

Abstract
Two new five-membered ring polyketide endoperoxides, epiplakinic acid F methyl ester (1) and epiplakinidioic acid (3), and a peroxide–lactone, plakortolide J (2), were isolated from the Puerto Rican sponge Plakortis halichondrioides, along with two previously reported cyclic peroxides, 4 and 5. The structures of the new metabolites were determined by spectroscopic and chemical analyses. The absolute stereostructures of 1, 2, and 5 were determined by degradation reactions followed by application of Kishi’s method for the assignment of absolute configuration of alcohols. Biological screening of cycloperoxides 1–5 and semisynthetic analogs 7–12 for cytotoxic activity against various human tumor cell lines revealed that compounds 3, 4 and 11 are very active. Upon assaying for antimalarial and antitubercular activity, some of the compounds tested showed strong activity against the pathogenic microbes Plasmodium falciparum and Mycobacterium tuberculosis.
Marine sponges of the family Plakinidae have been reported to be a rich source of cyclic peroxides, many of which often exhibit antimicrobial, ichthyotoxic, antineuroinflammatory, and antitumor properties.1 Recently, a number of such bioactive polyketides have also been shown to be active against the protozoan parasites Plasmodium falciparum, Leishmania chagasi, Trypanosoma brucei brucei, and Trypanosona cruzi.2 Many of these interesting natural products either contain a six-membered (1,2-dioxane) or a five-membered (1,2-dioxolane) peroxide ring.3 Representative examples of these groups of secondary metabolites, most of which have been isolated from sponges belonging to the two taxonomically-related genera Plakinastrella and Plakortis, include the plakinic acid and the plakortolide series of cyclic peroxides.4,5 Already in 2003, we reported two strongly cytotoxic 1,2-dioxanes, plakortide O and plakortide P, from the Caribbean marine sponge Plakortis halichondrioides (Wilson, 1902).6 Since that investigation, we have isolated three new endoperoxides from a recently re-collected sponge specimen using a bioassay-guided purification protocol. The first two compounds, trivially named epiplakinic acid F methyl ester (1) and plakortolide J (2), contain a conjugated triene along an unbranched C16 alkyl side chain. The least abundant of these metabolites, epiplakinidioic acid (3), is a rare dicarboxylic acid derivative with a shortened side chain compared to compounds 1 and 2. In this account, we report the isolation, structure determination, and biological evaluation of these natural products and those of several semisynthetic analogs.
Results and Discussion
A freeze-dried specimen of the sponge P. halichondrioides (395 g) was extracted with a 1:1 mixture of CHCl3–CH3OH, and after concentration the organic extract was partitioned between n-hexane and water. Significant antitumor activity was detected in the n-hexane extract (16.4 g); upon treatment with 1.0 mg/mL extract, reduction of cell viability was 70, 90, and 92% for DU-145 prostate cancer, A2058 melanoma, and MDA-MB-435 breast cancer cells, respectively. Subsequent analysis of this material by 1H and 13C NMR indicated the presence of polyketides; therefore a small portion of the n-hexane extract was subjected to Si gel flash chromatography. Only those compounds that eluted with mixtures of n-hexane–acetone of increasing polarity displaying ≤ 30% cells viability were investigated further. Additional separation steps of the active fractions by normal–phase column chromatography led to the isolation of new polyketide derivatives epiplakinic acid F methyl ester (1), plakortolide J (2), and epiplakinidioic acid (3), as well as the known polyketides epiplakinic acid F (4) and plakortolide F (5). As optical rotation data were lacking, identification of known compounds 4 and 5 was based exclusively on comparison of their spectroscopic properties (IR, MS, and NMR) with values already described in the literature.4d,7
The 13C NMR spectrum and HREIMS data for 1 suggested a molecular formula of C24H40O4. Inspection of the NMR spectra suggested that the compound was closely related to epiplakinic acid F (4) reported by Wright in 2001 from a Plakinastrella sponge species from the Seychelle Islands.4d A comparison of the NMR data of 1 (Table 1) with those of 4 quickly showed that 1 has the same 1,2-dioxolane ring (with trans-oriented methyl substituents at the C3 and C5 positions) and the same unbranched C16 alkyl chain (including assignment of geometry of the Δ14, Δ16, and Δ18 double bonds) of 4. Thus, the only difference between both compounds arises from the replacement of the carboxylic acid moiety at C2 in 4 by a methylcarboxylate group in 1. Methylation of 4 at 25 °C with diazomethane in ether yielded an inseparable mixture of compounds, the major one of which was shown to be ester 1 by comparison of 1H NMR spectra. Compound 1, however, was extremely unstable, decomposing in the presence of air over varying periods of time, to carboxylic acid 6, which was devoid of significant biological activity.8 The co-isolation of 1 and 4 from the same organism raises the possibility that the former compound is an isolation artifact due to the extraction with MeOH.
Table 1.
13C NMR (125 MHz), 1H NMR (500 MHz), HMBC and NOESY Spectroscopic Data for Epiplakinic Acid F Methyl Ester (1) in CDCl3a
| atom | δC, multb | δH, mult ( J in Hz) | HMBCc | NOESY |
|---|---|---|---|---|
| 1 | 171.1, C | |||
| 2α | 44.0, CH2 | 2.76, d (14.5) | 1, 3, 4, 22 | H-4β, H3-22 |
| 2β | 2.65, d (14.5) | 1, 3, 4, 22 | H-4β, H3-22 | |
| 3 | 83.9, C | |||
| 4α | 55.4, CH2 | 2.22, d (12.5) | 2, 5, 6, 7, 22 | H-6αβ, H3-22 |
| 4β | 2.46, d (12.5) | 2, 5, 6, 23 | H-2αβ, H3-23 | |
| 5 | 86.5, C | |||
| 6α | 39.6, CH2 | 1.69, br t (12.2) | H-4α | |
| 6β | 1.53, br t (12.2) | H-4α | ||
| 7α | 24.5, CH2 | 1.28, br s | ||
| 7β | 1.37, br m | |||
| 8 | 29.5, CH2 | 1.28, br s | ||
| 9 | 29.4, CH2 | 1.28, br s | ||
| 10 | 29.3, CH2 | 1.28, br s | ||
| 11 | 30.0, CH2 | 1.28, br s | ||
| 12 | 29.1, CH2 | 1.37, br m | 10 | |
| 13 | 32.8, CH2 | 2.05, m | 12, 14, 15 | |
| 14 | 134.5, CH | 5.65, dt (14.2, 7.0) | 13, 16 | |
| 15 | 130.4, CH | 6.03, br m | 16 | |
| 16 | 130.9, CH | 6.10, br m | 18 | |
| 17 | 130.8, CH | 6.08, br m | 18 | |
| 18 | 129.5, CH | 6.06, br m | 17 | |
| 19 | 135.9, CH | 5.71, dt (14.5, 7.5) | 17, 20, 21 | |
| 20 | 25.8, CH2 | 2.08, m | 18, 19, 21 | |
| 21 | 13.6, CH3 | 0.99, t (7.4) | 19, 20 | |
| 22 | 24.1, CH3 | 1.43, s | 2, 3, 4 | H-2αβ, H-4α |
| 23 | 23.3, CH3 | 1.28, s | 4, 5, 6, 7 | H-4β |
| −OCH3 | 51.7, CH3 | 3.69, s | 1 |
Chemical shift values are in parts per million relative to the residual CHCl3 (7.26 ppm) or CDCl3 (77.0 ppm) signals. Assignments were aided by 2D NMR experiments, spin-splitting patterns, number of attached protons, and chemical shift values.
