

Abstract
Objectives
We tested the hypothesis that bi-directional, gene-targeted regulation of cardiomyocyte cGMP-selective phosphodiesterase type 5 (PDE5) influences maladaptive remodeling in hearts subjected to sustained pressure-overload.
Background
PDE5 expression is up-regulated in human hypertrophied and failing hearts, and its inhibition (e.g. by sildenafil) stimulates protein kinase G activity, suppressing and reversing maladaptive hypertrophy, fibrosis, and contractile dysfunction. Sildenafil is currently being clinically tested for the treatment of heart failure. However, new studies have questioned the role of myocyte PDE5 and protein kinase G (PKG) to this process, proposing alternative targets and mechanisms.
Methods
Mice with doxycycline controllable myocyte-specific PDE5 gene-expression were generated (medium (me-TG) and high (hi-TG) expression lines), and subjected to sustained pressure-overload.
Results
Rest myocyte and heart function, histology, and molecular profiling were normal in both TG-lines versus controls at 2 months of age. However, upon exposure to pressure-overload (aortic banding), TG hearts developed more eccentric remodeling, maladaptive molecular signaling, depressed function, and amplified fibrosis with up-regulation of tissue growth factor signaling pathway. PKG activation was inhibited in TG myocytes versus control. After establishing a severe cardiomyopathic state, hi-TG mice received doxycycline to suppress PDE5 expression/activity only in myocytes. This in turn enhanced PKG activity, and reversed all of the previously amplified maladaptive responses despite sustained pressure-overload. Sildenafil was also effective in this regard.
Conclusions
These data strongly support a primary role of myocyte PDE5 regulation to myocardial pathobiology and PDE5 targeting therapy in vivo, and reveal a novel mechanism of myocyte-orchestrated extracellular matrix remodeling via PDE5/cGMP-PKG regulatory pathways
Keywords: phosphodiesterase-5, cyclic GMP, protein kinase G, heart failure, pressure overload, transgenic mouse models
Introduction
Cardiac hypertrophy and associated maladaptive cellular and molecular changes often develops from sustained pressure-overload, and is a major worldwide cause of morbidity and mortality. Current treatments target load reduction with diuretics and vasodilators, yet the disease often is refractory to these approaches. One alternative is to stimulate intrinsic negative regulators of hypertrophy, such as cyclic guanosine monophosphate (cGMP) and its downstream protein kinase G (PKG) (1). Cyclic GMP synthesis is triggered by nitric oxide and natriuretic peptides or by inhibiting cGMP-targeted phosphodiesterases (PDE) such as PDE5. The latter is currently used to treat erectile dysfunction and pulmonary hypertension, yet recent experimental studies show it also blunts cardiac disease induced by ischemia/re-oxygenation (2), pressure-overload(3;4), and doxorubicin toxicity(5). Such results helped foster the current NIH-sponsored multicenter clinical trial (RELAX, NCT00763867) testing sildenafil in patients with heart failure and a preserved ejection fraction.
The mechanisms by which sildenafil suppresses and/or reverses experimental cardiac disease remain somewhat controversial. Recent studies have suggested that PDE5 is not the primary target, proposing the more abundant dual-substrate PDE1 as an alternative (6;7). Furthermore, whether myocyte PDE5 and corresponding PKG regulation modulates cardiac hypertrophy/remodeling to counter cardiac stress has been recently questioned(6). Clarification of this signaling is important as this could argue for pursuing alternative molecular targets.
All prior studies in which PDE5 activity was suppressed employed inhibitors that are not perfectly selective and that influence multiple cell types. Global gene deletion is embryonic lethal, and successful attempts at conditional models have yet to be reported. One alternative model is to genetically modulate myocyte PDE5 in a bi-directional manner using a doxycycline-responsive α-MHC promoter driven over-expression. Up-regulation is relevant as myocardial PDE5 expression/activity rises several-fold in pressure-overload (4), and human cardiac hypertrophy(8) and dilated heart failure(9). Importantly, the transgene can be selectively re-suppressed in myocytes after disease is established. Here, we used this strategy to test the hypothesis that myocyte PDE5 suppression and associated PKG activation is an important modulator of pressure-overload maladaptation. The results support this role, and reveal novel crosstalk between myocyte cGMP/PKG and extracellular matrix (ECM) remodeling.
Methods
Cardiac specific PDE5A inducible transgenic mice
Tetracycline (tet)-controlled conditional PDE5 mice were generated by crossing mice expressing PDE5A (tagged with 3×Flag at the N-terminus) with a modified α-MHC promoter -Tet-off vector (gift from Jeffrey Robbins(10); Figure 1A), with mice expressing tetracycline transactivator (tTA, α-MHC promoter). Two founders (sv129xC57BL/6J background) were generated (me-TG, hi-TG) and littermate tTA served as controls. Mice were born without doxycycline (DOX), so PDE5A was over-expressed after birth, but it could be subsequently repressed by adding DOX (0.5 mg/ml) to drinking water.
Figure 1. Inducible cardiac specific PDE5 transgenic mouse model.
