) |
| n=78 | n=31 | n=40 | n=36 | n=46 | n=32 | n=15 | n=25 | |
| Width of parietal | 9.38 (0.37) | 9.85* (0.30) | 9.92* (0.32) | 9.82* (0.34) | 10.00§ (0.25) | 9.56† (0.35) | 9.62* (0.32) | 9.66* (0.65) |
| n=78 | n=29 | n=38 | n=31 | n=44 | n=30 | n=15 | n=25 | |
| Height of vault | 7.62 (0.25) | 6.83* (0.25) | 7.09† (0.16) | 6.73§ (0.26) | 6.93* (0.27) | 6.96* (0.15) | 7.25† (0.28) | 7.37† (0.17) |
| n=78 | n=31 | n=40 | n=32 | n=46 | n=29 | n=15 | n=25 | |
Sample sizes (n) vary within genotype as some skull structures could not be measured due to damage sustained during tissue removal and/or during handling.
Elements measured as indicated in Figure 3c,f.
Significantly different (p<0.05) from non-transgenic Mgrn1 null mutants.
Significantly different (p<0.05) from non-transgenic Mgrn1 null mutants and wild-type (Mgrn1md-nc/+, data in Table 1), with transgenic mice having a “more extreme” normal phenotype (i.e., longer or wider heads or shorter skull vaults than wild-type, where mutants are shorter, narrower or taller).
Significantly different (p<0.05) from both wild-type (Mgrn1md-nc/+, data in Table 1) and non-transgenic null mutant mice; intermediate phenotype.
Viability
Almost half of Mgrn1 null mutants die before or shortly after birth as a result of lethal congenital heart defects that arise as a consequence of abnormal patterning of the left-right body axis (Cota et al., 2006). To assess whether loss of a specific Mgrn1 isoform is responsible for this phenotype, we mated transgenic Mgrn1 null mutant mice to non-transgenic null mutants and determined the proportion of weaned pups that carried the transgene. If significantly more transgenic than non-transgenic Mgrn1 null mutant pups survived to weaning, this would indicate that presence of the transgene rescues the embryonic patterning defects to confer survival. If a transgene did not increase viability, equal numbers of transgenic and non-transgenic Mgrn1 null mutant pups should be observed at weaning age. As shown in Table 3, significantly more Mgrn1 null mutants survived when they carried an isoform I (line B7 or C3), II (line A3) or III (line A1) transgene. Isoform II line A1 and isoform IV line A3 appeared to partially rescue the phenotype as the number of transgenic pups weaned did not differ significantly from that expected if the transgene completely rescued the phenotype nor from that expected if the transgene had no effect on viability (although the small size of the isoform II line A1 population limits the power of the test) (Table 3). The isoform IV line A2 transgene appeared to further reduce the viability of Mgrn1 null mutant mice, suggesting disruption of an endogenous locus at the transgene insertion site. We did observe complete situs inversus in one adult Mgrn1 null mutant carrying the isoform IV (line A2) transgene, indicating that left-right patterning is still disrupted in those animals and would be expected to contribute to their reduced survival.
Table 3.
Viability of Mgrn1 transgenic mice.
