

Abstract
Plasma levels of high density lipoprotein cholesterol (HDL-C) have long been associated with protection against cardiovascular disease (CVD) in large populations. However, HDL-C has been significantly less useful for predicting CVD risk in individual patients. This has ignited a new debate on the merits of measuring HDL quantity versus quality in terms of protective potential. In addition, numerous recent studies have begun to uncover HDL functions that vary surprisingly from traditional lipid transport roles. In this paper, we review recent findings that point to important functions for HDL that go well beyond lipid transport. These discoveries suggest that HDL might be a platform that mediates protection from a host of disease states ranging from CVD to diabetes to infectious disease.
The traditional view
High density lipoprotein (HDL) is a circulating, non-covalent assembly of amphipathic proteins (~50% by mass) that stabilize lipid emulsions composed of a phospholipid monolayer (PL) (~25%) embedded with free cholesterol (~4%), with a core of triglycerides (TG) (~3%) and cholesteryl esters (CE) (~12%). Plasma levels of HDL cholesterol (HDL-C) are a well known negative risk factor for the development of cardiovascular disease (CVD). A widely accepted basis for the inverse relationship between human plasma HDL-C and CVD is the ability of HDL, and its major protein constituent apolipoprotein (apo)A-I, to mediate reverse cholesterol transport (RCT). In this process, excess cholesterol and other lipids (originally delivered from the liver via low density lipoproteins) are returned for catabolism. The primary targets are lipid-laden macrophages in the vessel wall, harbingers of the fatty streaks and atherosclerosis that can ultimately progress to myocardial infarction or stroke [1].
A large body of evidence supports the notion that HDL-mediated RCT is essential for human cardiovascular health. In addition to numerous in vitro studies that demonstrate cellular cholesterol efflux to HDL, there are well established biochemical pathways that process HDL lipid via numerous circulating enzymes and transfer proteins, and via receptor-mediated uptake into the liver. The in vivo RCT assay developed by Rader and colleagues clearly shows that apoA-I overexpression in mice promotes release of macrophage cholesterol first into the plasma compartment and then eventually to the feces, concomitant with a clear decrease in atherosclerosis susceptibility in this model [2]. However, recent studies have provided tantalizing clues that HDL might be more than it appears. The application of mass spectrometry (MS) based proteomic approaches has revealed unexpected diversity in the HDL proteome (Box 1). Interestingly, only about one-third of HDL proteins are known to mediate lipid transport. The rest play roles in such areas as protease inhibition, complement regulation and acute phase response. This suggests that HDL has broader functions (Figure 1). On an evolutionary scale, atherosclerosis is a relatively recent affliction that strikes well after reproductive age, and would not be expected to be amajor driving force for genetic evolution of either apoA-I or HDL. It follows that HDL probably evolved under selection pressure to support more basic survival functions.
Box 1. HDL proteome complexity.
Since the first isolation of HDL by ultracentrifugation over 60 years ago, hundreds of studies have aimed to characterize its protein complement. Until recently, HDL was believed to contain predominantly apoA-I, apoA-II and a limited number of less abundant proteins associated with (i) lipid transport or lipoprotein integrity, i.e., the apolipoproteins, (ii) lipid metabolism or transfer, and (iii) acute phase response. The known functions of these proteins fit well into the general dogma of a primary role for HDL in RCT. However, recent applications of MS based proteomics have provided a new appreciation for the complexity of the HDL proteome by detecting more than 50 distinct HDL associated proteins. Many of these have known functions that do not easily fit into the RCT paradigm. For example, several complement proteins, such as C3, C1 inhibitor and complement factor H were discovered. Additionally, protease inhibitors, including several members of the serine protease inhibitor (SERPIN) family were also found. These findings clearly link HDL to innate immunity and proteolytic pathways involved in inflammation and coagulation. The importance of these discoveries has been emphasized by the demonstration that the proteomic profiles of HDL are altered in patients with CVD, and can even be partially normalized by lipid modification therapies. These techniques have also been used to demonstrate that distinct clusters of proteins can segregate to specific HDL subfractions, including a very recent study using gel filtration as the mode of separation [67-69]. Such comparative proteomic studies offer the hope that new biomarkers can be identified to predict not only CVD, but possibly other disease states linked to the novel HDL functions reviewed in this paper.
Figure 1.