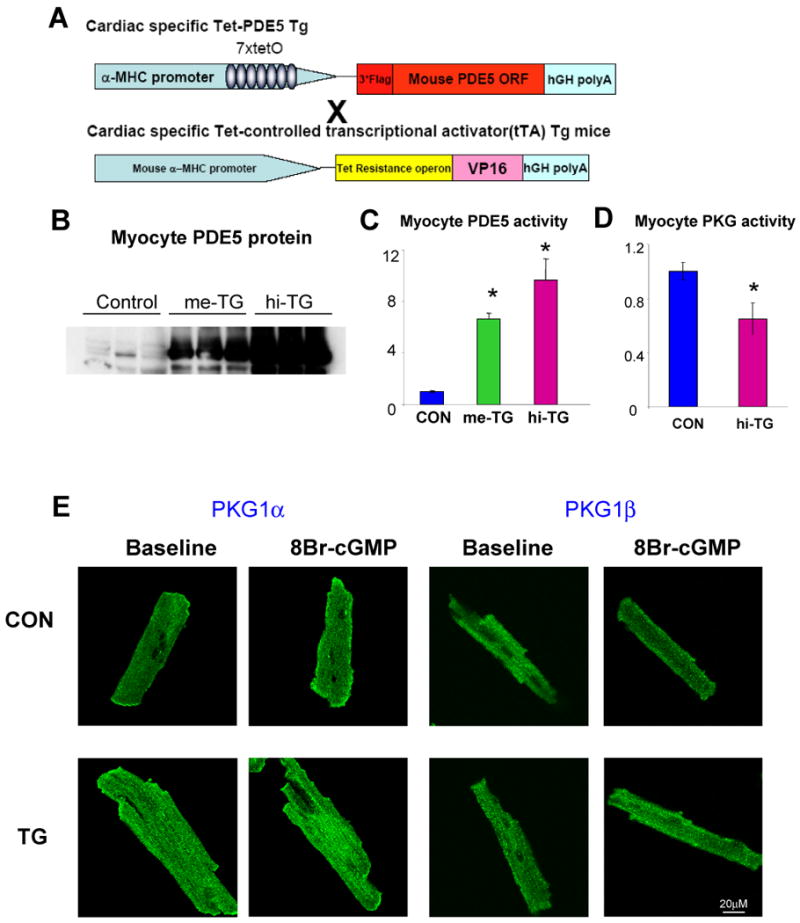
A) Schematic of cardiac specific Tet-off PDE5 Transgenic system. Two founder lines were developed. B) Western blot analysis of PDE5 expression in myocytes, C) Myocyte PDE5 activity from two founder lines (referred as me-TG and hi-TG based on expression level; *p<0.01 versus control, n=4-6 per group) D) Myocyte PKG activity of control and TG group at resting condition. *p<0.05 versus control, n=6-8 per group E) Activation of PKG1α (left) and PKG1β (right), stained green in cardiomyocyte exposed to 8Br-cGMP is reflected by rapid translocation to the plasma membrane. This occurred in control but not TG myocytes (original magnification 200×).
Cardiac and myocyte functional, biochemical, and molecular Analyses
These methods and statistical analysis are provided in detail in On-line Supplemental Methods.
Results
Cardiac myocyte specific PDE5 transgenic mice using tet-off induction system
Two independent conditional PDE5 overexpression lines were generated; (me-TG: 6.7×, and hi-TG:10× activity; Figure 1B, C), both born in normal Mendelian ratios and surviving into adulthood (to 16 months). PDE5 expression in lung was unchanged (Supplemental Fig 1A). Expression of alternative cGMP-PDEs (PDE1, PDE9) was also unchanged in TG mice (Supplemental Figure 1B), consistent with unaltered non-PDE5-dependent cGMP-PDE activity (Supplemental Fig 1C). Transgene PDE5 localized to myocyte z-disks, similar to native protein(11) (Supplemental Figure 2). Up-regulation of PDE5 activity in TG myocytes reduced resting PKG activity (Figure 1D) ≈35% (p<0.05) and inhibited stimulated PKG activity (Figure 1E). The latter was indexed by the lack of plasma membrane translocation of PKG1α and PKG1β in TG myocytes when exposed to cGMP(12). Basal myocyte cGMP was below detection in all models.
Resting cardiac chamber and myocyte function is normal in young adult PDE5-TG mice
Resting phenotype was comprehensively assessed at age 2-3 months. Both me-TG and hi-TG mice heart had normal structural morphology, myocardial histology, molecular signaling, and LV function (Supplemental Figures 3, 4). LV myocytes displayed similar resting sarcomere shortening and response to beta-adrenergic stimulation (isoproterenol, Supplemental Figure 5). The latter was suppressed by sildenafil, consistent with prior reports(13;14), confirming functional regulation by the transgene protein. In me-TG mice, cardiac function and histology remained similar to CON over 16-months observation whereas LV function declined after 6 months in hi-TG, with hypertrophy and interstitial fibrosis documented at 16 months (Supplemental Figure 3). For subsequent studies of pressure-overload with bi-directional myocyte-PDE5 gene regulation, 2-3 month animals were used.
Myocyte PDE5 upregulation worsens cardiac response to pressure-overload
To test the impact of PDE5 up-regulation on pressure-overload stress, 2-month old mice were subjected to 6-wks of trans-aortic constriction (TAC). TAC induced concentric and generally compensated hypertrophy in controls; however, both TG lines displayed substantial chamber dilation and reduced fractional shortening (Figure. 2A, B). PV relations confirmed dilation, particularly in hi-TG, and contractile depression reflected by load-independent indexes. Relaxation rate prolonged only in the TG lines (Figure 2C,D). Importantly, afterload increase (arterial elastance, Ea) was similar in all TAC groups. Worsened function was accompanied by hypertrophy at chamber and myocyte levels, increased heart and lung weight, and interstitial fibrosis (Figure 3). Apoptosis (activated caspase 3) was minimal and unaltered after TAC in all groups, and rare positive cells were interstitial and never myocytes (data not shown).