| # transgenic pups | # non-transgenic pups | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Transgene (line) | # pups weaned1 | Obs | Exp1 | Exp2 | Obs | Exp1 | Exp2 | χ2 (p), no rescue | χ2 (p) for full rescue |
| I(B7) | 133 | 88 | 66.5 | 86.45 | 45 | 66.5 | 46.55 | 13.90 (p<0.001) | 0.079 (p>0.75) |
| I(C3) | 217 | 130 | 108.5 | 141.05 | 87 | 108.5 | 75.95 | 8.52 (p<0.004) | 2.47 (p>0.1) |
| II(A1) | 30 | 20 | 15.0 | 19.50 | 10 | 15.0 | 10.50 | 3.33 (0.10>p>0.05) | 0.037 (p>0.8) |
| II(A3) | 23 | 17 | 11.5 | 14.95 | 6 | 11.5 | 8.05 | 5.26 (p<0.025) | 0.81 (p>0.35) |
| III(A1) | 126 | 79 | 63.0 | 81.90 | 47 | 63.0 | 44.10 | 8.13 (p<0.005) | 0.29 (p>0.5) |
| III(C6)3 | 120 | 69 | 60.0 | 78.00 | 51 | 60.0 | 42.00 | 2.70 (p=0.10) | 2.97 (0.1>p>0.05) |
| IV(A2)4 | 46 | 15 | 23.0 | 29.90 | 31 | 23.0 | 16.10 | 5.57 (p<0.02) | 21.21 (p<0.001) |
| IV(A3)3 | 46 | 27 | 23.0 | 29.90 | 19 | 23.0 | 16.10 | 1.39 (p>0.2) | 0.80 (p>0.35) |
From (Tg+/−; Mgrn1md-nc/md-nc × non-transgenic Mgrn1md-nc/md-nc) matings, where Tg+/− = transgene heterozygote.
Obs: number observed (actually weaned); Exp1: number expected if no rescue (50% for each genotype); Exp2: number expected if full rescue (based on prior observations, Cota et al., 2006)
Transgene partially rescues viability.
Excess of non-transgenic Mgrn1 null mutants indicates that the transgene significantly further reduces viability.
CNS vacuolation and mitochondrial dysfunction
Mgrn1 null mutant mice develop progressive, adult-onset spongiform neurodegeneration (He et al., 2003b). We first determined at what age vacuolation of the central nervous system (CNS) was sufficiently robust that we could unequivocally score whether vacuoles were present or absent in transgenic animals. Histological analysis of 6, 9 and 12 month-old non-transgenic Mgrn1 null mutant mice indicated that the brains from 12 month-old mutants had consistent and significant vacuolation. This age was therefore selected for subsequent analysis of transgenic mice. Consistent with previous observations (He et al., 2003b), spongiform changes were observed in many brain regions of the null mutant mice, including the cerebral cortex, hippocampus, thalamus, brain stem and cerebellum granule layer (Figure 4 and data not shown). The most prominent site of vacuolation was the thalamus.
Figure 4.

Brain histology of 12 month-old wild-type and non-transgenic and transgenic Mgrn1 null mutant mice. Wild-type mice did not display CNS vacuolation in any brain region. Non-transgenic Mgrn1 null mutant mice showed significant vacuolation in many brain regions, including the hippocampus, thalamus, cerebellum and pons. The brains of Mgrn1 null mutant mice carrying the isoform I line B7 transgene (Tg(Mgrn1I)B7) were vacuolated, while mutant mice carrying the isoform I line C3 transgene (Tg(Mgrn1I)C3) appeared normal. Mgrn1 null mutant mice carrying an isoform II transgene (both lines, Tg(Mgrn1II)A1 and Tg(Mgrn1II)A3) show variable CNS vacuolation, ranging from mild to severe. Mgrn1 null mutant mice carrying an isoform III transgene (Tg(Mgrn1III)A1) did not develop spongiform neurodegeneration, while those carrying an isoform IV transgene (both lines, Tg(Mgrn1IV)A2 and Tg(Mgrn1IV)A3) showed CNS vacuolation as severe as non-transgenic mutant mice. All images shown at same magnification. 63×31mm (400×400 DPI)
The brains of most twelve-month-old Mgrn1 null mutants carrying isoform I (C3 line) or isoform III (A1 line) transgenes showed no significant vacuolation, although one or two animals of each line displayed very mild vacuolation, usually in the cerebellum (Figure 4 and Table 4). At the same age, Mgrn1 null mutants heterozygous for the isoform I (B7 line) or isoform II (A1 or A3 line) transgenes developed vacuolation with the same distribution but less severe (fewer vacuoles present) than in age-matched non-transgenic null mutants (Figure 4 and Table 4). The brains of null mutants carrying the isoform I B7 line transgene generally contained fewer vacuoles than those of isoform II transgenic mice, but this phenotype tended to be more variable in isoform II transgenic mice, with some animals having no significant lesions in some brains regions while vacuolation in others was as severe as in the brains of non-transgenic null mutants. Twelve-month-old Mgrn1 null mutants carrying isoform IV (line A2 or A3) transgenes consistently developed extensive vacuolation, although not always as severe as in age-matched non-transgenic Mgrn1 null mutants (Figure 4 and Table 4). Mgrn1 expression in the brains of all transgenic lines was confirmed using RT-PCR (Figure 5).
