

Abstract
Oxidative stress is associated with vascular remodeling and increased preglomerular resistance that are both implicated in the pathogenesis of renal and cardiovascular disease. Angiotensin II induces superoxide production which is metabolized by superoxide dismutase (SOD) or scavenged by nitric oxide.
We investigated the hypothesis that SOD1 regulates renal microvascular remodeling, blood pressure and arteriolar responsiveness and sensitivity to angiotensin II, using SOD1-transgenic (SOD1-tg) and SOD1-knockout (SOD1-ko) mice.
Blood pressure, measured telemetrically, rose more abruptly during prolonged angiotensin II infusion in SOD1-ko mice. The afferent arteriole media-to-lumen ratios were reduced in SOD1-tg and increased in SOD1-ko mice. Afferent arterioles from non-treated wild-types had graded contraction to angiotensin II (sensitivity: 10-9 mol/l, responsiveness: 40%). Angiotensin II contraction were less sensitive (10-8 mol/l) and responsive (14%) in SOD1-tg, but more sensitive (10-13 mol/l) and responsive (89%) in SOD1-ko mice. Arterioles from SOD1-ko had 4-fold increased superoxide formation with angiotensin II at 10-9 mol/l. L-NAME reduced arteriole diameter of SOD1-tg, and enhanced angiotensin II sensitivity and responsiveness of wild-type and SOD1-tg to the level of SOD1-ko mice. Tempol increased arteriole diameter and normalized the enhanced sensitivity and responsiveness to angiotensin II of SOD1-ko, but did not affect wild-type or SOD1-tg mice. Neither SOD1-deficiency nor overexpression was associated with changes in nitrate/nitrite excretion, or renal mRNA expression of NOS-, NADPH oxidase-, SOD2/SOD3-isoforms, and angiotensin II receptors.
In conclusion, SOD1 limits afferent arteriole remodeling and reduces sensitivity and responsiveness to angiotensin II by reducing superoxide and maintaining nitric oxide bioavailability. This may prevent an early and exaggerated blood pressure response to angiotensin II.
Keywords: afferent arterioles, CuZnSOD, hypertension, ICSOD, oxidative stress, superoxide, superoxide dismutase
Introduction
Oxidative stress implies a shift in the balance between the production of reactive oxygen species (ROS) and the action of antioxidant systems, and has been implicated in the pathogenesis of renal and cardiovascular disease.1, 2 Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is an important source of superoxide (O2-) in the vasculature. O2- that escapes metabolism by superoxide dismutase (SOD) can inactivate nitric oxide (NO),3 and growing evidence demonstrate that oxidative stress and NO-deficiency in the kidney contribute to vascular dysfunction and hypertension.3 Furthermore, a potential crosstalk between NADPH oxidase and SOD activities has been suggested.4, 5
SOD exists as three different isoforms: copper-zinc-SOD (CuZnSOD; SOD1), predominately located in the cytoplasm; manganese-SOD (MnSOD; SOD2) in the mitochondria; and extracellular-SOD (ECSOD; SOD3) in the extracellular space.6 SOD1 accounts for 60-80% of SOD activity in the kidneys, and also has an important role in blood vessels where it preserves NO release from the endothelium.7, 8
The renal afferent arterioles are the major resistance vessels to the kidneys, and play an important role in blood pressure regulation.9 The afferent arteriolar tone and resistance are determined by the balance between constrictor agents such as angiotensin II (Ang II) and vasodilator pathways, notably NO.10 Infusions of Ang II induce NO release in afferent arterioles,11 but the response to this is limited by a concomitant increase in O2- produced by NADPH oxidase.8, 12 Indeed, an infusion of Ang II may produce sufficient ROS to overwhelm the antioxidant systems, with subsequent NO-deficiency, hyperreactivity, and remodeling of afferent arterioles.13, 14 Increased arteriolar reactivity may reduce renal perfusion and filtration, increase extracellular volume, and thus contribute to the development of Ang II-induced hypertension.15, 16
These properties of ROS underscore the importance of systems that limit O2- in adjusting renal arteriolar tone. However, there is less information regarding the functional significance of selected SOD-isoforms in the regulation of renal microcirculation and blood pressure. We tested the hypothesis that SOD1 regulates renal microvascular remodeling, and limits afferent arteriolar responsiveness and hypertension in response to Ang II.
Material and Methods
Experimental Design
The experiments were conducted on homozygous littermates from heterozygous breeding pairs of SOD1-tg (C57BL/6-Tg(SOD1)3Cje/J (stock# 002629)) or SOD1-ko mice (B6;129S7-Sod1tm1Leb/J (stock# 002972) from The Jackson Laboratory, Maine, USA. Wild-type littermates from the breeding colonies served as controls. Genotyping of the offspring were performed as previously described (Please see the Online Supplement at http://hyper.ahajournals.org).17 Both genders were used, with equal distribution and similar age (10-16 weeks) in every set of experiment. The animals were fed standardised mouse chow (0.7% NaCl, R36, batch SD389, Lactamin, Kimstad, Sweden) and tap water ad libitum. Telemetric blood pressure measurements were performed in wild-type and SOD1-ko mice, before and after subcutaneous infusion of a slow-pressor dose of Ang II for 14 days. Renal excretion of nitrate/nitrite was analysed to determine total NO production. All afferent arteriole experiments (i.e. isotonic contractions, changes in O2- levels) and gene expression studies were performed in non-treated mice (i.e. without prolonged Ang II infusion).
Blood pressure response to prolonged angiotensin II infusion
Telemetric devices (PA-C10, DSI™, St Paul, MN, USA) were implanted in wild-type and SOD1-ko mice. After surgery, the animals were allowed to recover for at least 10 days. Thereafter the blood pressure was measured continuously for 72 hours to determine basal levels (Please see the Online Supplement at http://hyper.ahajournals.org). The mice were anesthetised by spontaneous inhalation of isoflurane (Forene®, Abbot Scandinavia AB, Sweden) in air (~2.2%) and an osmotic minipump (Alzet®, Durect™, CA) was implanted subcutaneously, delivering Ang II (Sigma-Aldrich) at 400 μg/kg/24h for 14 days. After implantation, blood pressure was recorded continuously throughout the Ang II delivery period.