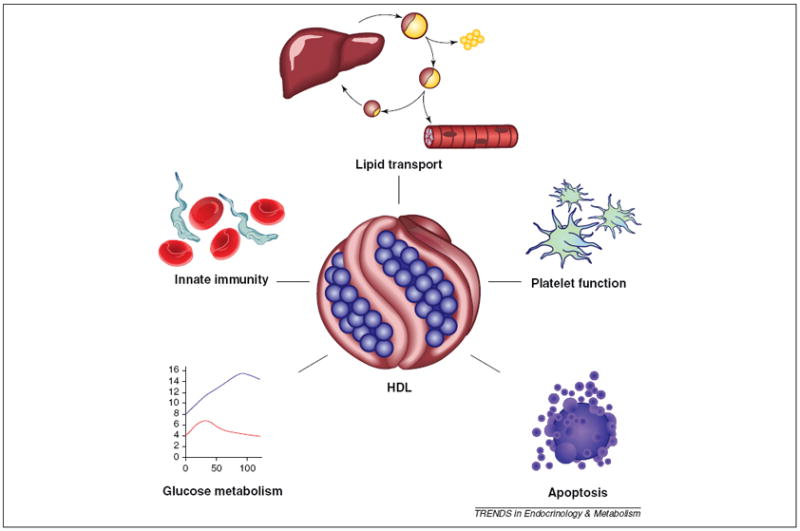
The increasing functional heterogeneity of high density lipoprotein. Numerous recent studies have begun to uncover HDL functions that vary surprisingly from traditionally recognized lipid transport roles. Further exploration of these functions will be useful for understanding the potential of HDL as a treatment option for non-cardiovascular pathologies.
Anti-inflammatory functions
Aside from RCT, the next best recognized HDL function is its role as an anti-inflammatory regulator. It accomplishes this through interactions with both the vascular endothelium and circulating inflammatory cells. For example, HDL limits the extent to which endothelial cells can become activated by proinflammatory cytokines, resulting in reduced expression of adhesion molecules [3]. This was elegantly demonstrated in vivo, in a study in which infusion of rabbits with reconstituted (r)HDL reduced vascular inflammation by inhibiting endothelial cell adhesion molecule expression [4]. The effect was diminished when the rHDL particle was generated with non-enzymatically glycated apoA-I [5] such as might occur in type II diabetes (T2D).
HDL can also inhibit production of chemoattractant molecules such as monocyte chemoattractant protein (MCP)-1 [6], a chemokine responsible for the recruitment of monocytes, dendritic cells and T lymphocytes to sites of injury and inflammation. Additionally, HDL can modulate vascular tone by affecting the production of nitric oxide (NO), a key mediator of vascular smooth muscle cell contraction. It accomplishes this by stimulating the activity of endothelial nitric oxide synthase (eNOS) to boost NO production, triggering vasorelaxation [7]. Earlier in vitro work demonstrated that apoA-I can increase eNOS activity via AMP-activated protein kinase (AMPK) activation [8], and via the phosphoinositide 3-kinase (PI3K)/AKT and mitogen activated protein kinase signaling pathways [9]. In vivo studies confirmed increased AKT and extracellular signal related kinase (ERK)1/2 phosphorylation in apoA-I transgenic animals, and decreased AKT and ERK1/2 phosphorylation in the aortas of apoA-I deficient mice [10].
Two cell surface proteins have also been implicated in HDL-mediated modulation of vascular tone, the scavenger receptor class B member (SR-B)I and the transporter ABCG1 (ATP-binding cassette, sub-family G, member 1). SR-BI colocalizes with eNOS in the caveolae of vascular endothelial cells, and interaction with HDL directly activates eNOS activity [11]. ABCG1 is strongly expressed in endothelial cells, and is known to promote the efflux of cholesterol and oxysterols to HDL, and to mediate various important intracellular processes [12]. It has been demonstrated that formation of eNOS dimers, which is necessary for proper enzyme function, is inhibited in the aortas of ABCG1 deficient (ABCG1−/−) mice fed high cholesterol or western diets [13]. The oxysterol 7-ketocholesterol tended to accumulate in the aortic endothelial cells, and the femoral arteries fromthese mice displayed impaired vasorelaxation in response to the agonist acetylcholine. Treatment with HDL prevented 7-ketocholesterol-induced disruption of eNOS dimer formation and restored eNOS activity in wild type aortic endothelial cells, but not in ABCG1−/− cells. Furthermore, ABCG1−/− mouse endothelial cells displayed increased surface expression of inflammatory markers such as E-selectin and intercellular adhesion molecule-1 and increased secretion of interleukin-6 and MCP-1 [14]. These findings correlated with increased endothelial cell activation in the ABCG1−/− mouse, suggesting that HDL-mediated lipid removal is an important mechanism in reducing vascular inflammation.
HDL also interacts directly with circulating leukocytes to limit inflammation. For example, apoA-I on HDL can inhibit activation of the monocyte cell surface protein CD11b, an integrin involved in vascular adhesion [15]. Interestingly, this process was dependent on another ATP binding cassette transporter, ABCA1 (member of subfamily A). In this gene lie the molecular defects responsible for Tangier’s disease, in which patients exhibit markedly depressed HDL levels. The transporter plays a crucial role in HDL formation and cholesterol efflux via an interaction with lipid-free forms of apoA-I, which are either secreted after synthesis in the liver and intestine, or displaced from intact lipoproteins. The involvement of two ABC transporters with established cholesterol efflux capacity is strongly suggestive of a mechanistic link between the cholesterol efflux and anti-inflammatory properties of HDL. This connection has recently been elegantly demonstrated at the molecular level (Box 2).