Figure 2. Exacerbated LV chamber dysfunction and dilation in me-TG and hi-TG mice subjected to 6-wk TAC.
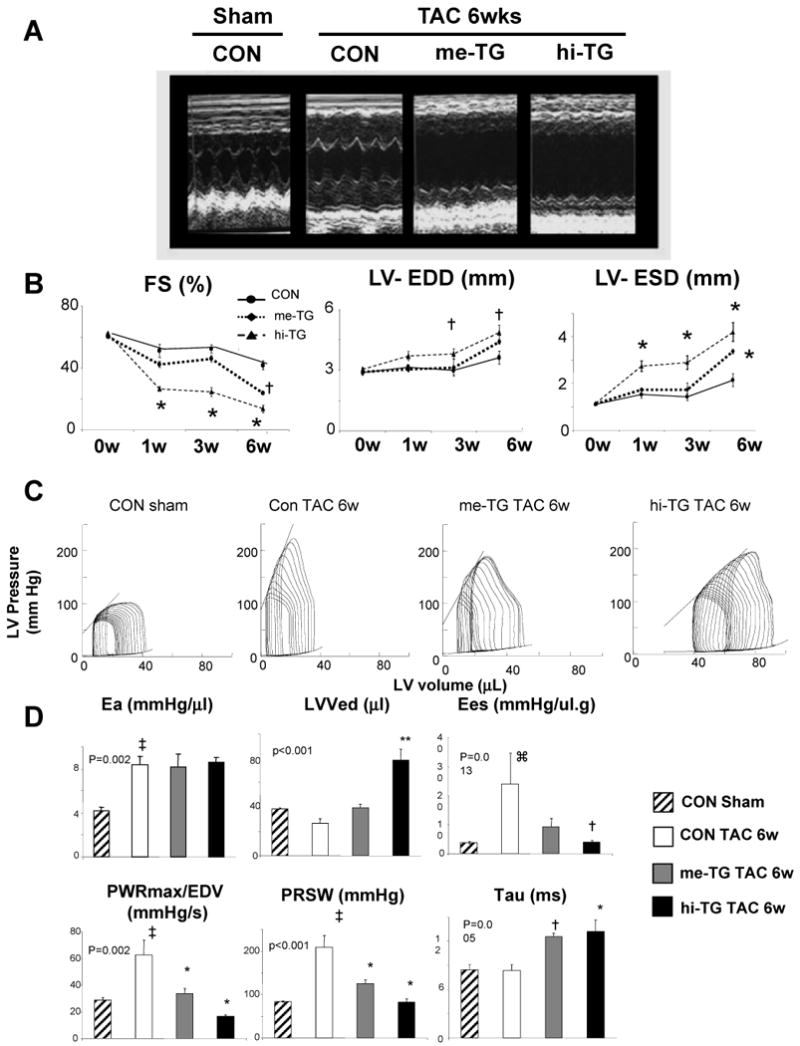
A) Representative echocardiograms and B) Summary data for fractional shortening (FS%), left ventricular end-diastolic and end-systolic dimension (LV-EDD, LV-ESD). (n=3-5/ group; 2-way ANOVA p<0.05 for interaction of group*time for each; † p<0.05 versus CON-TAC-6wk; * p<0.01 versus CON-TAC-6w. C) Representative left-ventricular pressure-volume loops and relations show a right shift and contractile depression, particularly in hi-TG mice vs controls. Slope of the end-systolic PV (left upper line) relationship (Ees) reflects contractile function. D) Summary data from PV loop analysis. Effective arterial elastance (LV afterload, Ea) increased similarly in all groups. LVEDV- LV end-diastolic volume, Tau – time constant of relaxation, Ees, - end-systolic elastance, PWRmax/EDV – maximal LV power divided by EDV, and PRSW, preload recruitable stroke work. The latter three are indexes of contractile function. (n=3∼4 per group; p-value for 1-way ANOVA shown for each. ⌘ p<0.05 versus control sham, Kruskal-Wallis test, ‡ p<0.01 versus control sham, † p<0.05 versus control TAC 6w, * p<0.01 versus control TAC 6w, **p<0.001 versus control TAC 6w.
Figure 3. Chamber, myocyte, and molecular remodeling induced by 6-wk transaortic constriction (TAC) are exacerbated in me-TG and hi-TG mice.