Table 4.
Effect of transgenic over-expression of individual Mgrn1 isoforms on CNS vacuolation.1
| Brain region2 | Wild-type (n=6) |
Non-transgenic Mgrn1md-nc/md-nc (n=3) |
Mgrn1md-nc/md-nc; Tg(Mgrn1I)B7 (n=5) |
Mgrn1md-nc/md-nc; Tg(Mgrn1I)C3 (n=5) |
Mgrn1md-nc/md-nc; Tg(Mgrn1II)A1 (n=5) |
Mgrn1md-nc/md-nc; Tg(Mgrn1II)A3 (n=5–6) |
Mgrn1md-nc/md-nc; Tg(Mgrn1III)A1 (n=5) |
Mgrn1md-nc/md-nc; Tg(Mgrn1IV)A2 (n=5) |
Mgrn1md-ncnc/md-nc; Tg(Mgrn1IV)A3 (n=5–6) |
|---|---|---|---|---|---|---|---|---|---|
 |
NSL (6) | +++ (1) | ++ (4) | NSL (4) | ++ (3) | + (2) | NSL (5) | ++ (2) | ++ (2) |
| ++++ (2) | +++ (1) | + (1) | +++ (2) | ++ (1) | +++ (2) | +++ (3) | |||
| +++ (3) | ++++ (1) | ++++ (1) | |||||||
 |
NSL (6) | ++++ (3) | ++ (3) | NSL (1) | ++ (2) | + (1) | NSL (3) | ++ (2) | +++ (3) |
| +++ (2) | + (4) | +++ (2) | ++ (3) | + (2) | +++ (2) | ++++ (2) | |||
| ++++ (1) | +++ (1) | ++++ (1) | |||||||
| ++++ (1) | |||||||||
 |
NSL (6) | +++ (3) | + (2) | NSL (2) | NSL (1) | + (3) | NSL (3) | ++ (4) | ++ (3) |
| ++ (3) | + (2) | + (3) | ++ (2) | + (1) | +++ (1) | +++ (3) | |||
| ++ (1) | ++ (1) | ++ (1) | |||||||
 |
NSL (6) | +++ (3) | + (4) | NSL (3) | ++ (4) | NSL (1) | NSL (5) | ||
| ++ (3) | ++ (3) | ||||||||
| ++ (1) | + (2) | +++ (1) | + (1) | +++ (2) | +++ (2) | ||||
| ++ (3) |
Scored as NSL for no significant lesions, + for rare, scattered vacuoles, ++ for mild (2–5 vacuoles/region/section); +++ for moderate (6–10 vacuoles/region/section), ++++ for severe (>10 vacuoles/region/section) vacuolation. Number of animals with given phenotype indicated in parentheses following +/− score
Images from C57BL/6J Mouse Brain Library, www.mbl.org (Williams, 2000). Boxes indicate region scored.
Figure 5.