13C NMR multiplicities were obtained from a DEPT-135 experiment.
Carbon atoms correlated to proton resonances in the 1H column. Parameters were optimized for 2,3JC–H = 6 and 8 Hz.
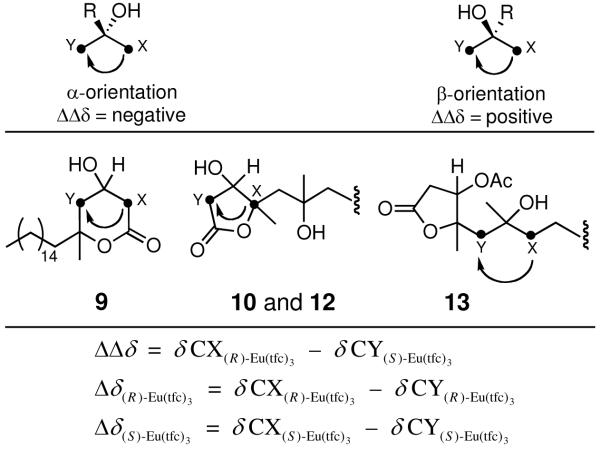
As the absolute stereostructure of epiplakinic acid F (4) was not determined during the 2001 investigation, we established the absolute configuration from its methyl ester 1 by reductive cleavage of the peroxide ring with H2 and 10% Pd/C in EtOAc to afford the diol–ester derivative 8 and a small amount of the separable peroxide–ester derivative 7 (Scheme 1). The former intermediate, obtained in 29% isolated yield, was intramolecularly transesterified to δ-lactone 9. Upon analysis of the 13C NMR chemical shifts of the carbons in 9 adjacent to the tertiary alcoholic center in the presence of chiral lanthanide shift reagents, as described by Kishi et al.,9 the absolute configuration at C-3 was found to be S (Table 2). Coupled with the relative geometry derived from the above analyses, this established the absolute configuration of the peroxide ring in compound 1 as 3S, 5R.
Scheme 1.

Conversion of endoperoxide 1 to derivatives 7–9.
Table 2.
Assignment of Absolute Configuration of Compounds 9, 10, 12, and 13 at 15 mol % per OH of (R)- and (S)-Eu(tfc)3 and Empirical Rules for the Determination of Absolute Configuration of Secondary and Tertiary Alcoholsa
| ΔδR = δCXR - δCYR |
Δδs = δCXS - δCYS |
||||||
|---|---|---|---|---|---|---|---|
| Compound | δCXR | δCYR | Δ δ R | δCXS | δCYS | Δ δ S | ΔΔδ = ΔδR - ΔδS |
| 9 | 44.16 | 44.73 | −0.57 | 44.23 | 44.75 | −0.52 | −0.05 |
| 10 | 90.41 | 38.87 | 51.54 | 90.32 | 38.73 | 51.59 | −0.05 |
| 12 | 90.53 | 38.15 | 52.38 | 90.53 | 38.14 | 52.39 | −0.01 |
| 13 | 46.01 | 43.39 | 2.62 | 45.97 | 43.34 | 2.63 | −0.01 |
The 13C NMR data of alcohols 9, 10, 12, and 13 were recorded in C6D6, CDCl3, acetone-d6, and CDCl3, respectively.
Plakortolide J [(2), (3S,4S,6S,15E,17E,19E)-4,6-dimethyl-4-hydroxy-3,6-peroxy-docosa-15,17,19-trienoic acid 1,4-lactone] was isolated as an extremely unstable colorless oil, which rendered its purification and characterization difficult. The molecular formula of C24H38O4, which requires six degrees of unsaturation, was established from HREIMS. The IR spectrum showed a strong absorbance at 1767 cm−1, suggestive of a γ-lactone, and no other carbonyl or hydroxy band. The 13C NMR data, together with the results of 1H NMR (Table 3) and HSQC experiments, indicated the presence of six sp2 methine, 11 sp3 methylene, and three methyl groups, two of which appeared as singlets (δ 1.38 and 1.28) and one as a triplet (δ 1.00, J = 5.0 Hz) in the 1H NMR spectrum. The remaining carbons were assigned as a lactone carbonyl (δ 174.3, C), two oxygenated quaternary carbons observed at δ 82.8 and 80.1, and one oxygenated methine carbon at δ 81.1. Analysis of the 1H–1H COSY and HSQC spectra of 2 allowed us to establish partial structures which in turn were linked by the 2,3JC–H long-range HMBC correlations and side-by-side comparisons to the spectroscopic data of related peroxide–lactones.5 In the HMBC spectrum, H-3 (δ 4.45) was coupled to C-1 (δ 174.3); H3-23 (δ 1.38) was coupled to C-3 (δ 81.1), C-4 (δ 82.8), and C-5 (δ 40.5); H3-24 (δ 1.28) was coupled to C-5 (δ 40.5), C-6 (δ 80.1), and C-7 (δ 41.0). While the carbon connectivity about the peroxide–lactone moiety of 2, established by COSY, HSQC, and HMBC experiments, was found to be identical to that of known plakortolide F (5), the relative configuration of the peroxide ring was different. A NOESY spectrum exhibited strong correlation between the H-5α resonance observed at δ 2.16 and H3-24 and between the H-5β signal observed at δ 1.70 and both H3-23 and H-3, suggesting that the relative configuration of the CH3-23 and CH3-24 groups of plakortolide J is trans.10 This contention was supported by the shift differences observed in the 13C NMR spectra of peroxide–lactones 2 and 5 for the signals due to the angular methyls at C-6 (δ 22.4 in 2 vs. 24.9 in 5) and the C-7 methylenes (δ 41.0 in 2 vs. 37.0 in 5).
Table 3.
13C NMR (125 MHz), 1H NMR (500 MHz), HMBC and NOESY Spectroscopic Data for Plakortolide J (2) in CDCl3a
| atom | δC, multb | δH, mult ( J in Hz) | HMBCc | NOESY |
|---|---|---|---|---|
| 1 | 174.3, C | |||
| 2α | 34.3, CH2 | 2.62, d (15.0) | 1, 3 | H-2β |
| 2β | 2.91, dd (15.0, 10.0) | 1, 3 | H-2α, H3-23 | |
| 3 | 81.1, CH | 4.45, d (10.0) | 1, 2, 3 | H-5β, H3-23 |
| 4 | 82.8, Cd | |||
| 5α | 40.5, CH2 | 2.16, d (15.0) | 3, 4, 23, 24 | H-5β, H3-24 |
| 5β | 1.70, d (15.0) | 6, 7, 23, 24 | H-3, H-5α, H-7αβ, H3-23 | |
| 6 | 80.1, C | |||
| 7α | 41.0, CH2 | 1.50, br m | ||
| 7β | 1.50, br m | |||
| 8 | 23.1, CH2 | 1.37, br m | ||
| 9 | 29.5, CH2 | 1.27, br m | ||
| 10 | 29.4, CH2 | 1.27, br s | ||
| 11 | 29.3, CH2 | 1.27, br s | ||
| 12 | 30.0, CH2 | 1.27, br m | ||
| 13 | 29.1, CH2 | 1.37, br m | ||
| 14 | 32.8, CH2 | 2.06, br m | 13, 15, 16 | |
| 15 | 134.4, CH | 5.65, dt (14.2, 7.0) | 14, 17 | |
| 16 | 130.4, CH | 6.04, br m | 17 | |
| 17 | 130.9, CH | 6.10, br m | 19 | |
| 18 | 130.8, CH | 6.07, br m | 19 | |
| 19 | 129.4, CH | 6.02, br m | 18 | |
| 20 | 135.9, CH | 5.71, dt (14.5, 7.5) | 18, 21, 22 | |
| 21 | 25.8, CH2 | 2.08, m | 19, 20, 22 | |
| 22 | 13.6, CH3 | 1.00, t (5.0) | 20, 21 | |
| 23 | 25.9, CH3 | 1.38, s | 3, 4, 5 | H-2β, H-3, H-5β |
| 24 | 22.4, CH3 | 1.28, s | 5, 6, 7 | H-5α |
Chemical shift values are in parts per million relative to the residual CHCl3 (7.26 ppm) or CDCl3 (77.0 ppm) signals. Assignments were aided by 2D NMR experiments, spin-splitting patterns, number of attached protons, and chemical shift values.