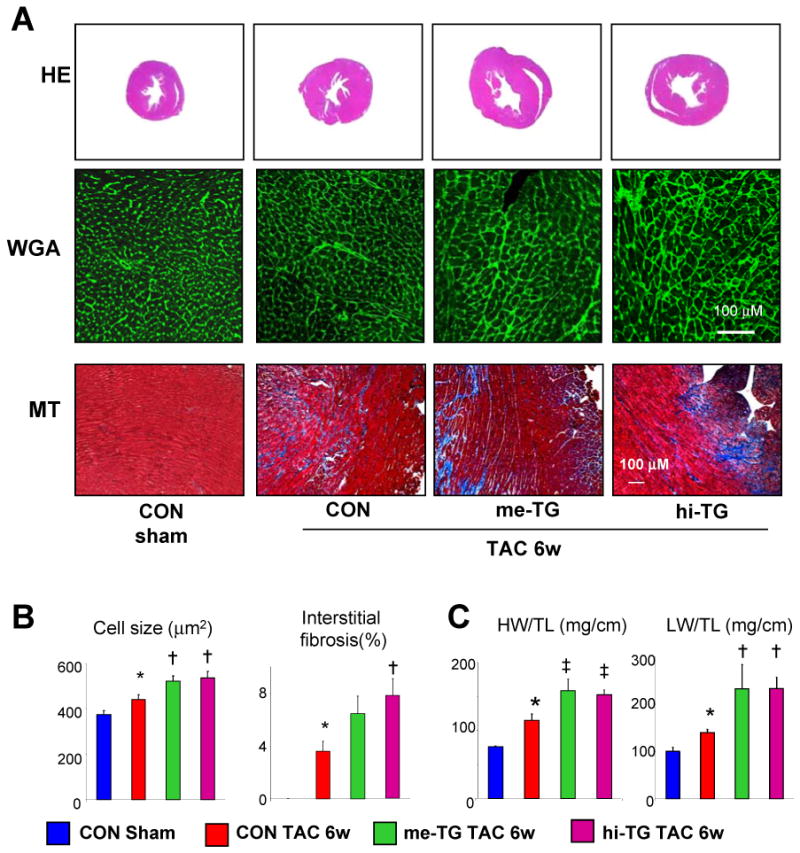
A) Representative cardiac cross sections (top: H&E stain, middle: Wheat germ agglutinin (WGA) stain; lower: Masson-trichrome (MT) stain). B) Summary data for myocyte cross-sectional area and interstitial fibrosis (analysis and sample size in supplementary methods). C) Heart weight/Tibia length(HW/TL) and Lung weight/Tibia length(LW/TL); p<0.005 for one way ANOVA, * p<0.01 versus CON-SHAM, † p<0.05 versus CON-TAC-6w; ‡ p<0.01 versus CON-TAC-6w;
Lack of PKG activation and enhanced maladaptive signaling pathways in TG-TAC hearts
Enhanced myocardial PDE5 activity was unaltered in TG myocardium before and after TAC, whereas it rose ∼80% over baseline in controls (about rest level in me-TG, Figure 4A). PDE1a-c expression was unaltered after TAC (Supplemental Fig 6). Basal myocardial PKG activity was similar, reflecting low basal cGMP-synthesis; however, TAC stimulated PKG activity in CON by 70%, but not at all in TG myocardium and isolated myocytes (Figure 4B). Increased PKG activity despite PDE5 up-regulation after TAC in controls likely reflects a balance and compartmentation of cGMP synthesis/hydrolysis(4). These data show this balance can tilt towards PKG suppression with sufficient PDE5 activity. Protein expression of PKG1α and PKG1β was less in TG at rest, the latter at a slightly lower weight suggesting post-translational change. Myocyte mRNA levels did not change with TAC (Supplemental Figure 7A); however, PKG1α protein levels rose similarly in both groups, and PKG1β increased in TG. Thus, reduced PKG activity in TG-TAC hearts could not be attributed to depressed PKGα/β expression, but rather to post-translational modulation.
Figure 4. Effect of PDE5 upregulation on TAC-stimulated PKG activity, calcineurin, and calcium cycling proteins.
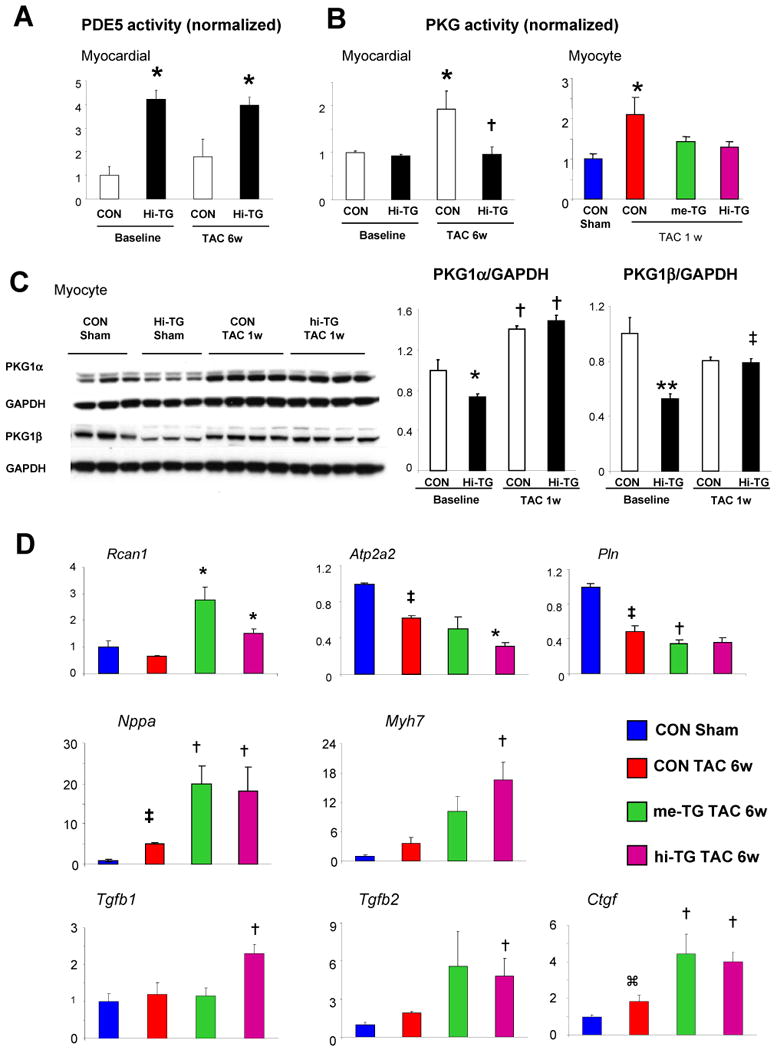
A) Myocardial PDE5 activity in CON versus hi-TG mice before and after TAC (* p<0.01 versus CON, same condition). B) Myocardial PKG activity in CON versus hi-TG mice before and after 6-wk TAC (*p<0.05 versus CON baseline, † p <0.05 versus CON-TAC 6w), Right: isolated myocyte PKG activity determined after 1-wk TAC (* p<0.01 versus CON, n=3-5 in each group. C) PKG1α and PKG1β protein expression at baseline and after TAC in CON and hi-TG mice. (2W-ANOVA,* p<0.05 vs CON baseline, ** P<0.005 vs CON baseline, ‡ p<0.05 vs hi-TG baseline, †p<0.001 vs same group baseline). D) Gene expression assessed by quantitative rtPCR for regulator of calcineurin (Rcan1), SERCA2A (Atp2a2), phospholamban (Pln), A-type natriuretic peptide (Nppa), β-myosin heavy chain (Myh7), tissue growth factor β1 and β2 (Tgfb1, Tgfb2), and connective tissue growth factor (Ctgf) – each normalized to Gapdh. ⌘ p<0.05 versus control sham, ‡ p<0.01 versus control sham, * p<0.01 versus control TAC 6w, † p<0.05 versus control TAC 6w (n=3-6 in each group).