Renal excretion of nitrate/nitrite
Mice were placed in metabolic cages for 24-hours, with food and water given ad libitum. Water consumption and urine production were measured gravimetrically. Samples of fresh urine were stored at -70°C until analysis. Nitrate and nitrite in urine were measured with a dedicated high performance liquid chromatography system (ENO-20; EiCom, Kyoto, Japan) described previously (Please see the Online Supplement at http://hyper.ahajournals.org).18
Afferent arteriole measurements
Dissection and perfusion of arterioles
Dissection and perfusion were performed as described previously (Please see the Online Supplement at http://hyper.ahajournals.org).
Measurements of arteriolar diameter
The technique for the renal afferent arterioles is demonstrated in Figure 1. The experiments were recorded by a video system, digitised off-line, and analysed as described previously.11 The inner luminal diameters of the arterioles were measured at the most active site to estimate the effect of vasoactive substances. Responsiveness (i.e. maximal change in arteriolar diameter) and sensitivity (i.e threshold concentration of Ang II mediating a significant change in arteriole diameter) was calculated to investigate the role of NO and O2- for the Ang II-induced contraction. In all series, the last 10 seconds of a control or treatment period were used for statistical analysis of steady state responses. Each experiment used a separate dissected afferent arteriole: only one arteriole was used per animal. The inner luminal diameter and media thickness were measured during baseline (before application of any substances), and the areas were calculated to compute the media-to-lumen ratios to assess vascular remodeling.
Figure 1. Setup for the renal afferent arteriole experiments.
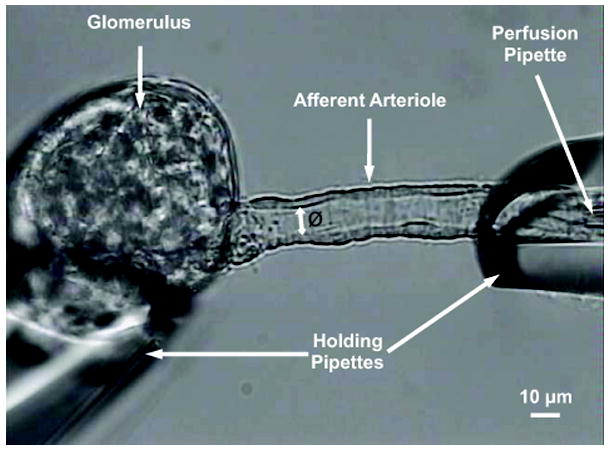
The microphotograph shows a glomerulus and its afferent arteriole held by two holding pipettes. The perfusion pipette (5-μm diameter), inserted into the holding pipette on the right hand side, was connected to a reservoir containing the perfusion solution to provide a pressure of 100 mmHg in the pressure head, which produced a flow of about 50 nl/min. The inner luminal diameter (Ø) of the arteriole was measured at the most active site to estimate the effect of vasoactive substances.
Superoxide measurements in afferent arterioles
Superoxide generation was assessed by fluorescence microscopy of perfused afferent arterioles with dihydroethidium (DHE) or tempo-9AC, as previously described (Please see the Online Supplement at http://hyper.ahajournals.org).19
Analysis of mRNA expression
Infusion of cold PBS (Phosphate Buffered Saline) was started once the vena cava was cut to remove the blood. The heart and kidneys were explanted, blotted and weighed. The renal cortex was separated, homogenised, and quantitative PCR analysis was performed (Please see the Online Supplement at http://hyper.ahajournals.org).
Drugs and Reagents
All drugs were applied to the bath solution in the arteriolar contraction experiments (Please see the Online Supplement at http://hyper.ahajournals.org).
Statistical Analysis
Values are presented as mean ± SEM. Repeated measures Analysis of Variance (ANOVA) was used to test time- or concentration-dependent changes in the arteriolar diameter and to assess differences between the groups. Post-hoc comparisons were performed with Fisher’s test. ANOVA followed by the Fisher’s post-hoc test, when appropriate, were used for analysis of blood pressure, changes in O2- levels, and vascular remodeling. Wilcoxon tests were applied for gene expression data. Differences were considered to be statistically different if P<0.05.
Ethics
The experiments were approved by the Uppsala Ethical Committee for Animal Experiments and Georgetown University Animal Care and Use Committee, and conducted in accordance with the National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals.
Results
All animals were in good condition, and at the time of euthanasia, there was no difference in body weight between the groups. The heterozygous SOD1-ko breeding generated a lower number of homozygous offspring [(+/+; 29%); (+/-; 52%); (-/-; 19%), but no differences in Mendelian distribution were found for the heterozygous SOD1-tg breeding. SOD1-ko mice displayed higher kidney and heart masses, adjusted for body weights (Table 1).
Table 1.
Renal and Cardiovascular Characteristics of SOD1 mice
| A: Kidney and Heart | wild-type | SOD1-ko | SOD1-tg |
|---|---|---|---|
| BW (g) | 30.1 ± 1.3 | 27.2 ± 1.8 | 26.5 ± 1.1 |
| TKW (g) | 0.328 ± 0.004 | 0.329 ± 0.017 | 0.279 ± 0.010*† |
| TKW/BW ×103 | 10.9 ± 0.1 | 12.2 ± 0.5* | 10.6 ± 0.2† |
| HW (g) | 0.155 ± 0.002 | 0.155 ± 0.004 | - |
| HW/BW ×103 | 5.1 ± 0.0 | 5.8 ± 0.3* | - |
| B: Afferent Arterioles | wild-type | SOD1-ko | SOD1-tg |
|---|---|---|---|
| Media Area (μm2) | 181 ± 10 | 202 ± 8* | 138 ± 10*† |
| Lumen Area (μm2) | 108 ± 8 | 77 ± 5* | 113 ± 9† |
| Media/Lumen | 1.83 ± 0.14 | 2.85 ± 0.18* | 1.35 ± 0.12*† |
| C: Afferent Arteriole O2- | wild-type | SOD1-ko | SOD1-tg |
|---|---|---|---|
| Change with Ang II (%) | 6.6 ± 1.9 | 28.9 ± 6.8* | - |
A) Body weight (BW), total kidney weight (TKW) and heart weight (HW) in wild-type (n=7), SOD1-knockout (SOD1-ko, n=7), and SOD1-transgenic (SOD1-tg; n=7) mice.