Mgrn1 is expressed in the brains of all transgenic lines. Top panel: RT-PCR was used to assess Mgrn1 expression in the brains of Mgrn1 null mutant mice carrying each transgene (abbreviated as Tg(isoform #)line#, i.e., Tg(I)C3 for isoform I line C3). Control reactions for wild-type and non-transgenic Mgrn1 null mutant brain RNA samples and no template (water) are also shown. Bottom panel: RT-PCR of the same samples for β-actin. 177×60mm (300×300 DPI)
We previously identified mitochondrial dysfunction and elevated oxidative stress in the brains of 1-month-old Mgrn1 null mutant mice (Sun et al., 2007). Significant reductions were detected in the expression of several mitochondrial proteins and levels of adenosine triphosphate (ATP) and mitochondrial cytochrome c oxidase activity in brains of Mgrn1 null mutant mice. The appearance of these biochemical phenotypes well before the onset of spongiform changes suggested that mitochondrial function may be a causative mechanism of neurodegeneration in these mice. We examined cytochrome c oxidase activity levels in mitochondria isolated from the brains of 1-month-old isoform I (lines B7 and C3) and III (line A1) transgenic mice to determine whether this could be used as an early marker of neurodegeneration. All of these transgenic mice had similar cytochrome c oxidase activity levels, which were intermediate to and not significantly different from those of Mgrn1 null mutants and heterozygous or wild-type mice (Figure 6).
Figure 6.

Cytochrome c oxidase activity levels in mitochondria isolated from wild-type and transgenic (isoforms I and III) and non-transgenic Mgrn1 null mutant mouse brains. Enzyme activity levels were significantly reduced (* p<0.05) in Mgrn1 null mutants relative to Mgrn1 heterozygotes and wild-type mice, indicating mitochondrial dysfunction. Enzyme activity levels for Mgrn1 null mutants carrying isoform I (line B7 or C3) and III transgenes were intermediate to and not significantly different from those of non-transgenic Mgrn1 null mutants and wild-type (Mgrn1md-nc/+ and Mgrn1+/+) mice. WT: wild-type, het: heterozygotes. 190×143mm (300×300 DPI)
MGRN1 isoform I and IV subcellular localization
The fact that isoform II and IV transgenes had either no effect or only a limited effect on most of the Mgrn1 null mutant phenotypes suggests that the 22 amino acids encoded by exon 12 affect the function of MGRN1. Bioinformatic analysis (www.expasy.ch/prosite) revealed a bipartite nuclear targeting sequence in the sequence encoded by exon 12, which is only present in isoforms II and IV (Figure 1). All MGRN1 isoforms also contain a putative nuclear export signal (Figure 1). This suggests that MGRN1 isoforms II and IV may be capable of translocation into and out of the nucleus, in which case they would be expected to have different targets and cellular function(s) than isoforms I and III. As there are no antibodies available that distinguish MGRN1 isoforms I and III from II and IV, we generated GFP-tagged expression constructs for isoforms I and IV, transiently transfected them into HEK293T cells, and examined the distribution of the GFP signal by confocal microscopy. GFP-tagged isoform I was widely distributed throughout the cytoplasm (Figure 7a), while GFP-tagged isoform IV was observed in the nucleus as well as in the cytoplasm (Figure 7b). The distribution of the two isoforms in the cytoplasm differed: while isoform I was broadly distributed, isoform IV was mostly localized to the perinuclear region and discrete vesicular structures. These observations suggest that the distribution of MGRN1 may regulate its cellular function(s), with little or no isoform II or IV being targeted to the cellular location(s) required for the essential physiological functions of MGRN1.
Figure 7.