13C NMR multiplicities were obtained from a DEPT-135 NMR experiment.
Carbon atoms correlated to proton resonances in the 1H column. Parameters were optimized for 2,3JCH = 6 and 8 Hz.
Broad resonance line of low intensity.
The remaining part of the molecule was established to be a conjugated triene along the unbranched C16 alkyl chain on the basis of interpretation of the UV, 1H NMR, and 1H–1H COSY spectra. The position of the triene at Δ15, Δ17, and Δ19 was determined from the COSY, HSQC, and HMBC data. Long-range correlations were observed in the HMBC experiment as follows: the terminal methyl protons observed at δ 1.00 showed coupling to C-21 (δ 25.8) and C-20 (δ 135.9); the allylic protons H2-21 (δ 2.08) showed coupling to C-19 (δ 129.4), C-20 (δ 135.9) and C-22 (δ 13.6). The 1H NMR spectrum indicated large J values (J ≈ 14 Hz) for the scalar coupling between H-15 and H-16 and between H-19 and H-20, allowing for the assignment of E configuration to the Δ15 and Δ19 double bonds. The configuration at Δ17 was deduced to be E as the 13C chemical shift values for C-15–C-20 were found to be almost identical to those of conjugated triene 1 in addition to the remarkably similar absorption maxima observed in the UV spectra of 1 and 2. Moreover, both C-16 and C-19 in 2 would be expected to resonate at higher field (ca. 125 ppm) should the double bond at C-17 have the opposite Z configuration.11,12
The absolute configuration of 2 was likewise determined upon reductive cleavage of the peroxide–lactone ring with H2 and 10% Pd/C in EtOAc to afford diol derivative 10 as the main product along with minor peroxide–lactone derivative 11 (Scheme 2). Assignment of the absolute configuration of 10 at C-3 following simple analysis of its 13C NMR behavior in CDCl3 in the presence of chiral lanthanide shift reagents established the absolute configuration of the peroxide ring as 3S, 4S, 6S (Table 2). In a similar manner, we also determined the absolute configuration of plakortolide F (5) as 3S, 4S, 6R by reductive cleavage of the peroxide ring in 5 to yield diol–lactone derivative 12 as the sole detectable isomer (Scheme 3) followed by application of Kishi’s method.9 Furthermore, as the ΔΔδ value obtained for 12 was arguably low (Table 2), we decided to confirm the latter assignment utilizing the diacetate 13, which was readily prepared from 12 after treatment with Ac2O/pyridine. Thus, while the outcomes of these measurements were comparable, insofar as both afforded the same absolute configuration for 5, we consider that the −0.01 value for ΔΔδ in these instances is predictive (Table 2).13
Scheme 2.

Conversion of plakortolide J (2) to lactones 10 and 11.
Scheme 3.

Conversions of plakortolide F (5) to diol–lactone derivative 12 and diacetate 13.
Epiplakinidioic acid [(3), (3S,5R)-3,5-dimethyl-3,5-peroxy-tetradecanedioic acid], C16H28O6 by HRESIMS, was isolated as a colorless oil that showed a shortened side chain compared to compound 4. The IR spectrum revealed a broad hydroxy band centered at 3416 cm−1 and a carbonyl band at 1715 cm−1. Inspection of the 1H and 13C NMR (see Experimental Section), COSY, HSQC, and HMBC spectra of 3 indicated the presence of two carboxylic acid units, located at the termini of the 14-carbon spin system starting from C-1. A comparison of the 1H and 13C NMR spectra with those of methyl ester 6 indicated that 3 was the corresponding diacid. All other spectral data, together with the [α]D value of +33.9 (c 1.2, CHCl3), support the absolute stereostructure assigned to 3. The observation that compound 4 was also extremely unstable, decomposing rapidly over time, suggests that 3 might be formed during the isolation process upon air-oxidation on the double bond at C-14 of 4.14,15
Naturally occurring cycloperoxides 1–5, as well as semisynthetic derivatives 7–12, were evaluated using MTS assays in DU-145 prostate cancer and melanoma cells in the City of Hope (COH) Comprehensive Cancer Center.16 Table 4 shows the behavior of these compounds in the DU-145 prostate cancer and A2058 melanoma cells. Fresh samples of 1 and 2, which might have partially decomposed in transit, were not very active. Aged samples of 1, which had previously rearranged to 6, were completely inactive.8 On the other hand, the prostate cancer and melanoma cells were significantly more sensitive to compounds 3 and 11, whereas epiplakininc acid F (4) was notably more toxic to the prostate cancer cells. Also, Table 4 shows that semisynthetic derivative 11 was surprisingly potent (less than 1 μM for the IC50 value) against the melanoma cells. The most abundant natural product, plakortolide F (5), was assayed in the NCI’s in vitro antitumor screen consisting of 60 human tumor cell lines selectively displaying potent cytotoxicity against LOX IMVI melanoma cancer, IGROV1 ovarian cancer, and UO-30 renal cancer (the percent of growth of the treated cells when compared to the untreated cells was approximately 0, 0, and 17%, respectively). Of the compounds tested for the inhibition of Plasmodium falciparum, the parasite responsible for the most severe forms of malaria, compounds 3, 10 and 11 demonstrated the most toxic effects (IC50’s < 1 μg/mL) (Table 4). Interestingly, the in vitro antimarial activity of 1,2-dioxanes 2 and 5 was quite modest compared to that of 1,2-dioxolanes 1 and 4. When taken together, these results alone cannot account for the strong antimalarial activity shown by 3, 10 and 11. In vitro antituberculosis screening of compounds 1, 2, 4, 5 and 11 against Mycobacterium tuberculosis H37Rv showed moderate to weak inhibitory activity (MIC ≥ 50 μg/mL), whereas hydroxy–lactones 9 and 10 significantly inhibited bacterial growth.
Table 4.