B) Evaluation of vascular properties, as a measure for vascular remodeling, in wild-type (n=22), SOD1-ko (n=23) and SOD1-tg (n=26).
C) Changes in superoxide (O2-) levels in response to extra-luminal application of angiotensin II (Ang II) at 10-9 mol/l in wild-type (n=7) and SOD1-ko (n=5) mice.
Values are presented as mean ± SEM.
P<0.05 compared to wild-type
P<0.05 compared to SOD1-ko
Blood pressure response to prolonged angiotensin II-infusion
There was no difference in basal blood pressure (i.e. 4 days mean) between the wild-type (107±1 mmHg) and SOD1-ko (110±5 mmHg) mice. SOD1-ko mice displayed a dramatic initial blood pressure response to Ang II, but reached a similar blood pressure level as wild-types after 10 days of treatment. During the first four days of Ang II infusion, blood pressure rose with 37±11 mmHg in SOD1-ko (P<0.05), while no significant change was observed in wild-type mice (3±1 mmHg). The wild-type mice displayed a typical slow-pressor response to Ang II with a significant blood pressure response after 10 days of treatment. Averaged blood pressure data for baseline period and for the whole Ang II treatment period are presented in Figure 2.
Figure 2. SOD1 prevents a rapid and exaggerated blood pressure response to angiotensin II.
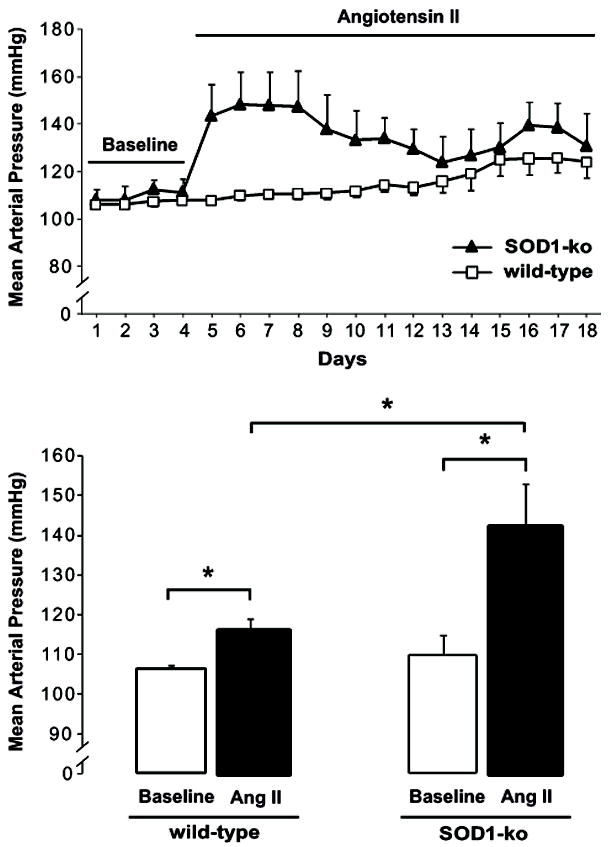
Upper Panel: Mean arterial pressure in conscious wild-type (n=5) and SOD1-knockout (SOD1-ko) mice (n=5). The telemetric measurements were conducted continuously under control conditions for 4 days (Baseline) and then with chronic angiotensin II (Ang II) infusion at a slow-pressor rate (400 μg/kg/24h) for 14 days.
Lower Panel: Mean arterial pressure during baseline period (4 days average) and the whole Ang II infusion period (14 days average). Basal blood pressures were similar in both genotypes; however, Ang II elevated blood pressure more in SOD1-ko mice than in wild-types. Values are presented as mean ± SEM.
* P<0.05
Renal nitrate/nitrite excretion
There were no differences in renal nitrate/nitrite excretion (nmol/24h/g bw) among SOD1-tg (37±4; n=7), wild-type (50±15; n=7), and SOD1-ko mice (53±20; n=7), indicating that the systemic NO production was similar in all three genotypes.
Afferent arteriole measurements
In order to investigate the hypothesis that increased arteriolar responsiveness/sensitivity contributes to the early and rapid blood pressure response observed in SOD1-ko mice, all afferent arteriole experiments were performed in non-treated mice (i.e. without prolonged Ang II infusion).
Basal arteriolar areas and vascular remodeling
The basal luminal area of afferent arterioles was significantly smaller in SOD1-ko mice than in wild-type mice and SOD1-tg (Table 1). No difference was found between the SOD1-tg and wild-type mice. Compared with wild-types, the arteriolar media thickness and media-to-lumen ratios were increased in SOD1-ko, and reduced in SOD1-tg (Table 1). To control for these differences in baseline luminal area, results are presented for absolute and fractional changes of the vessels. Please see the Online Supplement at http://hyper.ahajournals.org for relative changes in arteriole diameters.
Effect of angiotensin II
Concentration response curves were obtained by cumulative application of Ang II (10-14 to 10-6 mol/l; each for 2 min). Ang II constricted afferent arterioles from wild-type mice in a concentration dependent manner (threshold response: 10-9 mol/l), with a maximum response of 40%. Afferent arterioles of SOD1-ko mice were more sensitive (threshold response: 10-13 mol/l) and more responsive (89%) to Ang II, but arterioles from SOD1-tg mice were less sensitive (threshold response: 10-8 mol/l) and less responsive (14%) to Ang II (Figure 3).
Figure 3. SOD1 attenuates renal microvascular responses to angiotensin II.