Box 2. An anti-inflammatory receptor.
The ABCA1 transporter is an important regulator of cholesterol efflux from lipid laden cells in the periphery to lipid-free apoA-I, a crucial step in reverse cholesterol transport and HDL production. Interestingly, Oram and colleagues have published a series of papers demonstrating that the apoA-I/ABCA1 interaction not only results in a transfer of cellular lipid to form HDL, but also mediates outside-in signaling events. One consequence is a triggering of enhanced apoA-I binding to ABCA1 mediated by Janus kinase (JAK)2 to reinforce lipid transfer [72]. More recently, a second arm of this pathway has been identified in which JAK2 activation by apoA-I binding to ABCA1 promotes the association of the STAT3 (signal transducer and activator of transcription 3) transcription factor with ABCA1 [73]. Once bound, STAT3 is phosphorylated to its activated state and translocates to the cell nucleus, where it activates anti-inflammatory gene expression programs. An important result of this is a reduced inflammatory response to bacterial LPS. This discovery has solidified the relationship between the lipid transport functions of apoA-I and the inflammatory response pathways in macrophages. It also raises the intriguing possibility that HDL or its components might be beneficial in other chronic inflammatory conditions such as rheumatoid arthritis.
Finally, the binding of HDL to macrophage-derived cytokines and growth factors might limit the proinflammatory activity of these proteins. For example, progranulin is a proinflammatory protein secreted by macrophages, which has been shown to bind to plasma apoA-I. Incubation with apoA-I or HDL suppresses progranulin induced expression of inflammatory markers in HEK293 cells [16].
Innate immune functions
The innate immune system represents the first line of defense against invading microorganisms. Accumulating evidence supports the idea that HDL is an integral component of innate immunity, mediating diverse functions that defend against viral, bacterial and parasitic infections. The finding that HDL is host to several complement factors reinforces this view. and suggests that HDL acts as a platform for the assembly of potent immunomodulatory complexes that regulate antimicrobial activity [17,18]. In addition, because infection and inflammation are tightly linked processes, the ability of HDL to regulate the amplitude of the inflammatory response might work in conjunction with these direct antimicrobial effects to influence the outcome of the infection.
Bacterial infection
HDLs are conserved in lower vertebrates such as fish, where they are expressed at high levels in plasma, as well as in tissues that constitute the primary defense barriers to bacterial infection [19]. Recent studies have demonstrated that apoA-I has potent bactericidal and bacteriostatic effects against both Gram-positive and Gram-negative bacteria, and vaccination with bacterial preparations increased apoA-I levels and enhanced the antibacterial activity of serum [20]. This antimicrobial effect is also effective against pathogens that infect humans, suggesting that HDLs might have originally evolved as components of primitive innate immune systems [19,21]. In addition to their direct antibacterial effects, mammalian HDLs have also been shown to protect against the adverse consequences of a bacterial infection by limiting the toxicity of bacterial components, many of which are responsible for life-threatening pathophysiological changes in the host. This toxin-neutralizing activity is largely attributed to apoA-I, and has been shown to be effective against enterohemolysin [22], lipopolysaccharide (LPS), and lipoteichoic acid [23-27].
Viral infection
The antiviral effects of human serum have been known for some time, and HDLs are known to account for a significant proportion of this activity, neutralizing both DNA and RNA viruses, and both enveloped and non-enveloped viruses [28]. The precise mechanism for HDL-mediated antiviral activity is incompletely understood, but current evidence suggests that it can involve direct viral inactivation, interference with viral entry into the cell, or inhibition of virus-induced cell fusion [28,29].
Parasitic infestation
The most well understood mechanism for the antimicrobial effects of HDL involves Trypanosoma brucei, a eukaryotic parasite that is the causative agent of African sleeping sickness [30]. Humans are naturally resistant to infection by the subspecies T. brucei brucei because the parasite is highly susceptible to lysis by trypanosome lytic factor (TLF), present in human serum [31].
TLF is composed of at least two independent circulating complexes: TLF1 and TLF2 [32]. TLF1 is bound to a subfraction of HDL and contains the primate-specific proteins apoL-I and haptoglobin-related protein (Hpr) [31,33]. TLF2 contains the same two proteins, but is part of a lipid-poor complex comprising IgM and apoA-I [34,35]. Within the circulation, the Hpr component of TLF1 associates with hemoglobin (Hb), generating a Hb-charged TLF1 particle that is efficiently taken up by the parasite via a receptor-mediated endocytic process that trypanosomes use to acquire heme from the host bloodstream [36]. Once inside the parasite, TLF1 is delivered to the lysosome by the endocytic pathway, where progressive acidification dissociates apoL-I from the complex, allowing it to insert into the lysosomal membrane. The bcl2-related pore-forming domain in the apoL-I protein triggers an influx of chloride ions into the lysosome, followed by water, resulting in swelling and trypanosome lysis. The pathway by which TLF2 enters the parasite is less clear, but the lysis mechanism appears to be the same. Interestingly, parasites that reside within the phagolysomes of macrophages, such as Leishmania sp, are not protected from TLF because the infected macrophages deliver plasma TLF directly into the vacuole in which the Leishmania organisms reside [37]. HDL is a sufficiently important threat that invading pathogens have evolved defense mechanisms or infection strategies that directly target HDL (Box 3).