Biological Activities of Compounds 1–5 and 7–12
| Cancer cell line |
Infectious microorganism |
|||||
|---|---|---|---|---|---|---|
| (% Growth)a |
(IC50 μM)b |
|||||
| Compound | DU-145 | A2058 | DU-145 | A2058 | P. falciparum c | M. tuberculosis d |
| 1 | 92 | 86 | >10 | >10 | 4 | 62 |
| 2 | 112 | 106 | >10 | >10 | >10 | 71 |
| 3 | 31 | 23 | 4 | 3 | 0.3 | N.T.e |
| 4 | 15 | 51 | 1 | >10 | 3 | 92 |
| 5 | 62 | 71 | >10 | >10 | >10 | 59 |
| 7 | 38 | 69 | 8 | >10 | >10 | N.T.e |
| 8 | 81 | 79 | >10 | >10 | >10 | N.T.e |
| 9 | 102 | 60 | >10 | >10 | >10 | 30 |
| 10 | 79 | 75 | 24 | 24 | 0.6 | 13 |
| 11 | 13 | 8 | 2 | 0.8 | 0.3 | 68 |
| 12 | 95 | 81 | >10 | >10 | 7 | N.T.e |
| +Ctrl | – | – | – | – | 0.09 | 0.05 |
DU-145 prostate cancer and A2058 melanoma cells (5,000 cells/well, 96 well plates) were treated with compounds at 10μM for 48 h.
Cells were treated with compounds in a dose-dependent manner for 48 h. Then, the MTS assays assessed cell viability. Experiments in quadruplicate.
Chloroquine-resistant (CQ-R) W2 strains. IC50 in μg/mL. Chloroquine was used as a positive control.
Mtb H37Rv strain. Minimum inhibitory concentrations (MIC) in μg/mL. Rifampin was used as a positive control.
N.T. = Not tested.
Experimental Section
General Experimental Procedures
Optical rotations were obtained with an Autopol IV automatic polarimeter. Infrared and UV spectra were obtained with a Nicolet Magna FT-IR 750 spectrometer and a Shimadzu UV-2401 PC UV-Visible spectrophotometer, respectively. 1D- and 2D-NMR spectra were recorded with a Bruker DRX-500 FT-NMR spectrometer. Mass spectrometric data were generated at the Mass Spectrometry Laboratory of the University of Illinois at Urbana–Champaign. Column chromatography was performed using Si gel (35–75 mesh), and TLC analysis was carried out using glass pre-coated Si gel plates and the spots were visualized using a UV lamp at λ = 254 nm or by exposure to I2 vapor. All solvents used were either spectral grade or were distilled from glass prior to use. Commercially available chiral shift reagents (R)-Eu(tfc)3 and (S)-Eu(tfc)3 were purchased from Sigma Aldrich Co. and dried at 130 °C for 48 h prior to use.17 The percentage yields of compounds 1–5 are based on the weight of the dry sponge specimen. The trivial names assigned to compounds 1–3 are based on previous work by Rinehart and Davidson, respectively.4a,5a
Animal Material
All individuals, found along the ceiling of cave overhangs with a drop-shape, were massively encrusting with irregular conulose surface. Most specimens collected measured up to 20 cm long and 5 cm thick. Color in situ was olive green internally and externally with light gray markings on ridges. However, specimens turned into a brownish color when brought to the surface. Individuals were easily broken, with compressible consistency. Noticeable oscules along the surface were circular and measured 2.0–10.0 mm in diameter. The choanosome was compact with many cavities. Both choanosome and ectosome formed by high abundance of diods. However, there were no signs of alveolar arrangement among the diods. The ectosome was darker than the choanosome. Diods were curved with sharp edges. Straight triods with sharp edges were highly abundant. Nonetheless, many triods had rounded edges with a thick center. Triods and diods were variable in size. Minimum diod length varied from 110–160 μm and the maximum actine length of triod length varied between 20–60 μm. The ectosome measured between 450–650 μm thick. A high density of unusual cavities formed a mesh that ran perpendicular to the surface of the ectosome. An underwater photograph of one of the sponge specimens is available as Supporting Information.
Collection, Extraction, and Isolation
Fresh specimens of the sponge Plakortis halichondrioides (Wilson, 1902) (Phylum Porifera; Class Demospongiae; Subclass Homoscleromorpha; Order Homosclerophorida; family Plakinidae) were collected by hand using SCUBA at depths of 90–100 ft off Mona Island, Puerto Rico, in July 2006. A voucher specimen (No. IM06-09) is stored at the Chemistry Department of the University of Puerto Rico, Río Piedras Campus. The organism was frozen and lyophilized prior to extraction. The dry specimens (395 g) were cut into small pieces and blended in a mixture of CHCl3–MeOH (1:1) (11 × 1L). After filtration, the crude extract was concentrated and stored under vacuum to yield a dark gum (100 g) that was suspended in H2O (2L) and extracted with n-hexane (3 × 2L). Concentration under reduced pressure yielded 16.4 g of the n-hexane extract as a dark brown oil, a portion of which (3.7 g) was chromatographed over Si gel (130 g) using mixtures of n-hexane–acetone of increasing polarity (0–100%). A total of 11 fractions (I–XI) were generated on the basis of TLC and 1H NMR analysis. Further purification of fraction II (1.3 g) by Si gel (20.0 g) column chromatography in 2% acetone–n-hexane afforded 8 subfractions denoted as (A–H). Subfraction II(B) consisted of pure epiplakinic acid F methyl ester (1) (153.4 mg, 0.17% yield), and subfractions II(F) (47.0 mg) and II(G) (68.3 mg) were pooled together and re-chromatographed over Si gel (8.0 g) in 70% CHCl3–n-hexane containing several drops of glacial AcOH to yield pure plakortolide J (2) (18.6 mg, 0.02% yield). Purification of subfraction II(H) (659.1 mg) by Si gel (13.0 g) column chromatography using CHCl3 as eluant afforded epiplakinidioic acid (3) (4.6 mg, 0.005% yield) along with known epiplakinic acid F (4) (16.3 mg, 0.02% yield). Further scrutiny revealed that fraction IV consisted of pure plakortolide F (5) (196.3 mg, 0.22% yield).7
Methyl (3S,5R,14E,16E,18E)-3,5-dimethyl-3,5-peroxy-eneicosa-14,16,18-trienoate (epiplakinic acid F methyl ester) (1)
pale yellowish oil; [α]20D +34.4 (c 1.0, CHCl3); UV (CH3OH) λmax (log ε) 257 (4.54), 267 (4.66), 278 (4.57) nm; IR (neat) νmax 3013, 2928, 2854, 1740, 1455, 1437, 1375, 1346, 1209, 996 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 1; EIMS m/z [M]+ 392 (3), 173 (65), 141 (42), 117 (43), 99 (68), 81 (100), 57 (83), 55 (96); HREIMS m/z [M]+ 392.2918 (calcd for C24H40O4, 392.2927).
(3S,4S,6S,15E,17E,19E)-4,6-Dimethyl-4-hydroxy-3,6-peroxy-docosa-15,17,19-trienoic acid 1,4-lactone (plakortolide J) (2)
colorless oil; [α]20D +29.1 (c 1.1, CHCl3); UV (CH3OH) λmax (log ε) 258 (4.62), 267 (4.75), 278 (4.67) nm; IR (neat) νmax 3011, 2963, 2924, 2851, 1767, 1456, 1267, 1175, 1159, 993, 916, 725 cm−1; 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz), see Table 3; EIMS m/z [M]+ 390 (5), 262 (5), 222 (4), 177 (22), 149 (84), 109 (25), 85 (65), 83 (100); HREIMS m/z [M]+ 390.2756 (calcd for C24H38O4, 390.2770).