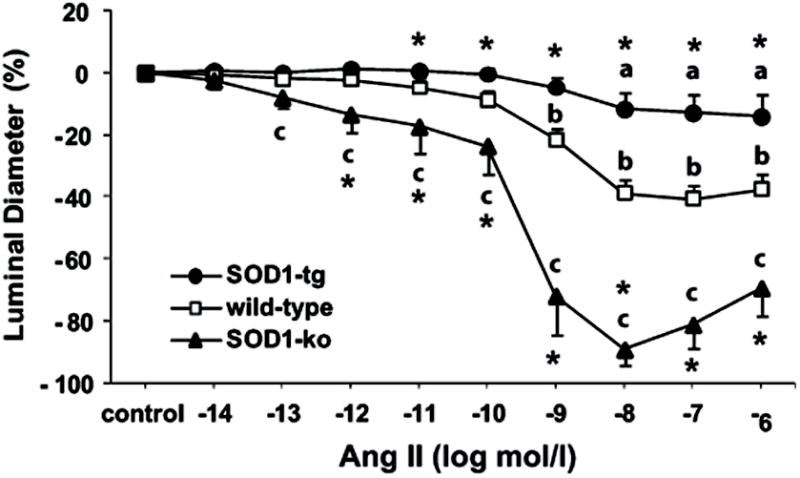
Angiotensin II (Ang II) concentration response curves in isolated and perfused afferent arterioles of non-treated SOD1-transgenic (SOD1-tg; n=7), wild-type (n=11), and SOD1-knockout (SOD1-ko, n=7) mice. Values are presented as mean ± SEM.
a P<0.05 compared to control period in SOD1-tg
b P<0.05 compared to control period in wild-type
c P<0.05 compared to control period in SOD1-ko
* P<0.05 compared to wild-type
Effect of NOS-inhibition with L-NAME
L-NAME (10-4 mol/l), applied for 15 min, constricted afferent arterioles of wild-type mice by 8%, but no contractile response was observed in the SOD1-ko mice. Arterioles from the SOD1-tg mice had a stronger contraction (-38%) after 15 min treatment (Figure 4).
Figure 4. SOD1 is associated with increased nitric oxide bioavailability in renal microvessels.
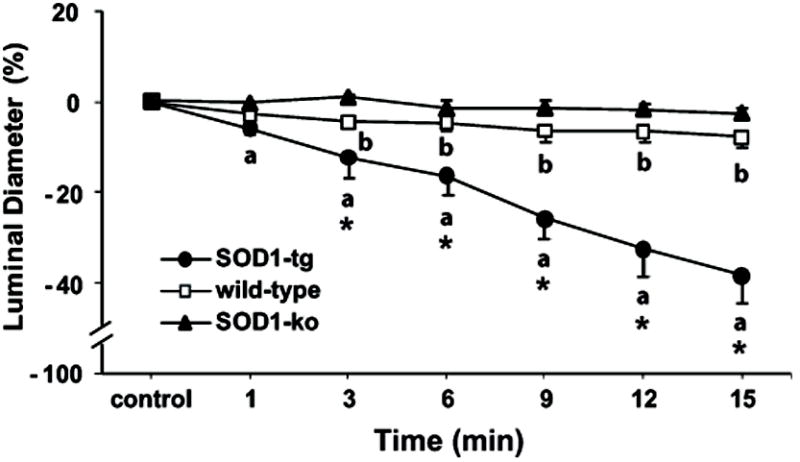
Effect of L-NAME (10-4 mol/l) on diameters of isolated and perfused afferent arterioles from non-treated SOD1-transgenic (SOD1-tg; n=7), wild-type (n=8), and SOD1-knockout (SOD1-ko; n=8) mice. Values are presented as mean ± SEM.
a P<0.05 compared to control period in SOD1-tg
b P<0.05 compared to control period in wild-type
* P<0.05 compared to wild-type and SOD1-ko
Effect of L-NAME treatment on angiotensin II-induced contractions
To investigate the role of NO in offsetting Ang II-induced arteriolar contraction, vessels were treated with L-NAME (10-4 mol/l) for 15 min before, and during treatment with Ang II (10-14 to 10-6 mol/l; 2 min each dose). L-NAME did not affect the responsiveness to Ang II in SOD1-ko mice, but enhanced the response of wild-type and SOD1-tg mice to the level of SOD1-ko mice (Figure 5). L-NAME treatment increased the sensitivity to Ang II in SOD1-ko (10-13 to 10-14 mol/l), but to a much higher degree in wild-types (10-9 to 10-14 mol/l) and in SOD1-tg mice (10-8 to 10-13 mol/l).
Figure 5. During nitric oxide synthase inhibition, the renal microvascular responses to angiotensin II of the different genotypes became indistinguishable.
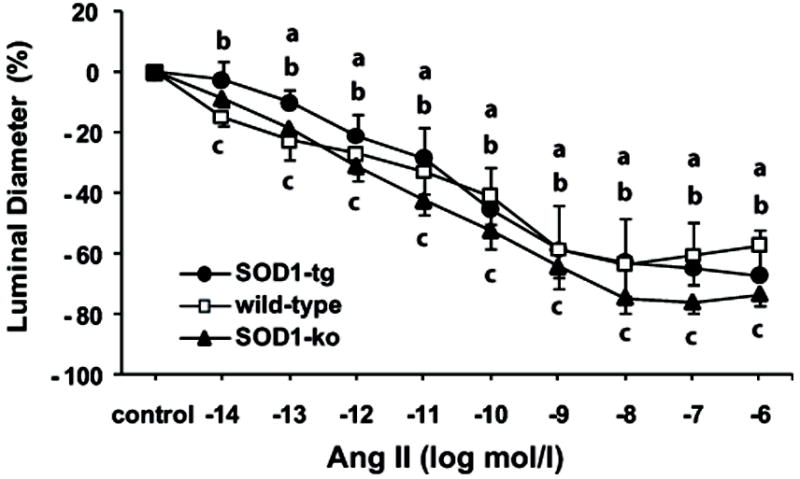
Effect of L-NAME (10-4 mol/l) on angiotensin II (Ang II) concentration response in isolated and perfused afferent arterioles of non-treated SOD1-transgenic (SOD1-tg; n=10), wild-type (n=8), and SOD1-knockout (SOD1-ko; n=7) mice. Control period represents the arteriolar diameter after L-NAME pre-treatment for 15 min. Values are presented as mean ± SEM.
a P<0.05 compared to control period in SOD1-tg
b P<0.05 compared to control period in wild-type
c P<0.05 compared to control period in SOD1-ko
Effect of tempol
Tempol (10-4 mol/l) was applied for 15 min to reduce basal levels of O2-. Tempol dilated afferent arterioles of SOD1-ko mice by 11%, but did not change the diameter of arterioles from wild-type or SOD1-tg mice (Figure 6).
Figure 6. SOD-mimic treatment dilates renal microvessels that lack SOD1.