Upon exposure to TAC, both TG models developed worsened molecular abnormalities consistent with maladaptive hypertrophy over controls. Fetal gene recapitulation (Nppa and Myh7) was more prominent, regulator of calcineurin-1 (Rcan1, reflecting calcineurin activity) expression rose more, while sarcoplasmic reticulum ATPase (Atp2a2) and phospholamban (Pln), both key calcium handling proteins, declined more (Figure 4D). Respective changes in protein expression/activation were also observed (data not shown). Given worsened fibrosis in TG mice, we determined expression of ECM regulating genes (Tgfb1,2 and Ctgf). All increased more in TG over CON. Prior to TAC, their expression was similar (Supplemental Figure 4).
Re-suppression of PDE5 in myocytes reverses maladaptive remodeling
TAC-induced cardiac abnormalities and early lethality were observed within 1 week in hi-TG mice (Supplemental Fig 7B,C), but not in me-TG mice. This provided a model to test the impact of subsequently down-regulating only cardiomyocyte PDE5. Both CON and hi-TG mice were exposed to 4-wks of TAC, with doxycycline (DOX) added to the drinking water after Day-7, inhibiting PDE5 transgene expression in TG mice thereafter. DOX eliminated the disparity in PDE5 activity between groups (Figure 5A) and PKG activity rose in hi-TG+DOX, matching CON levels (Figure 5B). Figure 5C shows echocardiography summary data. After 7-days TAC, hi-TG mice displayed worsened function, dilation, and hypertrophy. Without subsequent DOX, this progressed further over the ensuing 17 days, while DOX-treated TG animals displayed recovery to behavior in non-TG controls. DOX treatment did not alter controls.
Figure 5. Re-suppression of myocyte PDE5 expression reverses established maladaptive cardiac remodeling despite sustained pressure-overload.
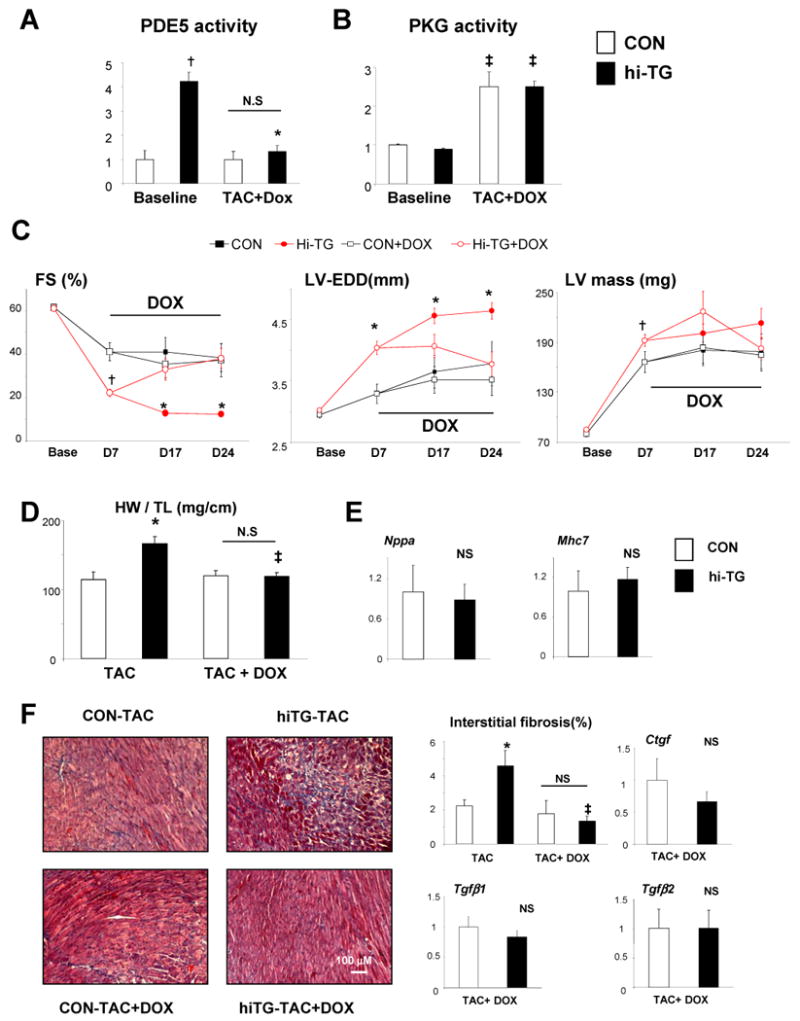
CON and hi-TG mice were subjected to 7-day TAC, then randomized to either doxycycline (DOX) to suppress PDE5 transgene expression or no treatment, while TAC continued for 17 more days. A) Myocardial PDE5 and B) PKG activity measured in CON and hi-TG at baseline and at end-of-study. DOX restored both to levels observed in CON; † p<0.001 versus CON; * p<0.01 versus hi-TG pre-DOX treatment; ‡ p<0.01 versus same genotype at pre-DOX treatment (n=3-12/group). C) Echocardiogram data for DOX-reversal study. Abbreviations are as in Figure 2, in addition, Left ventricle mass(LV mass). All indexes improved in hi-TG with DOX treatment and becoming indistinguishable from CON values by Day 24. † p<0.05 versus CON at same time point and treatment, * p<0.01 versus CON at same time point and treatment (n=5-16). D) Cardiac hypertrophy assessed by heart weight/tibia length rose in TG-TAC (end-of-study), but declined to same level as in CON with DOX treatment (one way ANOVA, p=0.002, * p<0.01 versus CON-TAC, ‡ p<0.01 versus hi-TG-TAC. E) Fetal gene expression (normalized to Gapdh) is similar in CON and hi-TG TAC groups with DOX treatment. F) Myocardial fibrosis was exacerbated in hi-TG after TAC without DOX, but fell to CON levels with DOX. Summary data for histologic analysis, and connective tissue genes (expression shown normalized to CON-TAC, determined at final time point; * p<0.01 versus CON-TAC, ‡ p<0.01 versus hi-TG TAC.