Subcellular localization of MGRN1 isoforms I and IV. HEK293T cells were transiently transfected with C-terminal GFP-tagged MGRN1 isoform I or IV expression constructs. (a) GFP-tagged isoform I was widely distributed throughout the cytoplasm but excluded from nuclei. (b) GFP-tagged isoform IV was present in the nucleus and in the cytoplasm. In the nucleus, it was broadly distributed and appeared to be excluded from nucleoli (arrowheads). Cytoplasmic isoform IV predominantly localized to the perinuclear region and discrete vesicular structures. 63×36mm (400×400 DPI)
DISCUSSION
The existence of several MGRN1 isoforms that are expressed at different levels in different tissues suggested that loss of specific isoforms may be responsible for different aspects of the Mgrn1 null mutant phenotype. We used transgenesis to examine the in vivo role of each isoform. Isoforms I and III rescued every Mgrn1 null mutant phenotype examined, while isoform II rescued the embryonic viability and craniofacial patterning phenotypes but only partially rescued the pigmentation and spongiform neurodegeneration phenotypes. Isoform IV had a small but significant effect on skull patterning and no discernable effect on the other phenotypes examined. These results indicate that Mgrn1 isoforms are not functionally equivalent. Isoforms I and III appear able to perform all the functions of Mgrn1 that lead to the known phenotypes of Mgrn1 null mutant mice. Isoform II only appears able to compensate for loss of MGRN1 when expressed at very high levels, while isoform IV does not appear able to replace other isoforms in the skin or brain. All MGRN1 isoforms contain a putative NES, while only isoforms II and IV contain a putative NLS. This suggested that isoforms II and IV may be targeted to the nucleus, in which case their cellular function would be expected to differ from that of isoforms I and III. Analysis of the expression patterns of GFP-tagged expression constructs confirmed that the subcellular distribution of isoforms I and IV differ, with isoform I being broadly distributed in the cytoplasm and isoform IV being found in the nucleus and on vesicles in the cytoplasm. Since isoforms I and III best rescue the null mutant phenotypes, it would appear that cytoplasmic MGRN1 performs all of its known essential functions.
The pigmentation, spongiform neurodegeneration, craniofacial and embryonic lethality phenotypes of some Mgrn1 transgenic mice were intermediate to those of wild-type and null mutant mice. This demonstrates that none of the Mgrn1 null mutant phenotypes are threshold (“all-or-none”) traits. In the past, spongiform neurodegeneration had only been observed in mice completely lacking Mgrn1 expression. The observation of less severe vacuolation in Mgrn1 null mutants carrying isoform I (B7 line) or II (both lines) transgenes suggests that expression of some Mgrn1 may delay the onset or progression of CNS vacuolation. Further studies will be needed to understand why they still develop vacuoles while hypomorphic mutants such as Mgrn1md and Mgrn1md-2J do not, but the fact that another isoform I line completely rescued this phenotype suggests that the level of Mgrn1 expression or the cell type in which it is expressed may be important.
Mitochondrial dysfunction has been implicated in the pathogenesis of many neurodegenerative disorders and Mgrn1 null mutant mice have previously been shown to exhibit mitochondrial dysfunction by 1 month of age (Sun et al., 2007). The fact that mitochondrial dysfunction could be detected well before the onset of histologically-evident vacuoles suggested that this may be a primary cause and early marker of spongiform neurodegeneration in Mgrn1 null mutant mice. Our present study suggests this is not the case, however, as isoform I-B7 mice developed spongiform neurodegeneration while isoform I-C3 and isoform III-A1 mice did not, yet they all had similar cytochrome c oxidase activity levels. The fact that CNS vacuolation was less severe in Mgrn1 null mutants carrying the isoform I line B7 transgene than in non-transgenic mutants suggests that improved mitochondrial function may delay either the age-of-onset or subsequent progression of spongiform neurodegeneration in these animals. The isoform I B7 and C3 lines provide a powerful system for assessing other potential causes of the spongiform neurodegeneration phenotype in the future.
Interestingly, the ability of Mgrn1 isoforms to rescue the spongiform neurodegeneration correlated with their level of expression in normal adult brain, where isoform I is most abundant, isoforms II and III are present at lower levels, and isoform IV is only very weakly expressed (Bagher et al., 2006). The ability of different isoforms to rescue the Mgrn1 null mutant pigmentation phenotype did not correlate with their normal levels of expression in adult skin, however. In normal adult skin, isoforms II and IV are most abundant, isoform III is weakly expressed and isoform I is not detectable. It was therefore unexpected that isoforms I and III could completely rescue the pigmentation phenotype of Mgrn1 null mutants while isoform II had an incomplete effect and isoform IV had none. As Mgrn1 acts genetically upstream of Mc1r, a likely role for MGRN1 in pigment type switching is to regulate MC1R down-regulation and/or desensitization to affect its activity. In that scenario, it is not surprising that MGRN1 isoforms that are predicted to localize to the nucleus do not rescue the Mgrn1 pigmentation phenotype. Subcellular localization could also explain why Mgrn1 null mutants homozygous for the isoform II line A1 transgene had more yellow pigment in their coat hairs than null mutants heterozygous for the transgene if the higher level of expression in homozygotes results in more protein being present in the cytoplasm. The high expression levels of isoforms II and IV in normal skin suggest they may have other, unknown role(s) in skin that do not affect pigment-type switching. Mgrn1 null mutant mice also have curly hair, but this phenotype is unlikely to be dependent upon isoform II or IV as some Mgrn1 null mutant mice carrying those transgenes still had curly hair and whiskers. It is possible that a more subtle hair or skin phenotype exists that has not yet been identified or that another role of MGRN1 in the skin that is redundant with another E3 ubiquitin ligase that compensates for loss of MGRN1 in Mgrn1 null mutant mice.