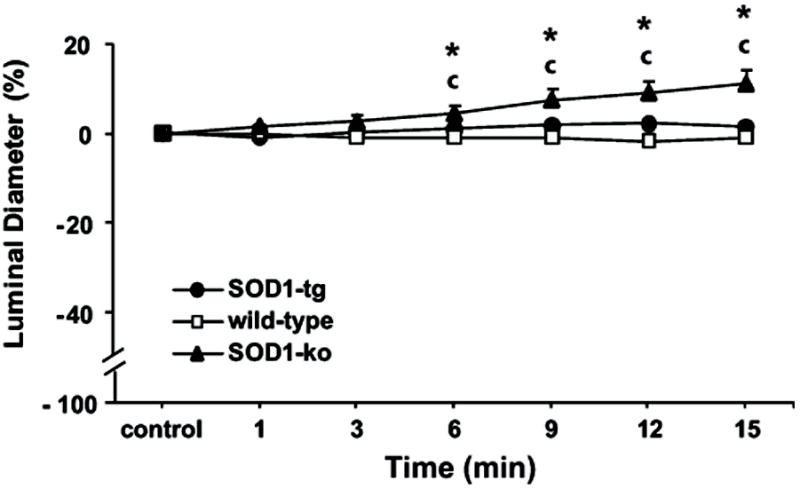
Effect of tempol (10-4 mol/l) on diameters of isolated and perfused afferent arterioles from non-treated SOD1-transgenic (SOD1-tg; n=6), wild-type (n=7), and SOD1-knockout (SOD1-ko; n=7) mice. Values are presented as mean ± SEM.
c P<0.05 compared to control period in SOD1-ko
* P<0.05 compared to wild-type and SOD1-tg
Effect of tempol treatment on angiotensin II-induced contractions
To investigate the role of O2- on Ang II-induced arteriolar contraction, vessels were treated with tempol (10-4 mol/l) for 15 min before, and during treatment with Ang II (10-14 to 10-6 mol/l; 2 min each dose). The addition of tempol did not influence the responsiveness to Ang II in SOD1-tg (18%) or wild-type (43%) mice (Figure 7), but strongly attenuated the contractile response to Ang II in the SOD1-ko mice, with a reduction in the maximal response from 89% to 47%. Tempol treatment did not change the sensitivity to Ang II in wild-types or in SOD1-tg mice, but normalized the enhanced sensitivity in SOD1-ko mice (10-13 to 10-9 mol/l).
Figure 7. SOD-mimic treatment normalizes the exaggerated angiotensin II response of renal microvessels that lack SOD1.
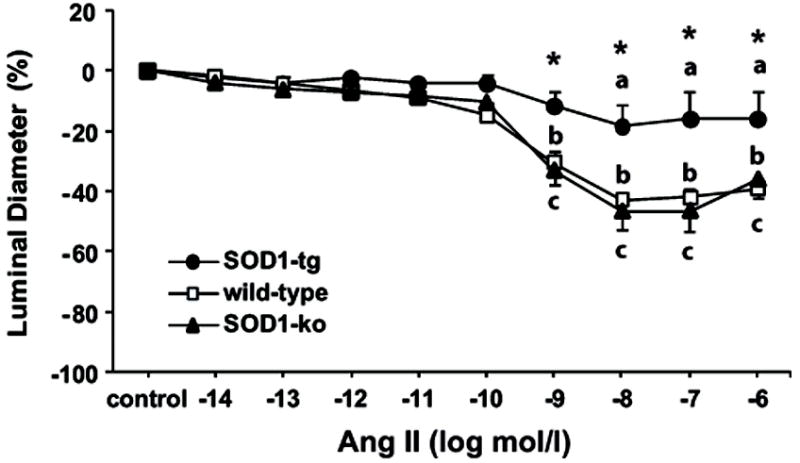
Effect of tempol (10-4 mol/l) on angiotensin II (Ang II) concentration response in isolated and perfused afferent arterioles of non-treated SOD1-transgenic (SOD1-tg; n=6), wild-type (n=7), and SOD1-knockout (SOD1-ko; n=7) mice. Control period represents the arteriolar diameter after tempol pre-treatment for 15 min. Values are presented as mean ± SEM.
a P<0.05 compared to control period in SOD1-tg
b P<0.05 compared to control period in wild-type
c P<0.05 compared to control period in SOD1-ko
* P<0.05 compared to wild-type and SOD1-ko
Afferent arteriolar superoxide levels
Effect of angiotensin II
To investigate if the enhanced contractile response to Ang II in SOD1-ko mice was related to compromised antioxidant defence with increased oxidative stress, O2- levels were measured in response to Ang II (10-9 mol/l). As shown in table 1, Ang II-induced changes in O2- levels were significantly higher in SOD1-ko than in wild-type mice.
Gene-Expression studies
Real-time PCR revealed no differences in renal cortical mRNA expression for NOS-, NADPH oxidase- or SOD2/SOD3-isoforms, or in Ang II receptors among the genotypes (Please see the Online Supplement at http://hyper.ahajournals.org).
Discussion
The main new findings are that the SOD1 attenuates responsiveness and sensitivity of renal afferent arterioles to Ang II, and moderates the early increase in blood pressure to prolonged Ang II. Furthermore, the study suggests that SOD1-deficiency is associated with renal and cardiac hypertrophy, and hypertrophic remodeling of renal microvessels.
SOD1 is the predominant isoform in the vascular wall and accounts for the majority of SOD activity in the kidney.8, 20 Didion and colleagues have demonstrated that SOD1-deficiency is associated with increased O2- levels in carotid arteries, and endothelial dysfunction,20 which can be prevented by a SOD mimetic.21 Furthermore, increased SOD1 expression markedly reduced Ang II-induced vascular O2- levels in the aorta.22 SOD1-ko mice are normotensive, but display renal oxidative stress and increased blood pressure during high sodium treatment, which can be reversed by treatment with a SOD mimetic.23
An attenuated blood pressure and vascular response to L-NAME has been reported in SOD3-ko mice24 and was ascribed to reduced NO bioactivity. The responsiveness of afferent arterioles to Ang II was reduced in SOD1-tg, but strongly augmented in SOD1-ko mice. NOS inhibition did not change arteriolar diameter in the SOD1-ko, but reduced the diameter in SOD1-tg mice. This suggests that the basal NO bioavailability in afferent arterioles was low in SOD1-ko, but high in SOD1-tg mice. This was confirmed by the finding that after L-NAME treatment, the contractile response to Ang II became similar among all three genotypes. Furthermore, L-NAME enhanced the sensitivity to Ang II in wild-types and SOD1-tg mice (105 times) to the level of SOD1-ko mice. This suggests that NO bioavailability was a critical determinant for the differences in responsiveness and sensitivity to Ang II among the groups.