Box 3. Anti-HDL warfare.
If there is any doubt that HDL plays an important role in host defense, it might be of interest to consider some of the adaptations that invading pathogens have evolved to specifically target HDL or its components. For example, Streptococcus pyogenes, a group A streptococcal bacterium responsible for tonsilitis, pharyngitis, and toxic shock syndrome, secretes a protein called serum opacity factor (SOF). Originally described for its ability to opacify the normally translucent plasma of infected individuals, SOF has recently been found to specifically target HDL particles by direct binding to apoA-I and apoA-II [70]. This causes a dramatic redistribution of the HDL neutral lipid cargo into large, protein-poor microemulsions. Although the exact effect of this reaction on the competitive fitness of the bacterium has not yet been identified, it is likely that SOF evolved as a virulence factor designed to subvert the antibacterial properties of intact HDL particles. Interestingly, parasites have also developed innovative strategies to frustrate HDL-based defenses. In contrast to T. b. brucei, humans can acquire African sleeping sickness by infection with the related subspecies Trypanosoma brucei rhodesiense. The ability of T. b. rhodesiense to infect humans has been attributed to its secretion of serum resistance-associated protein (SRA), a virulence factor that directly interacts with the C-terminal helix of HDL-associated apoL-I and inhibits its antitrypanosomal activity [33]. However, variants of apoL-I have recently been identified in the African population that do not bind SRA and thus retain lytic activity against T. b. rhodesiense [71]. These apoL-I variants are associated with high rates of renal disease in African Americans, suggesting that the selection pressure to acquire an SRA-resistant variant of apoL-I in Africa might have contributed to the development of a risk factor for kidney disease.
Modulation of glucose metabolism
T2D is characterized by a lack of glucose control due to the development of insulin resistance. Patients with T2D also display dyslipidemia with low HDL-C concentrations [38]. It has been shown by cell culture, animal and human studies that apoA-I gene expression is decreased by elevated glucose levels and increased by insulin [39], but there is emerging evidence that HDL, and apoA-I in particular, might also modulate glucose metabolism directly.
In ex vivo experiments, it was found that recombinant apoA-I improved glucose uptake associated with increased AMP-activated protein kinase (AMPK) activity in mouse skeletal muscle. It was also demonstrated that the absence of apoA-I in mice resulted in higher fasting blood glucose levels associated with reduced AMPK activity, and increased hepatic gluconeogenesis determined by increased phosphoenolpyruvate carboxykinase and glucose-6-phosphatase expression levels [40]. Furthermore, apoA-I was internalized through a clathrin-dependent endocytotic process before activating AMPK and acetyl-coenzyme A carboxylase in cell culture experiments. Recent observations indicate that infusions of rHDL particles reduced plasma glucose, increased insulin secretion and promoted glucose uptake in skeletal muscle of patients with T2D [41]. Based on cell culture experiments, those authors suggested that the AMPK signaling pathway might be involved in the rHDL-mediated increase in glucose uptake. Regarding the observed increased insulin levels in the patients with T2D, the report provided the first evidence, using a murine β cell line, that HDL directly stimulates insulin secretion from pancreatic β cells. In a follow-up study using transformed β cell lines and primary islets under basal and high-glucose conditions, it was determined that insulin secretion can be mediated by apoA-I and apoA-II, in their lipid-free forms, as constituents of rHDL or of HDL isolated from human plasma [42]. Furthermore these effects were calcium dependent and involved expression of ABCA1, ABCG1, and SR-BI. The importance of ABCA1 in modulating insulin secretion has been emphasized by the observation that mice with specific inactivation of ABCA1 in β cells had markedly impaired glucose tolerance and defective insulin secretion, but normal insulin sensitivity [43].