The craniofacial patterning phenotype of Mgrn1 null mutant mice is described here for the first time. Significant differences were observed in the length, width and height of the frontal and parietal bones of the skull vault, indicating a role for MGRN1 in calvarial development. These bones are thought to be derived at least in part from the cranial neural crest (Couly et al., 1993). Melanocytes are also derived from the neural crest, and we have observed that some Mgrn1 null mutants have a small white belly spot in addition to a pigment-type switching defect. Furthermore, some of the congenital heart defects observed in Mgrn1 null mutant mice, such as atrial ventricular septal defects and malposition of the great arteries, could result from abnormal left-right patterning or cardiac neural crest defects (Cota et al., 2006; Gittenberger-de Groot et al., 2005). This raises the possibility that MGRN1 may have a general function in neural crest cells that affects their migration, survival, differentiation or function. While examination of the consequences of loss of Mgrn1 specifically in neural crest cells would require generation of a conditional allele, Mgrn1 null mutant mice expressing isoform I or III under control of a neural crest specific promoter could be used to test whether restoration of Mgrn1 expression in these cells rescues the craniofacial patterning, pigmentation and/or outflow tract or artery defects.
E3 ubiquitin ligases have important roles in regulating the trafficking, turnover and/or function of their targets. For MGRN1, only one target (TSG101) has been identified to date, but it is likely to ubiquitinate multiple proteins. All MGRN1 isoforms contain the RING domain and are expected to have ubiquitin ligase activity, but the fact that different isoforms had different effects on the phenotypes of Mgrn1 null mutant mice suggests that isoforms I and III may have different targets from isoforms II and IV. Moreover, loss of MGRN1-mediated ubiquitination of different targets is likely to underlie different aspects of the Mgrn1 null mutant phenotype. Knowing that isoforms I and III can rescue all known aspects of the Mgrn1 null mutant phenotype provides a foundation for future structure-function experiments that will provide insight into the physiological importance of MGRN1-dependent ubiquitination of specific targets.
METHODS
Mice
All mice were maintained under standard conditions in AALAC approved facilities at Cornell University. All experiments were IACUC approved. Mice carrying the null allele, Mgrn1md-nc, were originally obtained as frozen heterozygous F1 embryos (C3H X 101) from Harwell Mammalian Genetics Unit and have been maintained by brother-sister inbreeding for over 21 generations. Mice were genotyped for the Mgrn1md-nc mutation (T to A transversion in the second position of intron 9) by Transnetyx, Inc. (Cordova, TN). Transgenic mice were generated as previously described (Bagher et al., 2006). Briefly, full-length Mgrn1 isoform I, II, III and IV cDNAs were amplified by RT-PCR (isoform I and II primers: GGCTGGCCGGTAGAAAC and GGACAACTGGCCTGGGAAAGACC; isoform III and IV primers: TTTTGGGCCATGTCCGTGTGA and CACGTGGCCCATGGAGACAGT) and cloned into pCR2.1-TOPO (Invitrogen, Carlsbad, CA). Clones were sequenced to confirm their identity (isoform) and sequence. Inserts were subcloned into pBluescript II KS(+/−), then into the transgene vector downstream of a 4.2-kb fragment consisting of the human ACTB promoter and first intron and upstream of a 460-bp SV40-derived fragment containing the small t intron and major polyadenylation site. After sequence verification, the final transgene cassettes were excised by digestion with XbaI and PsiI, gel purified (QIAEX II gel extraction kit; Qiagen, Valencia, CA, USA), eluted into DNA injection buffer (10 mM Tris, 0.1 mM ethylenediaminetetraacetic acid, pH 7.5), and injected into B6D2F2 embryos by the Cornell Transgenic Mouse Core Facility. Pups were genotyped by PCR using forward primer TGGTGAACATTCGCAAAGA in exon 3 and reverse primer CCGCCTGACAATAGATG in exon 4 to amplify a 669-bp band from the Mgrn1 genomic locus and a 156-bp band from each transgene.