SOD1-ko mice have enhanced bioinactivation of NO and increased formation of peroxynitrite25 and lipid peroxidation.23 The proposal that increased microvascular O2- levels were responsible for the enhanced responses to Ang II in SOD1-ko mice was supported by measurements of Ang II-induced changes in O2-, and experiments with tempol in afferent arterioles. Administration of Ang II (10-9 mol/l) caused a 4-fold greater arteriolar contraction in SOD1-ko than in wild-types, accompanied by a 4-fold greater increase in microvascular O2-. Tempol is a redox-cycling nitroxide that effectively dismutases O2-.26 Tempol dilated afferent arterioles in SOD1-ko mice, and prevented their exaggerated contractile response to Ang II. Furthermore, tempol normalized the enhanced sensitivity to Ang II in SOD1-ko (reduced by 104 times), but had no effects in wild-types or SOD1-tg mice. This finding is consistent with previous studies that the renal vascular resistance of normal mice is little affected by tempol, but is reduced in mice with oxidative stress due to prolonged Ang II infusion.27 Taken together, this demonstrates that SOD1-ko mice have increased microvascular oxidative stress and reduced NO bioavailability that enhances their response to Ang II. However, the diminished contraction to Ang II in SOD1-tg mice persisted after tempol, indicating the operation of another mechanism besides reduced O2- that limited responsiveness in the group.
Oxidative stress and NO-deficiency have been implicated in microvascular remodeling and endothelial dysfunction in cardiovascular disease.28 Scavenging of ROS or overexpression of SOD suppresses Ang II-induced hypertrophic remodeling in aortic smooth muscle cells.29 Furthermore, eNOS-deficient mice and rats with prolonged NOS blockade displayed vascular remodeling that is independent of blood pressure,28 demonstrating that basal NO regulates vascular growth. Thus renal microvascular hypertrophic remodeling in SOD1-ko mice and reduced wall thickness of SOD1-tg mice may relate to O2- dependent changes in NO bioavailability. We have previously demonstrated that kidneys from SOD1-ko mice displayed hypertrophy, fibrotic and inflammatory changes.23 The present study confirms renal hypertrophy, and showed further that SOD1-deficiency caused cardiac hypertrophy.
Our findings that SOD1 limits vascular remodeling of afferent arterioles might have contributed to the differences in contractile behaviour. However, our results dissociate the effects of vascular remodeling from arteriole contractile responses to Ang II, since the responses became indistinguishable by short-term inhibition of NOS. This indicates that the effects of SOD1 on the response to Ang II are due to modulation of the offsetting effect of bioactive NO. Furthermore, the finding that Ang II caused a 4-fold greater increase in afferent arteriolar O2- in SOD1-ko mice, and that tempol normalized their enhanced contractile responses, demonstrates an important role of SOD1 to limit the vascular accumulation of O2- during challenge with Ang II.
Evaluation of mRNA expression in the renal cortex did not reveal any differences between SOD1-tg and SOD1-ko mice for gene expression of NOS-, NADPH oxidase- or SOD2/SOD3-isoforms, or Ang II receptors. Even though expressional studies in afferent arterioles would have been more conclusive, our renal cortical measurements indicate that differences among the genotypes are mainly due to the ability of SOD1 to metabolize O2-, and modulate NO bioavailability, rather than changes in other key enzymes/receptors. Studies in SOD3-knockout mice also have determined that a lack of this SOD-isoform is not compensated by other antioxidative enzymes.24 Moreover, unchanged expression of NOS isoforms in the kidney and similar rates of nitrate/nitrite excretion among the genotypes are consistent with similar overall NO production, and suggest that the major effects of SOD1 expression are to increase NO bioavailability. A recent study demonstrated that glutathione S-transferase Alpha 4 (GSTA4) is compensatory induced in young SOD1-ko mice and may have a protective role against oxidative stress.30 However, the present study suggests that changes in GSTA4 do not, or only partly, compensate for SOD1-deficiency in adult mice.
Ang II treatment can activate NADPH oxidases and increase O2- production with subsequent renal NO-deficiency. The slow-pressor response to Ang II is suggested to be of renal origin,31 because the blood pressure is salt-sensitive,32 and is associated with elevated renal microvascular resistance,33 enhanced Ang II induced responses of afferent arterioles,12, 34 and salt retention.35 In the present study, a slow-pressor infusion of Ang II was converted into a pressor infusion in SOD1-ko mice in which it elevated blood pressure within the first day. Interestingly, SOD3-ko mice also displayed an exaggerated hypertensive response to Ang II at pressor rate, which was attributed to reduced NO bioavailability.8, 24, 36 In contrast, Ang II infused at a slow-pressor rate led to similar increases in blood pressure in SOD3-ko and wild-type mice8 which was attributed to down-regulating of SOD3 and up-regulation of SOD1 in the kidney.8 Thus, SOD1 appears to provide a major defence in the kidney against Ang II-induced ROS accumulation. A limitation of the present study is that blood pressure responses to Ang II were not investigated in SOD1-tg mice. We have previously demonstrated that tempol treatment in wild-type mice reduced the slow-pressor response to Ang II.27 Based on this finding we would expect SOD1-tg mice to have an attenuated response compared with wild-types given Ang II. The mechanisms responsible for returning blood pressure in SOD1-ko to levels of wild-types during prolonged infusion of Ang II remain to be investigated.
Our findings provide evidence that reduced antioxidant capacity, due to lack of SOD1, rendered the renal microvascular system sensitive to changes in O2- production. Increased O2- availability may contribute to the exaggerated blood pressure response to Ang II in SOD1-ko mice by preventing buffering by NO in blood vessels or in the kidney.3, 37 Both effects can increase the sensitivity of the tubuloglomerular feedback response which may reduce the glomerular filtration rate and cause volume retention, which has been demonstrated in the development of hypertension in several experimental models.16, 38-40
In conclusion, the present study provides evidence that SOD1 attenuates renal microvascular responses to Ang II by ROS scavenging and maintaining NO bioavailability. This feature of SOD1 may prevent an early and exaggerated blood pressure response to Ang II. Furthermore, SOD1 plays an important role in regulating renal microvascular hypertrophic remodeling.