Besides its direct effect on glucose homeostasis, there is recent evidence that HDL might improve insulin resistance and obesity through its anti-inflammatory actions. The apoA-I mimetic peptide L-4F, known to have antioxidant properties, was investigated to determine if it would ameliorate insulin resistance and diabetes in genetically obese mice. It was found that L-4F treatment reduced adipocity and inflammation, and improved glucose tolerance in genetically obese ob/ob mice [44]. The observed reduction in glucose levels and prevention of fat mass accumulation was later attributed to upregulation of heme oxygenase expression and downregulation of endocannabinoid receptor-1 expression, resulting in adipose tissue remodeling [45]. In addition, the authors of that report detected increased AKT and AMPK phosphorylation in the aortas of L-4F treated mice, which could be prevented by inhibitors of PI3K activity. However, pharmacological inhibition only partially prevented the glucose lowering effect of L-4F in ob/ob mice, indicating the possible existence of alternative mechanisms. Another recent study in mice showed that apoA-I and its mimetic D-4F, an absorbable L-4F stereoisomer, increased energy expenditure by upregulating expression of UCP-1 in brown adipose tissue, thus adding an ulterior antiobesity function to HDL [46].
Antiapoptotic functions
HDL has been demonstrated to inhibit apoptotic activity in at least six different cell types including vascular endothelial and smooth muscle cells, some leukocytes, pancreatic β cells, cardiomyocytes and even bone-forming osteoblasts. Several different modes of action have been identified, involving both the protein and lipid components of HDL, which can act directly to influence cellular signaling or by various indirect mechanisms to prevent apoptosis. The protein component of HDL, consisting primarily of apoA-I, has been shown to be responsible for about 70% of HDL mediated inhibition of oxidized LDL-induced apoptosis in human microvascular endothelial cell line-1 [47].
There is evidence that some of the less abundant HDL proteins might also be involved. One minor HDL protein called paraoxonase (PON) 1 has been found to be important for HDL binding and protection of macrophages from apopotic events. This effect is the result of increased expression of SR-BI via PON 1 mediated activation of ERK1/2 and PI3K signaling pathways [48]. Another HDL associated protein, alpha-1-antitrypsin, can prevent apoptosis in a more indirect manner. This protein inhibits elastase-induced degradation of the extracellular matrix, thus preventing detachment and subsequent apoptosis of vascular smooth muscle cells [49].
The lipid composition of HDL can also play roles in antiapoptotic signaling. Recently, reports have indicated important roles for sphingosine 1 phosphate (S1P), a common component of HDL, in the protection from apoptosis of cardiomyocytes, pancreatic β cells and endothelial cells [50-53]. In addition to SR-BI, these protein and lipid components of HDL are likely to interact with other cell surface proteins to produce these effects. For example, the ATP binding cassette transporters ABCA1 and ABCG1 have both been implicated in the antiapoptotic effects of HDL on macrophages [54,55]. Furthermore, a known receptor for apoA-I and HDL, called cell surface F1-ATPase (an enzyme related to mitochondrial F1-ATPase), can inhibit apoptosis independent of ABC transporters and SRBI. Interaction of F1-ATPase with HDL not only protects against apoptotic signaling but also stimulates endothelial cell proliferation [56]. Different density fractions of HDL can have varying antiapoptotic capacities, with small dense HDL3c having the most potent effects, shown on endothelial cells and osteoblasts [47,57]. Interestingly, this activity is impaired in patients with metabolic syndrome, possibly due to the changes in HDL particle protein and lipid composition seen in these patients [58].
Influence on stem cells and embryogenesis
Another functional property of HDL currently under investigation is a role in the maturation of stem cells. Bone marrow cells (BMCs) are a key source of vascular progenitor cells that contribute to vessel repair upon endothelial denudation. BMCs are thought to be constantly shuffling back and forth between the bone marrow and the circulation, allowing them to migrate to sites of injury in response to proinflammatory cytokines. Once there, they can differentiate into the cell types that are needed to effect endothelial repair.
It was observed that treatment of lineage-negative BMCs with lipid-free apoA-I induced a change in their morphology and promoted expression of CD31, an adhesion molecule present in endothelial cells. This increased their ability to bind to both fibronectin and cultured endothelial cells [59]. A mutant of apoA-I lacking a key lipid binding site failed to promote these transformations, suggesting that apoA-I might mediate these effects via lipid efflux. The authors of that report suggested that apoA-I stimulation of BMC differentiation might be a mechanism by which HDL mediates vessel repair. The ability of apoA-I to mobilize cellular lipids also appears to play a role in the proliferation of hematopoietic stem and progenitor cells (HSPCs). For example, it was found that ABCA1 and ABCG1 deficient (ABCA1−/−ABCG1−/−) mice, which have extremely low levels of circulating HDL, displayed a five-fold increase in HSPCs compared with controls [60]. When ABCA1−/− ABCG1−/− bone marrow was transplanted into apoA-I transgenic mice, which display elevated levels of HDL, these levels decreased significantly. These results suggest that HDL might inhibit leukocytosis (elevated levels of circulating leukocytes) and reduce monocyte infiltration into vascular lesions.