Analysis of transgene expression
Brain RNA was isolated from at least two transgenic Mgrn1md-nc/md-nc (null mutant) mice for each transgenic line, non-transgenic Mgrn1 null mutants, and wild-type mice using Trizol reagent (Invitrogen). RNA samples were treated with DNase I to remove any potential DNA contamination since the presence of the transgene in the genomic DNA would produce the same size PCR product for Mgrn1 as the mRNA. Removal of DNA was confirmed by performing a PCR reaction for Mgrn1 (using the genotyping primers) without a reverse transcription (RT) step. RNA samples were then diluted to equal concentrations in diethylpyrocarbonate (DEPC) treated water and used as template for RT-PCR reactions using the Titanium One-Step RT-PCR kit (Clontech, Mountain View, CA) following the manufacturer’s instructions. PCR conditions (RNA concentration and cycle number) were optimized to allow for relative quantification of the products. The same primers described above for genotyping were used to amplify a 156 bp product from Mgrn1 mRNA, and the β-actin primers included with the Titanium kit (540 bp product) were used for a control reaction to assess quality and concentration of template RNAs.
Skull preparation and measurements
Mice were euthanized by CO2 inhalation, decapitated and the skin removed from heads prior to boiling them in water for 1 minute. After cooling, heads were incubated in papain solution (a “pinch” of papain in 0.85% NaCl) at 37°C overnight. Soft tissues and the brain were removed under running water and the skulls were then left to dry at room temperature. The length and width of the frontal and parietal bones (width measured at their posterior ends) and height of skull vault (at its highest point) were measured using a digital vernier caliper. Averages were calculated by sex for each genotype and statistical analyses performed using Student’s t-test. As no significant differences were observed between males and females of a given genotype, data for both sexes was pooled.
Brain histology
Brains of 6, 9 and/or 12 month-old transgenic Mgrn1md-nc/md-nc mice and their non-transgenic Mgrn1md-nc/md-nc littermates were dissected, fixed in 10% formalin, ethanol dehydrated and paraffin embedded for serial sectioning at 5 μm. Brain sections were stained with hematoxylin and eosin (H&E) following standard protocols and examined by light microscopy.
Cytochrome c oxidase activity
Cytochrome c oxidase activity was determined in mitochondria isolated from brains of 1 month-old Mgrn1 mutant, non-mutant and isoform I and III transgenic mice using the Cytochrome c Oxidase Assay Kit (Sigma, MO). A decrease in absorbance at 550 nm of ferrocytochrome c is caused by its oxidation to ferricytochrome c by cytochrome c oxidase. Absorbance at 550 nm was measured by spectrophotometry (Beckman DU640). Equal amounts of protein were assayed for each mitochondrial extract.
Expression constructs and cell culture
Full-length MGRN1 isoforms I and IV were amplified by RT-PCR from C3H/HeJ mouse brain RNA using forward primer GGGGACAAGTTTGTACAAAAAAGCAGGCTCCGGCTGGCCGGTAGAAAC and reverse primer GGGGACCACTTTGTACAAGAAAGCTGGGTACTATACCAACAGAGCACGAC (isoform I) or GGGGACCACTTTGTACAAGAAAGCTGGGGGTCAGCTCAGCAACTCCA (isoform IV). The products were cloned into a Gateway-converted pEGFP-N1 vector (Clontech). HEK293T cells were transiently transfected using a standard calcium phosphate protocol and imaged on a Zeiss LSM 510 Meta Confocal Microscope.