Perspectives
Oxidative stress has been implicated in the pathogenesis of many human diseases including hypertension. Emerging evidence suggests that increased O2- levels and NO-deficiency in the kidney play a crucial role in the development and persistence of hypertension. SOD-1 deficiency leads to endothelial dysfunction and, and in the present study we demonstrate that SOD-1 deficiency enhances preglomerular and blood pressure responses to Ang II, and is associated with renovascular remodeling and cardiac hypertrophy. From the present study one could speculate that increased O2- levels in the kidney, due to compromised antioxidant defense, can cause hypertension by increasing preglomerular vascular resistance. Studies have shown that patients with endothelial dysfunction, vascular remodeling, or cardiac hypertrophy are at increased risk for subsequent cardiovascular events. Therefore, antioxidant strategies specifically targeting SOD-isoforms could have therapeutic benefit in preventing a wide spectrum of adverse cardiovascular outcomes.
Supplementary Material
Acknowledgments
Carina Nihlén, Karolinska Institute (Stockholm, Sweden), and Ulrike Neumann, Charité-Universitätsklinikum (Berlin, Germany), are acknowledged for technical assistance.
Sources of Funding
This study was financially supported by the Swedish Research Council (K2009-64X-03522-38-2 and K2009-54X-21117-01-3), the Wallenberg Foundation, the Swedish Heart and Lung Foundation (20090264), the Wenner-Gren Foundation, the Swedish Society of Medicine, Magnus Bergvall Foundation, The Swedish Society of Medical Research (SSMF), the Heart, Lung and Blood Institute of the National Institute of Health (HL68686), and the Diabetes, Digestive Disorder and Kidney Institute (DK-036079 and DK-049870) of the of the National Institute of Health.
Footnotes
Disclosures
None
References
- 1.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 2.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 3.Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol. 2005;289:R913–935. doi: 10.1152/ajpregu.00250.2005. [DOI] [PubMed] [Google Scholar]
- 4.Khan MA, Islam MT, Castillo A, Majid DS. Attenuation of renal excretory responses to ANG II during inhibition of superoxide dismutase in anesthetized rats. Am J Physiol Renal Physiol. 2010;298:F401–407. doi: 10.1152/ajprenal.00511.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez-Iturbe B, Sepassi L, Quiroz Y, Ni Z, Wallace DC, Vaziri ND. Association of mitochondrial SOD deficiency with salt-sensitive hypertension and accelerated renal senescence. J Appl Physiol. 2007;102:255–260. doi: 10.1152/japplphysiol.00513.2006. [DOI] [PubMed] [Google Scholar]
- 7.Mugge A, Elwell JH, Peterson TE, Harrison DG. Release of intact endothelium-derived relaxing factor depends on endothelial superoxide dismutase activity. Am J Physiol. 1991;260:C219–225. doi: 10.1152/ajpcell.1991.260.2.C219. [DOI] [PubMed] [Google Scholar]
- 8.Welch WJ, Chabrashvili T, Solis G, Chen Y, Gill PS, Aslam S, Wang X, Ji H, Sandberg K, Jose P, Wilcox CS. Role of extracellular superoxide dismutase in the mouse angiotensin slow pressor response. Hypertension. 2006;48:934–941. doi: 10.1161/01.HYP.0000242928.57344.92. [DOI] [PubMed] [Google Scholar]
- 9.Smeda JS, Lee RM, Forrest JB. Structural and reactivity alterations of the renal vasculature of spontaneously hypertensive rats prior to and during established hypertension. Circ Res. 1988;63:518–533. doi: 10.1161/01.res.63.3.518. [DOI] [PubMed] [Google Scholar]
- 10.Patzak A, Persson AE. Angiotensin II-nitric oxide interaction in the kidney. Curr Opin Nephrol Hypertens. 2007;16:46–51. doi: 10.1097/MNH.0b013e328011a89b. [DOI] [PubMed] [Google Scholar]
- 11.Patzak A, Lai EY, Mrowka R, Steege A, Persson PB, Persson AE. AT1 receptors mediate angiotensin II-induced release of nitric oxide in afferent arterioles. Kidney Int. 2004;66:1949–1958. doi: 10.1111/j.1523-1755.2004.00981.x. [DOI] [PubMed] [Google Scholar]
- 12.Carlstrom M, Lai EY, Ma Z, Patzak A, Brown RD, Persson AE. Role of NOX2 in the regulation of afferent arteriole responsiveness. Am J Physiol Regul Integr Comp Physiol. 2009;296:R72–79. doi: 10.1152/ajpregu.90718.2008. [DOI] [PubMed] [Google Scholar]
- 13.Fellner SK, Arendshorst WJ. Angiotensin II, reactive oxygen species, and Ca2+ signaling in afferent arterioles. Am J Physiol Renal Physiol. 2005;289:F1012–1019. doi: 10.1152/ajprenal.00144.2005. [DOI] [PubMed] [Google Scholar]
- 14.Skov K, Mulvany MJ. Structure of renal afferent arterioles in the pathogenesis of hypertension. Acta Physiol Scand. 2004;181:397–405. doi: 10.1111/j.1365-201X.2004.01311.x. [DOI] [PubMed] [Google Scholar]
- 15.Wilcox CS. Redox regulation of the afferent arteriole and tubuloglomerular feedback. Acta Physiol Scand. 2003;179:217–223. doi: 10.1046/j.0001-6772.2003.01205.x. [DOI] [PubMed] [Google Scholar]
- 16.Ollerstam A, Pittner J, Persson AE, Thorup C. Increased blood pressure in rats after long-term inhibition of the neuronal isoform of nitric oxide synthase. J Clin Invest. 1997;99:2212–2218. doi: 10.1172/JCI119394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zabihi S, Wentzel P, Eriksson UJ. Maternal blood glucose levels determine the severity of diabetic embryopathy in mice with different expression of copper-zinc superoxide dismutase (CuZnSOD) Toxicol Sci. 2008;105:166–172. doi: 10.1093/toxsci/kfn101. [DOI] [PubMed] [Google Scholar]