At 4 wks TAC, DOX treated hi-TG had similar LV hypertrophy, fetal gene expression, interstitial fibrosis, and corresponding expression of connective tissue genes as observed in controls (Figure 5D-F). We also examined genome-wide modifications in these models using microarray analysis. CON and hi-TG mice had generally similar profiles pre-TAC, but transcriptional changes after TAC were more pronounced in hi-TG. KEGG pathway analysis found greater upregulation of TGF-β, focal adhesion, and extracellular matrix-receptor interaction genes, and reduced oxidative phosphorylation and metabolic pathway genes (Supplemental Figure 8). These disparities were largely reversed when PDE5 expression was reduced by DOX treatment.
Lastly, we compared these results to non-cell specific and less selective inhibition with sildenafil (100 mg/kg/day; yielding free plasma concentration 10-30 nM, similar to humans with standard doses). TAC was instituted, and sildenafil started on day-8, by which time hi-TG already displayed much more hypertrophy and dysfunction, and was then continued for 3-weeks. LV function improved and chamber dilation diminished in hi-TG-TAC +sildenafil (Figure 6A,B), lowering PDE5 activity and enhanced PKG activity (Figure 6B), all matching levels observed in the CON-TAC group. Importantly, sildenafil was applied when there was already far more cardiac disease in the TG group, so it indeed reversed severity as observed before, and this occurred despite continued myocyte PDE5 up-regulation and sustained TAC.
Figure 6. Improvement in cardiac function with PDE5 inhibitor treatment (sildenafil, SIL) in hi-TG mice.

A) Data for sham-control, hi-TG (untreated), and hi-TG-SIL treated is shown at baseline (2-month age mice), 7 days TAC (pre-treatment), and at day 17 and 24 (10 and 17 days after starting treatment during continued TAC). SIL blocked progressive dilation and depressed function observed in untreated hi-TG mice. P<0.01 by 2-way ANOVA for fractional shortening (FS) and end-systolic dimension (ESD). * p<0.01 versus same time point CON-TAC group, † p<0.01 versus same time point hiTG-TAC group, ‡ p<0.05 versus same time point hi-TG-TAC group. B) Sildenafil treatment reduced PDE5 activity to levels observed in CON, and enhanced PKG activity also to levels comparable to controls.
Discussion
The present study addressed the question of whether bi-directional regulation of myocyte PDE5 influences myocardial function and remodeling in hearts subjected to stress. Enhancing PDE5 expression/activity suppressed PKG activation and worsened responses to pressure-overload in a dose-dependent manner. Subsequent gene-targeted down-regulation of myocyte PDE5 reversed pre-established chamber remodeling and dysfunction despite continued pressure-overload, and raised PKG activity to control levels. Similar reversal was obtained with sildenafil. These data support a role for myocyte PDE5 in heart disease, and show reduction of such activity in myocytes is sufficient to observe profound reversal of maladaptive cardiac stress responses including cross-talk between myocytes and the extracellular matrix.