Acknowledgments
We thank Christina Cota for providing mouse skull photos and sketches, Dr. SeungWoo Jung for assistance with RNA extractions, and Ke-Yu Deng and the Cornell University Transgenic Core Facility for generation of the transgenic mice. This work was supported by the National Institute on Aging RO1AG022058 and AHA Scientist Development Grant 0330010N to T.M.G. The content is solely the responsibility of the authors and does not necessarily represent the official views of these agencies.
LITERATURE CITED
- Bagher P, Jiao J, Owen Smith C, Cota CD, Gunn TM. Characterization of Mahogunin Ring Finger-1 expression in mice. Pigment Cell Res. 2006;19:635–643. doi: 10.1111/j.1600-0749.2006.00340.x. [DOI] [PubMed] [Google Scholar]
- Cota CD, Bagher P, Pelc P, Smith CO, Bodner CR, Gunn TM. Mice with mutations in Mahogunin ring finger-1 (Mgrn1) exhibit abnormal patterning of the left-right axis. Dev Dyn. 2006;235:3438–3447. doi: 10.1002/dvdy.20992. [DOI] [PubMed] [Google Scholar]
- Couly GF, Coltey PM, Le Douarin NM. The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development. 1993;117:409–429. doi: 10.1242/dev.117.2.409. [DOI] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Bartelings MM, Deruiter MC, Poelmann RE. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr Res. 2005;57:169–176. doi: 10.1203/01.PDR.0000148710.69159.61. [DOI] [PubMed] [Google Scholar]
- He L, Eldridge AG, Jackson PK, Gunn TM, Barsh GS. Accessory proteins for melanocortin signaling: attractin and mahogunin. Ann N Y Acad Sci. 2003a;994:288–298. doi: 10.1111/j.1749-6632.2003.tb03192.x. [DOI] [PubMed] [Google Scholar]
- He L, Gunn TM, Bouley DM, Lu XY, Watson SJ, Schlossman SF, Duke-Cohan JS, Barsh GS. A biochemical function for attractin in agouti-induced pigmentation and obesity. Nat Genet. 2001;27:40–47. doi: 10.1038/83741. [DOI] [PubMed] [Google Scholar]
- He L, Lu XY, Jolly AF, Eldridge AG, Watson SJ, Jackson PK, Barsh GS, Gunn TM. Spongiform degeneration in mahoganoid mutant mice. Science. 2003b;299:710–712. doi: 10.1126/science.1079694. [DOI] [PubMed] [Google Scholar]
- Kim BY, Olzmann JA, Barsh GS, Chin LS, Li L. Spongiform neurodegeneration-associated E3 ligase Mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol Biol Cell. 2007;18:1129–1142. doi: 10.1091/mbc.E06-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KA, Gunn TM, Carrasquillo MM, Lamoreux ML, Galbraith DB, Barsh GS. Genetic studies of the mouse mutations mahogany and mahoganoid. Genetics. 1997;146:1407–1415. doi: 10.1093/genetics/146.4.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Johnson BS, Gunn TM. Mitochondrial dysfunction precedes neurodegeneration in Mahogunin (Mgrn1) mutant mice. Neurobiol Aging. 2007;28:1840–1852. doi: 10.1016/j.neurobiolaging.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker WP, Aradhya S, Hu CL, Shen S, Zhang W, Azarani A, Lu X, Barsh GS, Gunn TM. Genetic analysis of attractin homologs. Genesis. 2007;45:744–756. doi: 10.1002/dvg.20351. [DOI] [PubMed] [Google Scholar]
- Wolff GL. Regulation of yellow pigment formation in mice: a historical perspective. Pigment Cell Res. 2003;16:2–15. doi: 10.1034/j.1600-0749.2003.00012.x. [DOI] [PubMed] [Google Scholar]