(3S,5R)-3,5-Dimethyl-3,5-peroxy-tetradecanedioic acid (epiplakinidioic acid) (3)
colorless oil; [α]20D +33.9 (c 1.2, CHCl3); IR (neat) νmax 3416, 2930, 2855, 1715, 1456, 1376, 1306, 1217, 1091, 756 cm−1; 1H NMR (CDCl3, 500 MHz) δ 2.78 (d, 1H, J = 15.0 Hz, H-2α), 2.71 (d, 1H, J = 15.0 Hz, H-2β), 2.45 (d, 1H, J = 12.5 Hz, H-4β), 2.34 (t, 2H, J = 7.5 Hz, H-13αβ), 2.24 (d, 1H, J = 12.5 Hz, H-4α), 1.70–1.49 (broad m, 4H, H-6αβ and H-12αβ), 1.46 (s, 3H, H3-15), 1.29 (broad envelope, 13H, H-7-H-11 and H3-16); 13C NMR (CDCl3, 125 MHz) δ 172.2 (C, C-14), 172.1 (C, C-1), 86.6 (C, C-5), 83.9 (C, C-3), 55.6 (CH2, C-4), 44.0 (CH2, C-2), 39.6 (CH2, C-6), 29.9-28.9 (6 X CH2, C-8–C-13), 24.4 (CH2, C-7), 23.9 (CH3, C-15), 23.2 (CH, C-16); HRESIMS m/z [M + Na]+ 339.1777 (calcd for C16H28O6Na, 339.1784).18
Plakortolide F (5)
yellowish oil; [α]20D +5.7 (c 1.1, CHCl3); UV (CH3OH) λmax (log ε) 224 (3.82), 269 (3.43) nm; IR (neat) νmax 3417, 2925, 2853, 1763, 1614, 1515, 1464, 1263, 1218, 1172, 955 cm−1. Neither the specific rotation nor the UV data for 5 were previously published. In addition, the original IR spectral data lack some of the key absorption bands described here.4d,7
Spontaneous Decomposition of Epiplakinic Acid F Methyl Ester (1)
Compound 1 was very unstable, decomposing rapidly in CDCl3 solution in the presence of air and light. Following purification of the decomposition mixture by Si gel column chromatography using 10% EtOAc in hexane as eluant compound 6 was identified as the sole product: colorless oil; [α]20D +32.3 (c 1.6, CHCl3); IR (neat) νmax 3700–3000 (broad), 2926, 2854, 1732, 1715–1682 (broad), 1456, 1374, 1260, 1091 cm−1; 1H NMR (CDCl3, 300 MHz) δ 3.69 (s, 3H, –OCH3), 2.77 (d, 1H, J = 14.5 Hz, H-2α), 2.65 (d, 1H, J = 14.5 Hz, H-2β), 2.47 (d, 1H, J = 12.3 Hz, H-4β), 2.34 (t, 2H, J = 7.8 Hz, H-13αβ), 2.23 (d, 1H, J = 12.3 Hz, H-4α), 1.72–1.48 (broad m, 4H, H-6αβ and H-12αβ), 1.43 (s, 3H, H3-15), 1.28 (broad envelope, 13H, H-7-H-11 and H3-16); 13C NMR (CDCl3, 75 MHz) δ 171.1 (2 X C, C-1 and C-14), 86.5 (C, C-5), 83.9 (C, C-3), 55.4 (CH2, C-4), 51.7 (CH3, –OCH3), 44.0 (CH2, C-2), 39.6 (CH2, C-6), 30.9 (CH2, C-13), 29.9–29.4 (5 X CH2, C-8–C-12), 24.5 (CH2, C-7), 24.1 (CH3, C-15), 23.2 (CH3, C-16); HRESIMS m/z [M + Na]+ 353.1930 (calcd for C17H30O6Na, 353.1940).
Reduction of Epiplakinic Acid F Methyl Ester
A mixture of compound 1 (52.8 mg, 0.134 mmol) and 10% Pd on charcoal in EtOAc (15 mL) was stirred under H2 (1 atm) at 25 °C for 24 h. The reaction mixture was filtered, concentrated in vacuo, and the residue obtained was eluted through a short plug of Si gel (1.0 g) with 100% CHCl3 affording two fractions. The least polar fraction (23.9 mg) was purified further by flash CC over Si gel (0.7 g) using a 9:1 mixture of hexane–EtOAc to afford peroxide-ester derivative 7 (3.8 mg, 7%): yellow oil; [α]20D +36.0 (c 0.8, CHCl3); IR (neat) νmax 2917, 2849, 1738, 1463, 1375, 1220, 1163, 1097, 1011 cm−1; 1H NMR (CDCl3, 500 MHz) δ 3.69 (s, 3H, –OCH3), 2.76 (d, 1H, J = 15.0 Hz, H-2α), 2.65 (d, 1H, J = 15.0 Hz, H-2β), 2.46 (d, 1H, J = 15.0 Hz, H-4β), 2.22 (d, 1H, J = 15.0 Hz, H-4α), 1.59 (broad m, 2H, H-6αβ), 1.43 (s, 3H, H3-22), 1.28 (s, 3H, H3-23), 1.25 (broad envelope, 28H), 0.88 (t, 3H, J = 5.0 Hz, H3-21); 13C NMR (CDCl3, 125 MHz) δ 171.1 (C, C-1), 86.5 (C, C-5), 83.9 (C, C-3), 55.4 (CH2, C-4), 51.8 (CH3, –OCH3), 44.0 (CH2, C-2), 39.7 (CH2, C-6), 31.9 (CH2, C-19), 30.1 (CH2, C-7), 29.7–29.4 (11 X CH2, C-8–C-18), 24.2 (CH3, C-22), 23.3 (CH3, C-23), 22.7 (CH2, C-20), 14.1 (CH3, C-21); ESIMS m/z [M + H]+ 399.3 (calcd for C24H47O4, 399.3). The more polar fraction was identified as diol–ester derivative 8 (15.4 mg, 29%): colorless oil; [α]20D +8.9 (c 1.4, CHCl3); IR (neat) νmax 3400, 2920, 2851, 1738, 1467, 1439, 1377, 1342, 1206, 1013 cm−1; 1H NMR (CDCl3, 500 MHz) δ 3.71 (s, 3H, –OCH3), 2.83 (d, 1H, J = 15.0 Hz, H-2α), 2.50 (d, 1H, J = 15.0 Hz, H-2β), 1.80 (d, 1H, J = 15.0 Hz, H-4β), 1.70 (d, 1H, J = 15.0 Hz, H-4α), 1.52 (broad m, 2H, H-6αβ), 1.35 (s, 3H, H3-22), 1.27 (s, 3H, H3-23), 1.25 (broad envelope, 28H), 0.87 (t, 3H, J = 5.0 Hz, H -21); 13C NMR (CDCl3, 125 MHz) δ 173.4 (C, C-1), 73.4 (C, C-5), 72.3 (C, C-3), 51.7 (CH3, –OCH3), 49.0 (CH2, C-4), 46.1 (CH2, C-2), 44.8 (CH2, C-6), 31.9 (CH2, C-19), 30.2 (CH2, C-7), 29.7–29.3 (11 X CH2, C-8–C-18), 29.6 (CH3, C-22), 28.8 (CH3, C-23), 22.7 (CH2, C-20), 14.1 (CH3, C-21); ESIMS m/z [M + H]+ 401.3 (calcd for C24H49O4, 401.3).