Additionally, HDL is the predominant lipoprotein in follicular fluid, and might play a role in oocyte development and embryogenesis [61]. For example, a negative correlation has been found between human follicular HDL-C levels and embryo fragmentation during in vitro fertilization [62]. HDL metabolism might also affect embryo viability in vivo. Liver specific SR-BI knockout female mice with significantly decreased HDL uptake by the liver and large dysfunctional circulating HDL were infertile. However, reconstitution of SR-BI expression in the knockout animals by adenovirus mediated gene delivery restored both HDL morphology and fertility [63], consistent with a role for HDL in female reproductive physiology.
Effects on platelet function
HDL has been known to affect platelet function for many years, although the effects have been complex, and varied between experimental systems [64]. Recent evidence has revealed that the cholesterol homeostatic functions of HDL and apoA-I might significantly affect platelet function. Mice that lack SR-BI are known to be thrombocytopenic (low platelet count). Their platelets exhibit reduced ability to aggregate and this has been correlated to abnormally high levels of free cholesterol in the cells [65]. This suggests that platelet SR-BI interactions with HDL can modulate cellular cholesterol levels for optimal platelet function.
This assertion was supported by a more recent study in which patients with T2D were infused with reconstituted HDL preparations [66]. The infused patients exhibited a 50% decrease in platelet aggregation response versus controls. The effect was attributed to the phospholipid fraction of the particles with apoA-I having no role, consistent with the idea that SR-BI-mediated cholesterol efflux can beneficially modulate platelet function.
The discovery of several HDL proteins with known roles in platelet function such as the complement family and platelet activating factor acylhydrolase (Box 1), strongly suggest that more ties between HDL and platelet function exist, which go beyond cholesterol efflux. More work in this area is clearly needed.
Conclusions and future perspectives
Taking into account the diverse set of functions described above, and adding in the increasing appreciation of the compositional and structural heterogeneity of HDL, it is difficult to imagine that all these functions are mediated by the relatively limited number of HDL subspecies that are currently characterized. It is becoming clear that the term ‘HDL’ refers to an ensemble of discrete particles, each with their own complement of proteins and lipids that endow the host particle with distinct and varied functionalities. Indeed, many of these particles might play important physiological roles that have little to do with RCT or protection from heart disease. Some of these functions are closely related to the ability of HDL to modify the behavior of a target cell or organism by removing lipids. However, an increasing number appear to be mediated via interactions with cell surface proteins to trigger distinct signaling pathways to alter cell function. Although many of these studies are still in the initial stages, it would seem that it is an exciting time for the study of HDL metabolism.
In our view, two key challenges lie before the field of HDL research with respect to these alternative functions. First, we need a better understanding of the subparticle makeup of the fractions classically referred to as ‘HDL’. New technologies for alternative particle isolation and analysis and clever strategies for identifying distinct particle functions on the background of staggering compositional complexity will have to be developed to meet this important challenge. Once these subspecies are identified and characterized, it will be easier to correlate specific functions for these particles. Second, research needs to be directed at identifying additional roles for HDL outside of the classic purview of cardiovascular disease. The strong ties between inflammation and innate immunity make HDL a promising target for treatment or prevention of diseases such as those caused by opportunistic pathogens or chronic inflammatory states. This knowledge will not only be useful for understanding the potential of HDL as a treatment option for non-cardiovascular pathologies, but it could also be crucial for understanding the consequences of pharmacological manipulation of HDL-C levels. Indeed, current pharmacological therapies such as niacin or those in development including cholesteryl ester transfer protein inhibition and apoA-I transcription stimulation aim to raise plasma HDL cholesterol in the generic sense without direct knowledge of the functionality (or lack thereof) of the elevated species. It will be important to ascertain the effect of these manipulations, not only on those subspecies linked to CVD protection, but also on these peripheral functions, which might be just as important.
Acknowledgments
We thank Lisa Link (www.LisaLink.com) for her skillful rendering of Figure 1.