Resting myocyte PDE5 expression and activity is low; indeed some laboratories have detected neither, and attributed the sildenafil effect to its off target suppression of PDE1(6;7),. PDE1 is a dual-substrate esterase requiring Ca2+-calmodulin for activation, and while PDE1 blockade inhibited cell and organ hypertrophy induced by isoproterenol in a PKG-dependent manner, this was additive to effects from PDE5inhibition, suggesting they regulate different cGMP pools (15). Ours and other laboratories have observed myocyte PDE5 expression(8;9;14;16), and gene-silencing studies further support the specificity and selectivity of these results(16). Importantly, PDE5 is up-regulated 2-6-fold in experimental mice and human heart disease(4;7-9), potentially increasing its influence and potential impact from its subsequent inhibition. In contrast, PDE1 changes were small and insignificant. PDE5 gene up-regulation worsens cardiac responses to myocardial infarction in experimental animals(9), though reversibility was not tested in this prior study. Here we show that myocyte targeted PDE5 gene up- and down-regulation potently re-oriented cardiac stress responses, the latter showing indicating that myocyte PDE5 targeting itself confers potent cardiac protective effects.
In addition to expression and activity of PDE5 protein itself, its regulatory effects critically depend upon cGMP-cyclase activity that provides it substrate, and co-stimulation of pathways targeted by PKG (or cGMP) signaling. The cyclase most directly involved is soluble guanylate cyclase coupled to nitric oxide stimulation, as mice with NOS inhibited or eNOS genetically deleted lost myocardial modulation by PDE5 inhibitors(14). Both cGMP synthesis and cGMP-PKG targeted signaling (such as calcineurin) are stimulated by pathological stress coupled to Gαq cascades (12;17-19), even as PDE5 is up-regulated, setting the stage whereby subsequent PDE5 inhibition provides greater impact. Based on such data, we and others have proposed that PDE5 inhibition acts like a targeted intracellular brake, with minimal impact in normal hearts but an enhanced impact in those under stress(17;18).
Though sustained PKG activation is traditionally thought to benefit stressed hearts, direct proof remains lacking(20). Genetic models involving global PKG-1 deletion induce early lethality coupled to intestinal dilation and malabsorption(21). Recently, Lukowski et al. (6) studied mice in which PKG1β coupled to the smooth muscle promoter (sm22) was overexpressed under control of the smooth muscle promoter sm22 in mice globally lacking both PKG1 (α and β). PKG was lacking in myocytes, yet pressure-overload or ISO induced hypertrophy was similar to controls, leading the authors to conclude myocyte PKG is unimportant to hypertrophic regulation. The current study provides a counter-argument in that myocardial (and myocyte) PKG activity was inversely correlated to myocyte PDE5 activation and maladaptive remodeling after TAC. Several factors could explain these differences. The model used by Lukowski exhibits striking premature mortality whereas even our hi-TG mice had a normal lifespan. In addition, disparities in the severity of the hypertrophic stress and associated PKG activation, and as well as the nature of targeted intracellular cGMP/PKG regulation, may have contributed.
Cardiac remodeling in response to pressure-overload involves complex communication between myocytes, myofibroblasts, and vascular cells. Each component informs the other of changes in the stress environment, and targeted signaling in one compartment can potently impact the others(22). One prominent communicator is the cytokine TGFβ which is synthesized and has potent activity on multiple cell types. Importantly, TGFβ signaling can be blunted by PKG by its phosphorylation of Smads at unique cites to inhibit their nuclear translocation and thereby transcriptional activity (23). ANP and NO block TGFβ (24;25) and myocyte-secreted CTGF(26), likely underlying anti-fibrotic effects. PDE5 is more highly expressed in myofibroblast and vascular cells, and its inhibition in such cells rather than myocytes has been suggested to underlie the cardiac effects of sildenafil(6). However, our data reveal that targeted genetic bi-directional control of myocyte PDE5 is sufficient to regulate interstitial fibrosis and associated signaling cascades.
Several potential study limitations should be noted. Transgenic models can be subject to non-specific effects from excessive protein expression. However, normal cardiac PDE5 expression is low and its overexpression still resulted in low levels of protein displaying normal sub-cellular localization and activity in a pathophysiologically relevant range. Secondly, 2-month hi-TG mice had no abnormal molecular profiles, cardiac morphology, or functional changes, but responded markedly and adversely to TAC within only a few days. Lastly, TAC-induced changes were reversible with either subsequent transgene silencing or sildenafil treatment (maintaining PDE5 overexpression), both despite persistent pressure-overload. Another potential limitation is that our analysis relied on reversing an overexpression model rather than using conditional gene-deletion. However, we contend that TAC responses in hi-TG rapidly invoked multiple cascades, and the capacity to broadly reverse these by targeted gene down-regulation supports the role of myocyte PDE5 and potential role for PDE5 inhibitors in heart failure patients.
In conclusion, we provide strong support for regulatory control of cardiac stress remodeling and PKG activity by myocyte PDE5. The data counter recent suggestions that neither play a role in cardiac hypertrophy. Furthermore, we show that myocyte PDE5 modulation can itself suppress fibrosis-related genes and fibrosis, highlighting a novel mechanism for myocyte-orchestrated extracellular matrix remodeling via PDE5/cGMP-PKG regulatory pathways. Reports that PDE5 expression rises in human heart diseases coupled with the present findings that myocyte gene targeting can bi-directionally impact maladaptive remodeling responses provides new insight into the role of this pathway and supports ongoing clinical therapeutic efforts to modulate it.