Synthesis of δ-Lactone 9
A solution of diol–ester derivative 8 (15.4 mg, 0.038 mmol) in hexane (15 mL) was treated with glacial acetic acid (3 drops) and heated to 60 °C for 2 h. Evaporation in vacuo afforded pure δ-lactone 9 (8.0 mg, 56%): white solid; [α]20D +14.6 (c 1.0, CHCl3); IR (neat) νmax 3470, 2920, 2851, 1703, 1468, 1379, 1145, 1115, 1036, 800 cm−1; 1H NMR (CDCl3, 500 MHz) δ 2.63 (dd, 1H, J = 16.5, 2.0 Hz, H-2α), 2.51 (d, 1H, J = 16.5 Hz, H-2β), 2.08 (dd, 1H, J = 14.5, 2.0 Hz, H-4α), 1.73 (d, 1H, J = 14.5 Hz, H-4β), 1.80 (br m, 2H, H2-6), 1.41 (s, 6H, H3-22 and H3-23), 1.30–1.20 (broad envelope, 28H), 0.89 (t, 3H, J = 6.5 Hz, H3-21); 13C NMR (CDCl3, 125 MHz) δ 170.3 (C, C-1), 83.7 (C, C-5), 69.3 (C, C-3), 45.1 (CH2, C-4), 43.9 (CH2, C-2), 42.5 (CH2, C-6), 31.9 (CH2, C-7), 30.9 (CH3, C-23), 29.9–29.3 (11 X CH2, C-8–C-18), 28.9 (CH3, C-22), 24.1 (CH2, C-19), 22.7 (CH2, C-20), 14.1 (CH3, C-21); EIMS m/z [M-H2O]+ 350 (4), 335 (3), 290 (4), 266 (10), 143 (100), 125 (52), 103 (45), 101 (90), 85 (15), 83 (20).
Reduction of Plakortolide J (2)
A mixture of plakortolide J (28.0 mg, 0.071 mmol) and 10% Pd on charcoal in EtOAc (15 mL) was stirred under H2 (1 atm) at 25 °C for 24 h. The reaction mixture was filtered and concentrated in vacuo to afford an oil that was chromatographed through a short plug of Si gel (0.8 g) using a 4:1 mixture of CHCl3–EtOAc as eluant thus providing pure 10 and 11; compound 10 (14.0 mg, 49%): white solid; [α]20D +3.7 (c 0.8, CHCl3); IR (neat) νmax 3487, 3345, 2915, 2850, 1767, 1470, 1379, 1292, 1201, 1170, 1070, 941 cm−1; 1H NMR (CDCl3, 500 MHz) δ 4.20 (d, 1H, J = 10.0 Hz, H-3), 2.92 (dd, 1H, J = 20.0, 10.0 Hz, H-2β), 2.55 (d, 1H, J = 20.0 Hz, H-2α), 2.17 (d, 1H, J = 15.0 Hz, H-5α), 2.09 (d, 1H, J = 15.0 Hz, H-5β), 1.60 (broad m, 2H, H-7αβ), 1.44 (s, 3H, H3-23), 1.35 (s, 3H, H3-24), 1.28–1.25 (broad envelope, 28H), 0.88 (t, 3H, J = 5.0 Hz, H3-22); 13C NMR (CDCl3, 125 MHz) δ 175.3 (C, C-1), 90.1 (C, C-4), 73.8 (CH, C-3), 73.0 (C, C-6), 43.9 (CH2, C-5), 43.6 (CH2, C-7), 38.1 (CH2, C-2), 31.9 (CH2, C-20), 30.0 (CH3, C-24), 29.7–29.3 (11 X CH2, C-9–C-19), 26.8 (CH3, C-23), 24.4 (CH2, C-8), 22.7 (CH2, C-21), 14.1 (CH3, C-22); EIMS m/z [M–H2O–CH3]+ 365 (3), 309 (17), 292 (5), 269 (14), 173 (38), 155 (87), 130 (13), 113 (100), 95 (19), 71 (26); HRESIMS m/z [M + H]+ 399.3483 (calcd for C24H47O4, 399.3474); compound 11 (5.0 mg, 18%): white solid; [α]20D +27.5 (c 1.2, CHCl3); IR (film) υmax 2920, 2851, 1769, 1468, 1379, 1268, 1176, 1080, 1057, 954, 917 cm−1; 1H NMR (CDCl3, 500 MHz) δ 4.46 (d, 1H, J = 5.0 Hz, H-3), 2.91 (dd, 1H, J = 20.0, 5.0 Hz, H-2β), 2.62 (d, 1H, J = 20.0 Hz, H-2α), 2.16 (d, 1H, J = 15.0 Hz, H-5α), 1.70 (d, 1H, J = 15.0 Hz, H-5β), 1.51 (m, 2H, H-7αβ), 1.39 (s, 3H, H3-23), 1.28 (s, 3H, H3-24), 1.25 (broad envelope, 28H), 0.88 (t, 3H, J = 5.0 Hz, H3-22); 13C NMR (CDCl3, 125 MHz) δ 174.3 (C, C-1), 82.8 (C, C-4), 81.1 (CH, C-3), 80.1 (C, C-6), 41.0 (CH2, C-7), 40.5 (CH2, C-5), 34.3 (CH2, C-2), 31.9 (CH2, C-20), 30.0 (CH2, C-8), 29.7–29.4 (11 X CH2, C-9–C-19), 25.9 (CH3, C-23), 23.1 (CH2, C-21), 22.7 (CH3, C-24), 14.1 (CH3, C-22); EIMS m/z [M–C16H33]+ 171 (50), 155 (48), 101 (42), 85 (68), 83 (100), 71 (88); HRESIMS m/z [M + H]+ 397.3316 (calcd for C24H45O4, 397.3318).
Reduction of Plakortolide F (5)
A mixture of plakortolide F (70.0 mg, 0.179 mmol) and 10% Pd on charcoal in EtOAc (15 mL) was stirred under H2 (1 atm) at 25 °C for 24 h. The reaction mixture was filtered and concentrated in vacuo to afford a homogeneous oil that was passed through a short plug of Si gel (1.0 g) using a 4.5:0.5 mixture of CHCl3–EtOAc to obtain diol–lactone derivative 12 (17.4 mg, 25%): white solid; [α]20D +31 (c 0.2, CHCl3); UV (CH3OH) (log ε) λmax 224 (4.16), 279 (3.56) nm; IR (neat) νmax 3381, 2918, 2848, 1729, 1615, 1518, 1465, 1384, 1249, 1200, 1077, 934 cm−1; 1H NMR (acetone-d6, 500 MHz) δ 7.00 (d, 2H, J = 5.0 Hz, H-17), 6.72 (d, 2H, J = 5.0 Hz, H-18), 5.05 (broad s, 1H, exchangeable, 3-OH), 4.39 (broad s, 1H, exchangeable, 6-OH), 4.22 (d, 1H, J = 10.0 Hz, H-3), 2.98 (dd, 1H, J = 20.0, 5.0 Hz, H-2β), 2.49 (t, 2H, J = 5.0 Hz, H-15αβ), 2.25 (d, 1H, J = 20.0 Hz, H-2α), 2.22 (d, 1H, J = 15.0 Hz, H-5α), 1.85 (d, 1H, J = 15.0 Hz, H-5β), 1.56–1.53 (broad m, 4H, H-7α, H-8α, H-14αβ), 1.47 (s, 3H, H3-20), 1.40 (broad m, 2H, H-7β, H-8β), 1.35 (s, 3H, H3-21), 1.30–1.28 (broad envelope, 10H); 13C NMR (acetone-d6, 125 MHz) δ 175.0 (C, C-1), 156.0 (C, C-19), 134.1 (C, C-16), 129.8 (2 X CH, C-17), 115.7 (2 X CH, C-18), 90.6 (C, C-4), 74.0 (CH, C-3), 72.0 (C, C-6), 47.0 (CH2, C-7), 45.2 (CH2, C-5), 38.2 (CH2, C-2), 35.5 (CH2, C-15), 32.5 (CH2, C-14), 30.8 (CH2, C-13), 29.0 (4 X CH2, C-9-C-12), 27.9 (CH3, C-20), 25.5 (CH3, C-21), 24.4 (CH2, C-8); EIMS m/z [M–C15H23O]+ 173 (6), 155 (100), 113 (20), 107 (98); ESIMS m/z [M + Li]+ 399.3 (calcd for C23H36O5Li, 399.3).