References
- 1.Lund-Katz S, Phillips MC. High density lipoprotein structure-function and role in reverse cholesterol transport. Subcell Biochem. 2010;51:183–227. doi: 10.1007/978-90-481-8622-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y, et al. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 3.Cockerill GW, et al. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:1987–1994. doi: 10.1161/01.atv.15.11.1987. [DOI] [PubMed] [Google Scholar]
- 4.Patel S, et al. Anti-inflammatory effects of apolipoprotein A-I in the rabbit. Atherosclerosis. 2010;2:392–397. doi: 10.1016/j.atherosclerosis.2010.05.035. [DOI] [PubMed] [Google Scholar]
- 5.Nobecourt E, et al. Nonenzymatic glycation impairs the antiinflammatory properties of apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2010;30:766–772. doi: 10.1161/ATVBAHA.109.201715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tolle M, et al. HDL-associated lysosphingolipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production. Arterioscler Thromb Vasc Biol. 2008;28:1542–1548. doi: 10.1161/ATVBAHA.107.161042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mineo C, et al. Endothelial and antithrombotic actions of HDL. Circ Res. 2006;98:1352–1364. doi: 10.1161/01.RES.0000225982.01988.93. [DOI] [PubMed] [Google Scholar]
- 8.Drew BG, et al. High-density lipoprotein and apolipoprotein AI increase endothelial NO synthase activity by protein association and multisite phosphorylation. Proc Natl Acad Sci U S A. 2004;101:6999–7004. doi: 10.1073/pnas.0306266101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mineo C, et al. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J Biol Chem. 2003;278:9142–9149. doi: 10.1074/jbc.M211394200. [DOI] [PubMed] [Google Scholar]
- 10.Norata GD, et al. High-density lipoproteins induce transforming growth factor-beta2 expression in endothelial cells. Circulation. 2005;111:2805–2811. doi: 10.1161/CIRCULATIONAHA.104.472886. [DOI] [PubMed] [Google Scholar]
- 11.Yuhanna IS, et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med. 2001;7:853–857. doi: 10.1038/89986. [DOI] [PubMed] [Google Scholar]
- 12.Tarr PT, et al. Emerging new paradigms for ABCG transporters. Biochim Biophys Acta. 2009;1791:584–593. doi: 10.1016/j.bbalip.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terasaka N, et al. ATP-Binding Cassette Transporter G1 and High-Density Lipoprotein Promote Endothelial NO Synthesis Through a Decrease in the Interaction of Caveolin-1 and Endothelial NO Synthase. Arterioscler Thromb Vasc Biol. 2010;11:2219–2225. doi: 10.1161/ATVBAHA.110.213215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whetzel AM, et al. ABCG1 deficiency in mice promotes endothelial activation and monocyte-endothelial interactions. Arterioscler Thromb Vasc Biol. 2010;30:809–817. doi: 10.1161/ATVBAHA.109.199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy AJ, et al. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol. 2008;28:2071–2077. doi: 10.1161/ATVBAHA.108.168690. [DOI] [PubMed] [Google Scholar]
- 16.Okura H, et al. HDL/apolipoprotein A-I binds to macrophage-derived progranulin and suppresses its conversion into proinflammatory granulins. J Atheroscler Thromb. 2010;17:568–577. doi: 10.5551/jat.3921. [DOI] [PubMed] [Google Scholar]
- 17.Murch O, et al. Lipoproteins in inflammation and sepsis. I. Basic science. Intensive Care Med. 2007;33:13–24. doi: 10.1007/s00134-006-0432-y. [DOI] [PubMed] [Google Scholar]
- 18.Jahangiri A. High-density lipoprotein and the acute phase response. Curr Opin Endocrinol Diabetes Obes. 2010;17:156–160. doi: 10.1097/MED.0b013e328337278b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villarroel F, et al. Apolipoprotein A-I, an antimicrobial protein in Oncorhynchus mykiss: evaluation of its expression in primary defence barriers and plasma levels in sick and healthy fish. Fish Shellfish Immunol. 2007;23:197–209. doi: 10.1016/j.fsi.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Caipang CM, et al. Intraperitoneal vaccination of Atlantic cod, Gadus morhua with heat-killed Listonella anguillarum enhances serum antibacterial activity and expression of immune response genes. Fish Shellfish Immunol. 2008;24:314–322. doi: 10.1016/j.fsi.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 21.Johnston LD, et al. Apolipoprotein A-I from striped bass (Morone saxatilis) demonstrates antibacterial activity in vitro. Comp Biochem Physiol B Biochem Mol Biol. 2008;151:167–175. doi: 10.1016/j.cbpb.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 22.Figueiredo PM, et al. Serum high-density lipoprotein (HDL) inhibits in vitro enterohemolysin (EHly) activity produced by enteropathogenic Escherichia coli. FEMS Immunol Med Microbiol. 2003;38:53–57. doi: 10.1016/S0928-8244(03)00125-1. [DOI] [PubMed] [Google Scholar]
- 23.Grunfeld C, et al. Lipoproteins inhibit macrophage activation by lipoteichoic acid. J Lipid Res. 1999;40:245–252. [PubMed] [Google Scholar]
- 24.Ma J, et al. Role of apolipoprotein A-I in protecting against endotoxin toxicity. Acta Biochim Biophys Sin (Shanghai) 2004;36:419–424. doi: 10.1093/abbs/36.6.419. [DOI] [PubMed] [Google Scholar]