Supplementary Material
Acknowledgments
The authors thank Marissa Hildebrandt for assistance with WGA analysis, and Dr. Carla Ellis for histology assistance.
Supported by: NIH-NHLBI grants: HL-089297; HL-084946 (DAK); HL-093432 (ET, DAK); T32-HL-07227 (MZ, AB) and a fellowship grant from the American Heart Association (MZ).
Abbreviations
- cGMP
cyclic guanosine monophosphate
- PKG
protein kinase G
- PDE5
phosphodiesterase type 5
- PDE1
phosphodiesterase type 1
- TAC
trans-aortic constriction
- TG
transgenic
- TGFβ
transforming growth factor beta
- ECM
extracellular matrix
- αMHC
alpha myosin heavy chain
- ANP
atrial natriuretic peptide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2009;122:216–238. doi: 10.1016/j.pharmthera.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Das A, Xi L, Kukreja RC. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem. 2008;283:29572–29585. doi: 10.1074/jbc.M801547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagayama T, Hsu S, Zhang M, et al. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy due to pressure-overload. J Am Coll Cardiol. 2009;53:207–215. doi: 10.1016/j.jacc.2008.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 5.Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005;111:1601–1610. doi: 10.1161/01.CIR.0000160359.49478.C2. [DOI] [PubMed] [Google Scholar]
- 6.Lukowski R, Rybalkin SD, Loga F, et al. Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase I in cardiomyocytes. Proc Natl Acad Sci U S A. 2010;107:5646–5651. doi: 10.1073/pnas.1001360107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vandeput F, Krall J, Ockaili R, et al. cGMP-hydrolytic activity and its inhibition by sildenafil in normal and failing human and mouse myocardium. J Pharmacol Exp Ther. 2009;330:884–891. doi: 10.1124/jpet.109.154468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagendran J, Archer SL, Soliman D, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116:238–248. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- 9.Pokreisz P, Vandenwijngaert S, Bito V, et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation. 2009;119:408–416. doi: 10.1161/CIRCULATIONAHA.108.822072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanbe A, Gulick J, Hanks MC, et al. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–616. doi: 10.1161/01.RES.0000065442.64694.9F. [DOI] [PubMed] [Google Scholar]
- 11.Takimoto E, Champion HC, Belardi D, et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005;96:100–109. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- 12.Takimoto E, Koitabashi N, Hsu S, et al. Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J Clin Invest. 2009;119:408–420. doi: 10.1172/JCI35620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee DI, Vahebi S, Tocchetti CG, et al. PDE5A suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to PKG-mediated troponin I phosphorylation. Basic Res Cardiol. 2010;105:337–347. doi: 10.1007/s00395-010-0084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takimoto E, Champion HC, Belardi D, et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005;96:100–109. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- 15.Miller CL, Oikawa M, Cai Y, et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res. 2009;105:956–964. doi: 10.1161/CIRCRESAHA.109.198515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang M, Koitabashi N, Nagayama T, et al. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell Signal. 2008:2231–2236. doi: 10.1016/j.cellsig.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagayama T, Hsu S, Zhang M, et al. Pressure-overload magnitude-dependence of the anti-hypertrophic efficacy of PDE5A inhibition. J Mol Cell Cardiol. 2009;46:560–567. doi: 10.1016/j.yjmcc.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tokudome T, Kishimoto I, Horio T, et al. Regulator of G-protein signaling subtype 4 mediates antihypertrophic effect of locally secreted natriuretic peptides in the heart. Circulation. 2008;117:2329–2339. doi: 10.1161/CIRCULATIONAHA.107.732990. [DOI] [PubMed] [Google Scholar]
- 19.Koitabashi N, Aiba T, Hesketh GG, et al. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2009;48:713–724. doi: 10.1016/j.yjmcc.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- 21.Weber S, Bernhard D, Lukowski R, et al. Rescue of cGMP kinase I knockout mice by smooth muscle specific expression of either isozyme. Circ Res. 2007;101:1096–1103. doi: 10.1161/CIRCRESAHA.107.154351. [DOI] [PubMed] [Google Scholar]
- 22.Kakkar R, Lee RT. Intramyocardial fibroblast myocyte communication. Circ Res. 2010;106:47–57. doi: 10.1161/CIRCRESAHA.109.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P, Wang D, Lucas J, et al. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102:185–192. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]
- 24.Li P, Oparil S, Novak L, et al. ANP signaling inhibits TGF-beta-induced Smad2 and Smad3 nuclear translocation and extracellular matrix expression in rat pulmonary arterial smooth muscle cells. J Appl Physiol. 2007;102:390–398. doi: 10.1152/japplphysiol.00468.2006. [DOI] [PubMed] [Google Scholar]
- 25.Saura M, Zaragoza C, Herranz B, et al. Nitric oxide regulates transforming growth factor-beta signaling in endothelial cells. Circ Res. 2005;97:1115–1123. doi: 10.1161/01.RES.0000191538.76771.66. [DOI] [PubMed] [Google Scholar]
- 26.Koitabashi N, Arai M, Kogure S, et al. Increased connective tissue growth factor relative to brain natriuretic peptide as a determinant of myocardial fibrosis. Hypertension. 2007;49:1120–1127. doi: 10.1161/HYPERTENSIONAHA.106.077537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.