Acetylation of Diol–lactone Derivative 12
A mixture of diol–lactone 12 (40.0 mg, 0.102 mmol), dry pyridine (2.0 mL), and acetic anhydride (0.2 mL) was stirred at 25 °C for 24 h. The reaction mixture was concentrated in vacuo and the oily residue obtained was passed through a short plug of Si gel (0.7 g) using a 7:3 mixture of n-hexane–acetone to afford diacetate 13 (38 mg, 95%): yellowish oil; [α]20D +8.2 (c 1.2, CHCl3); UV (CH3OH) (log ε) λmax 202 (3.78), 265 (2.58), 271 (2.55) nm; IR (neat) νmax 3516, 2926, 2854, 1747, 1508, 1464, 1372, 1234, 1046, 944, 913 cm−1; 1H NMR (CDCl3, 500 MHz) δ 7.16 (d, 2H, J = 5.0 Hz, H-17), 6.97 (d, 2H, J = 5.0 Hz, H-18), 5.18 (d, 1H, J = 10.0 Hz, H-3), 3.03 (dd, 1H, J = 20.0, 5.0 Hz, H-2β), 2.57 (t, 2H, J = 5.0 Hz, H-15αβ), 2.45 (d, 1H, J = 20.0 Hz, H-2α), 2.27 (s, 3H, OCOCH3), 2.09 (s, 3H, OCOCH3), 1.92 (dd, 2H, J = 20.0, 15.0 Hz, H-5αβ), 1.58 (s, 3H, H3-20), 1.47 (m, 2H, H-7αβ), 1.31 (s, 3H, H3-21), 1.29–1.25 (broad envelope, 14H); 13C NMR (CDCl3, 125 MHz) δ 173.7 (C, C-1), 169.8 (C, OCOCH3), 169.6 (C, OCOCH3), 148.5 (C, C-19), 140.4 (C, C-16), 129.2 (2 X CH, C-17), 121.1 (2 X CH, C-18), 88.1 (C, C-4), 75.8 (CH, C-3), 72.2 (C, C-6), 45.8 (CH2, C-7), 43.2 (CH2, C-5), 35.7 (CH2, C-2), 35.3 (CH2, C-15), 31.4 (CH2, C-14), 30.0 (CH2, C-13), 29.6–29.2 (4 X CH2, C-9–C-12), 27.9 (CH3, C-20), 24.6 (CH3, C-21), 23.7 (CH2, C-8), 21.1 (CH3, OCOCH3), 20.9 (CH3, OCOCH3); ESIMS m/z [M–H2O+Na]+ 481.3 (calcd for C27H38O6Na, 481.3) and m/z [M–H2O+H]+ 459.3 (calcd for C27H39O6, 459.3).
Determination of Absolute Configuration of Alcohols 9, 10, 12, and 13
Samples were prepared individually in an oven-dried vial with ~ 3.5–4.5 mg of alcohols 9, 10, 12, and 13 in 0.5 mL of C6D6, CDCl3, acetone-d6, and CDCl3, respectively, containing 15% per OH of chiral shift reagent. The samples were transferred to oven-dried NMR tubes (cooled to room temperature under a stream of nitrogen) via syringe.17 Following acquisition, each substrate was recovered by filtration through a pipette column of Si gel (Sep-Pak Si gel cartridge). For each filtration CH2Cl2 was used as the solvent. To determine the absolute configuration of the alcohols in 9, 10, 12, and 13 according to a method described by Kishi and coworkers, the NMR behaviors of the carbons adjacent to the alcoholic center (CX and CY) were measured in the presence of (R)- and (S)-Eu(tfc)3 (see Table 2).9,19
Cytotoxicity Assay
DU-145 human prostate cancer, A2058 malanoma, and MDA-MB-435 breast cancer cell lines were obtained from ATCC. These cells were cultured in RPMI-1640 or DMEM medium containing 10% fetal bovine serum (FBS), 100 units/mL of penicillin, and 100 μg/mL streptomycin. All cells were maintained in a 5% CO2 atmosphere at 37 °C. To determine the viability of the cells, Promega CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assays (MTS) were performed as described by the supplier (Promega; Madison, WI).20 Briefly, cells (5,000/well) were seeded in 96-well plates and incubated overnight at 37 °C in 5% CO2. Cells were treated for 48 h with each compound. The concentration used was 10 μM. Dimethyl sulfoxide (DMSO) was used as the vehicle control. IC50 values of compounds were determined in a dose-dependent manner (0.1, 0.5, 1, 5, 10, 20, and 50 μM). Cell viability was determined by tetrazolium conversion to its formazan dye and absorbance of formazan was measured at 490 nm using an automated ELISA plate reader. The production of formazan dye was directly proportional to the number of living cells. Each experiment was done in quadruplicate in the absence of a positive control.
Antituberculosis and Antiplasmodial Assays
Biological activity tests against the pathogenic microbes Mycobacterium tuberculosis and Plasmodium falciparum were performed as previously described.21 Rifampin and chloroquine were used as positive controls, respectively.
Supplementary Material
Acknowledgment
We thank J. Marrero, J.C. Asencio, R. Rios, J.J. La Clair, M.J. Lear, and the crew of the R/V Pez Mar for providing logistic support during the sponge collection. The National Cancer Institute (NCI), the Institute for Tuberculosis Research at the University of Illinois at Chicago, and the INDICASAT (Clayton, Panama) provided in vitro cytotoxicity, antituberculosis, and antimalarial activity data, respectively. Mass spectral determinations were provided by the Mass Spectrometry Laboratory of the University of Illinois at Urbana-Champaign. The expert assistance of Dr. V. Vicente for sponge taxonomy determination is gratefully acknowledged. Financial support to C. J.-Romero was provided by the IFARHU-SENACYT Program of the Republic of Panama. This work was supported by a grant from the NIH-SC1 Program (Grant 1SC1GM086271-01A1) awarded to A.D.R.