- 18.Jansson EA, Huang L, Malkey R, Govoni M, Nihlen C, Olsson A, Stensdotter M, Petersson J, Holm L, Weitzberg E, Lundberg JO. A mammalian functional nitrate reductase that regulates nitrite and nitric oxide homeostasis. Nat Chem Biol. 2008;4:411–417. doi: 10.1038/nchembio.92. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Jose P, Wilcox CS. beta(1) Receptors protect the renal afferent arteriole of angiotensin-infused rabbits from norepinephrine-induced oxidative stress. J Am Soc Nephrol. 2006;17:3347–3354. doi: 10.1681/ASN.2006030212. [DOI] [PubMed] [Google Scholar]
- 20.Didion SP, Ryan MJ, Didion LA, Fegan PE, Sigmund CD, Faraci FM. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ Res. 2002;91:938–944. doi: 10.1161/01.res.0000043280.65241.04. [DOI] [PubMed] [Google Scholar]
- 21.Didion SP, Kinzenbaw DA, Schrader LI, Faraci FM. Heterozygous CuZn superoxide dismutase deficiency produces a vascular phenotype with aging. Hypertension. 2006;48:1072–1079. doi: 10.1161/01.HYP.0000247302.20559.3a. [DOI] [PubMed] [Google Scholar]
- 22.Didion SP, Kinzenbaw DA, Faraci FM. Critical role for CuZn-superoxide dismutase in preventing angiotensin II-induced endothelial dysfunction. Hypertension. 2005;46:1147–1153. doi: 10.1161/01.HYP.0000187532.80697.15. [DOI] [PubMed] [Google Scholar]
- 23.Carlstrom M, Brown RD, Sallstrom J, Larsson E, Zilmer M, Zabihi S, Eriksson UJ, Persson AE. SOD1 deficiency causes salt sensitivity and aggravates hypertension in hydronephrosis. Am J Physiol Regul Integr Comp Physiol. 2009;297:R82–92. doi: 10.1152/ajpregu.90843.2008. [DOI] [PubMed] [Google Scholar]
- 24.Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res. 2003;93:622–629. doi: 10.1161/01.RES.0000092140.81594.A8. [DOI] [PubMed] [Google Scholar]
- 25.Cooke CL, Davidge ST. Endothelial-dependent vasodilation is reduced in mesenteric arteries from superoxide dismutase knockout mice. Cardiovasc Res. 2003;60:635–642. doi: 10.1016/j.cardiores.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 26.Wilcox CS, Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev. 2008;60:418–469. doi: 10.1124/pr.108.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J Am Soc Nephrol. 2002;13:2860–2868. doi: 10.1097/01.asn.0000035087.11758.ed. [DOI] [PubMed] [Google Scholar]
- 28.Feihl F, Liaudet L, Levy BI, Waeber B. Hypertension and microvascular remodelling. Cardiovasc Res. 2008;78:274–285. doi: 10.1093/cvr/cvn022. [DOI] [PubMed] [Google Scholar]
- 29.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 30.Yoshihara D, Fujiwara N, Ookawara T, Kato S, Sakiyama H, Yokoe S, Eguchi H, Suzuki K. Protective role of glutathione S-transferase A4 induced in copper/zinc-superoxide dismutase knockout mice. Free Radic Biol Med. 2009;47:559–567. doi: 10.1016/j.freeradbiomed.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 31.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Csiky B, Simon G. Synergistic vascular effects of dietary sodium supplementation and angiotensin II administration. Am J Physiol. 1997;273:H1275–1282. doi: 10.1152/ajpheart.1997.273.3.H1275. [DOI] [PubMed] [Google Scholar]
- 33.Imig JD. Afferent arteriolar reactivity to angiotensin II is enhanced during the early phase of angiotensin II hypertension. Am J Hypertens. 2000;13:810–818. doi: 10.1016/s0895-7061(00)00264-8. [DOI] [PubMed] [Google Scholar]
- 34.Wang D, Chen Y, Chabrashvili T, Aslam S, Borrego Conde LJ, Umans JG, Wilcox CS. Role of oxidative stress in endothelial dysfunction and enhanced responses to angiotensin II of afferent arterioles from rabbits infused with angiotensin II. J Am Soc Nephrol. 2003;14:2783–2789. doi: 10.1097/01.asn.0000090747.59919.d2. [DOI] [PubMed] [Google Scholar]
- 35.Hall JE. Control of sodium excretion by angiotensin II: intrarenal mechanisms and blood pressure regulation. Am J Physiol. 1986;250:R960–972. doi: 10.1152/ajpregu.1986.250.6.R960. [DOI] [PubMed] [Google Scholar]
- 36.Gongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S, Fukai T, Harrison DG. Role of extracellular superoxide dismutase in hypertension. Hypertension. 2006;48:473–481. doi: 10.1161/01.HYP.0000235682.47673.ab. [DOI] [PubMed] [Google Scholar]
- 37.Liu R, Ren Y, Garvin JL, Carretero OA. Superoxide enhances tubuloglomerular feedback by constricting the afferent arteriole. Kidney Int. 2004;66:268–274. doi: 10.1111/j.1523-1755.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- 38.Wilcox CS, Welch WJ. Interaction between nitric oxide and oxygen radicals in regulation of tubuloglomerular feedback. Acta Physiol Scand. 2000;168:119–124. doi: 10.1046/j.1365-201x.2000.00668.x. [DOI] [PubMed] [Google Scholar]
- 39.Carlstrom M, Brown RD, Edlund J, Sallstrom J, Larsson E, Teerlink T, Palm F, Wahlin N, Persson AE. Role of nitric oxide deficiency in the development of hypertension in hydronephrotic animals. Am J Physiol Renal Physiol. 2008;294:F362–370. doi: 10.1152/ajprenal.00410.2007. [DOI] [PubMed] [Google Scholar]
- 40.Thorup C, Persson AE. Impaired effect of nitric oxide synthesis inhibition on tubuloglomerular feedback in hypertensive rats. Am J Physiol. 1996;271:F246–252. doi: 10.1152/ajprenal.1996.271.2.F246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.