Footnotes
Supporting Information Available: An underwater photograph of Plakortis halichondrioides and copies of the 1H NMR, 13C NMR, HSQC, HMBC, NOESY, and MS spectra of cycloperoxides 1, 2, and 6. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1).(a) Casteel DA. Nat. Prod. Rep. 1992;9:289–312. doi: 10.1039/np9920900289. [DOI] [PubMed] [Google Scholar]; (b) Casteel DA. Nat. Prod. Rep. 1999;16:55–73. [Google Scholar]; (c) Rahm F, Hayes PY, Kitching W. Heterocycles. 2004;64:523–575. [Google Scholar]; (d) Dembitsky VM, Gloriozova TA, Poroikov VV. Mini-Rev. Med. Chem. 2007;7:571–589. doi: 10.2174/138955707780859396. [DOI] [PubMed] [Google Scholar]
- (2).(a) Taglialatela-Scafati O, Fattorusso E, Romano A, Scala F, Barone V, Cimino P, Stendardo E, Catalanotti B, Persico M, Fattorusso C. Org. Biomol. Chem. 2010;8:846–856. doi: 10.1039/b918600j. [DOI] [PubMed] [Google Scholar]; (b) Feng Y, Davis RA, Sykes M, Avery VM, Camp D, Quinn RJ. J. Nat. Prod. 2010;73:716–719. doi: 10.1021/np900535z. [DOI] [PubMed] [Google Scholar]; (c) Kossuga MH, Nascimento AM, Reimão JQ, Tempone AG, Taniwaki NN, Veloso K, Ferreira AG, Cavalcanti BC, Pessoa C, Moraes MO, Mayer AMS, Hajdu E, Berlinck RGS. J. Nat. Prod. 2008;71:334–339. doi: 10.1021/np0705256. [DOI] [PubMed] [Google Scholar]
- (3).Blunt JW, Copp BR, Munro MHG, Northcote PT, Prinsep MR. Nat. Prod. Rep. 2010;27:165–237. doi: 10.1039/b906091j. and previous articles in this series.
- (4).(a) Phillipson DW, Rinehart KL., Jr. J. Am. Chem. Soc. 1983;105:7735–7736. [Google Scholar]; (b) Davidson BS. J. Org. Chem. 1991;56:6722–6724. [Google Scholar]; (c) Horton PA, Longley RE, Kelly-Borges M, McConnell OJ, Ballas LM. J. Nat. Prod. 1994;57:1374–1381. doi: 10.1021/np50112a006. [DOI] [PubMed] [Google Scholar]; (d) Chen Y, Killday KB, McCarthy PJ, Schimoler R, Chilson K, Selitrennikoff C, Pomponi SA, Wright AE. J. Nat. Prod. 2001;64:262–264. doi: 10.1021/np000368+. [DOI] [PubMed] [Google Scholar]; (e) Dalisay DS, Quach T, Molinski TF. Org. Lett. 2010;12:1524–1527. doi: 10.1021/ol100249v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Davidson BS. Tetrahedron Lett. 1991;32:7167–7170. [Google Scholar]; (b) Varoglu M, Peters BM, Crews P. J. Nat. Prod. 1995;58:27–36. doi: 10.1021/np50115a003. [DOI] [PubMed] [Google Scholar]; (c) Qureshi A, Salvá J, Harper MK, Faulkner DJ. J. Nat. Prod. 1998;61:1539–1542. doi: 10.1021/np9802724. [DOI] [PubMed] [Google Scholar]; (d) Perry TL, Dickerson A, Khan AA, Kondru RK, Beratan DN, Wipf P, Kelly M, Hamann MT. Tetrahedron. 2001;57:1483–1487. [Google Scholar]; (e) Rudi A, Afanii R, Gravalos LG, Aknin M, Gaydou E, Vacelet J, Kashman Y. J. Nat. Prod. 2003;66:682–685. doi: 10.1021/np020589a. [DOI] [PubMed] [Google Scholar]
- (6).(a) del Sol Jiménez M, Garzón SP, Rodríguez AD. J. Nat. Prod. 2003;66:655–661. doi: 10.1021/np030021h. [DOI] [PubMed] [Google Scholar]; (b) del Sol Jiménez M, Garzón SP, Rodríguez AD. J. Nat. Prod. 2003;66:1404. doi: 10.1021/np030021h. [DOI] [PubMed] [Google Scholar]
- (7).The trivial name plakortolide F was claimed simultaneously by Hamann (ref. 5d) and Wright (ref. 4d) for two peroxide–lactones isolated from distinct Plakinastrella sponge species. The compound herein referred to as plakortolide F (i.e. 5) was identical to the material reported by the Wright group.
- (8).Compound 6 showed no cytotoxicity against DU-145 prostate cancer or A2058 melanoma cells at a concentration of 10 μM, nor was it active against Mycobacterium tuberculosis H37Rv at concentrations ≥ 64 μg/mL.
- (9).(a) Ghosh I, Zeng H, Kishi Y. Org. Lett. 2004;6:4715–4718. doi: 10.1021/ol048061f. [DOI] [PubMed] [Google Scholar]; (b) Adams CM, Ghosh I, Kishi Y. Org. Lett. 2004;6:4723–4726. doi: 10.1021/ol048059o. [DOI] [PubMed] [Google Scholar]
- (10).For related peroxide–lactones having trans methyl substituents at the C-4 and C-6 positions, see refs. 5b and 5d.
- (11).Yasuda I, Takeya K, Itokawa H. Phytochemistry. 1982;21:1295–1298. [Google Scholar]
- (12).Guella G, Mancini I, Pietra F. Helv. Chim. Acta. 1989;72:1121–1124. [Google Scholar]
- (13).The lowest value for ΔΔδ in the article by Kishi and coworkers (ref. 9a) is −0.029.
- (14).The conjugated polyene chain of compounds 1, 2 and 4 is a reactive, electron-rich system that is likely susceptible to attack by electrophilic reagents such as hydroxy and peroxy radicals, and thus is responsible for the instability of these compounds towards oxidation. `
- (15).Given their extreme chemical instability, we speculate that Nature uses unsaturated conjugated polyenes such as 1, 2 and 4 as biogenetic precursors to the phenyl polyketide peroxide series of Plakortis/Plakinastrella metabolites (i.e. the plakortolides) via a cascade of efficient E/Z-isomerizations, electrocyclizations, and further dehydrogenations to account for aromatic ring formation. For articles suggesting that some of these conversions are chemically viable, see: Jacobsen MF, Moses JE, Adlington RM, Baldwin JE. Tetrahedron. 2006;62:1675–1689. Rodríguez R, Adlington RM, Eade SJ, Walter MW, Baldwin JE, Moses JE. Tetrahedron. 2007;63:4500–4509.
- (16).Wright and coworkers reported that free acid 4 showed moderate antifungal activity against Candida albicans, whereas 5 was inactive against both C. albicans and Aspergillus fumigatus; see: ref. 4d.
- (17).In order to obtain reliable results, it is essential that both (R)- and (S)-enantiomers of the shift reagent be of similar quality (purity and moisture content), and that NMR data with both the (R)- and (S)-shift reagent be collected on the same instrument.
- (18).The 13C NMR chemical shifts assigned to the carbonyl carbons of 3 are not very accurate as these signals were weak and broad. The assignments shown can be interchanged.
- (19).It has been noted that Δδ are always greater with chiral Pr-based shift reagents than with the corresponding Eu-based shift reagents. Thus, the former lanthanide shift reagents have a superior capacity for enantiotopic discrimination of carbons within a molecule; see: ref. 9a.
- (20).(a) Malich G, Markovic B, Winder C. Toxicology. 1997;124:179–192. doi: 10.1016/s0300-483x(97)00151-0. [DOI] [PubMed] [Google Scholar]; (b) Nam S, Williams A, Vultur A, List A, Bhalla K, Smith D, Lee FY, Jove R. Mol. Cancer Ther. 2007;6:1400–1405. doi: 10.1158/1535-7163.MCT-06-0446. [DOI] [PubMed] [Google Scholar]
- (21).Wei X, Rodríguez AD, Baran P, Raptis RG. J. Nat. Prod. 2010;73:925–934. doi: 10.1021/np100074r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.