- 25.Parker TS, et al. Reconstituted high-density lipoprotein neutralizes gram-negative bacterial lipopolysaccharides in human whole blood. Infect Immun. 1995;63:253–258. doi: 10.1128/iai.63.1.253-258.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiao YL, Wu MP. Apolipoprotein A-I diminishes acute lung injury and sepsis in mice induced by lipoteichoic acid. Cytokine. 2008;43:83–87. doi: 10.1016/j.cyto.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, et al. Human ApoA-I overexpression diminishes LPS-induced systemic inflammation and multiple organ damage in mice. Eur J Pharmacol. 2008;590:417–422. doi: 10.1016/j.ejphar.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 28.Singh IP, et al. Lipoproteins account for part of the broad non-specific antiviral activity of human serum. Antiviral Res. 1999;42:211–218. doi: 10.1016/s0166-3542(99)00032-7. [DOI] [PubMed] [Google Scholar]
- 29.Srinivas RV, et al. Inhibition of virus-induced cell fusion by apolipoprotein A-I and its amphipathic peptide analogs. J Cell Biochem. 1991;45:224–237. doi: 10.1002/jcb.240450214. [DOI] [PubMed] [Google Scholar]
- 30.Wheeler RJ. The trypanolytic factor-mechanism, impacts and applications. Trends Parasitol. 2010;9:457–464. doi: 10.1016/j.pt.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 31.Hajduk SL, et al. Lysis of Trypanosoma brucei by a toxic subspecies of human high density lipoprotein. J Biol Chem. 1989;264:5210–5217. [PubMed] [Google Scholar]
- 32.Vanhollebeke B, Pays E. The trypanolytic factor of human serum: many ways to enter the parasite, a single way to kill. Mol Microbiol. 2010;76:806–814. doi: 10.1111/j.1365-2958.2010.07156.x. [DOI] [PubMed] [Google Scholar]
- 33.Vanhamme L, et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. 2003;422:83–87. doi: 10.1038/nature01461. [DOI] [PubMed] [Google Scholar]
- 34.Raper J, et al. Characterization of a novel trypanosome lytic factor from human serum. Infect Immun. 1999;67:1910–1916. doi: 10.1128/iai.67.4.1910-1916.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomlinson S, et al. High-density-lipoprotein-independent killing of Trypanosoma brucei by human serum. Mol Biochem Parasitol. 1995;70:131–138. doi: 10.1016/0166-6851(95)00019-w. [DOI] [PubMed] [Google Scholar]
- 36.Vanhollebeke B, et al. A haptoglobin-hemoglobin receptor conveys innate immunity to Trypanosoma brucei in humans. Science. 2008;320:677–681. doi: 10.1126/science.1156296. [DOI] [PubMed] [Google Scholar]
- 37.Samanovic M, et al. Trypanosome lytic factor, an antimicrobial high-density lipoprotein, ameliorates Leishmania infection. PLoS Pathog. 2009;5:e1000276. doi: 10.1371/journal.ppat.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van LS, et al. High-density lipoprotein at the interface of type 2 diabetes mellitus and cardiovascular disorders. Curr Pharm Des. 2010;16:1504–1516. doi: 10.2174/138161210791051031. [DOI] [PubMed] [Google Scholar]
- 39.Mooradian AD, et al. Transcriptional control of apolipoprotein A-I gene expression in diabetes. Diabetes. 2004;53:513–520. doi: 10.2337/diabetes.53.3.513. [DOI] [PubMed] [Google Scholar]
- 40.Han R, et al. Apolipoprotein A-I stimulates AMP-activated protein kinase and improves glucose metabolism. Diabetologia. 2007;50:1960–1968. doi: 10.1007/s00125-007-0752-7. [DOI] [PubMed] [Google Scholar]
- 41.Drew BG, et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation. 2009;119:2103–2111. doi: 10.1161/CIRCULATIONAHA.108.843219. [DOI] [PubMed] [Google Scholar]
- 42.Fryirs MA, et al. Effects of high-density lipoproteins on pancreatic beta-cell insulin secretion. Arterioscler Thromb Vasc Biol. 2010;30:1642–1648. doi: 10.1161/ATVBAHA.110.207373. [DOI] [PubMed] [Google Scholar]
- 43.Brunham LR, et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med. 2007;13:340–347. doi: 10.1038/nm1546. [DOI] [PubMed] [Google Scholar]
- 44.Peterson SJ, et al. L-4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J Lipid Res. 2008;49:1658–1669. doi: 10.1194/jlr.M800046-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peterson SJ, et al. The L-4F mimetic peptide prevents insulin resistance through increased levels of HO-1, pAMPK, and pAKT in obese mice. J Lipid Res. 2009;50:1293–1304. doi: 10.1194/jlr.M800610-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruan X, et al. Apolipoprotein A-I possesses an anti-obesity effect associated with increase of energy expenditure and upregulation of UCP1 in brown fat. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Souza JA, et al. Small, dense HDL 3 particles attenuate apoptosis in endothelial cells: pivotal role of apolipoprotein A-I. J Cell Mol Med. 2010;14:608–620. doi: 10.1111/j.1582-4934.2009.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fuhrman B, et al. Paraoxonase 1 (PON1) deficiency in mice is associated with reduced expression of macrophage SR-BI and consequently the loss of HDL cytoprotection against apoptosis. Atherosclerosis. 2010;211:61–68. doi: 10.1016/j.atherosclerosis.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 49.Ortiz-Munoz G, et al. HDL antielastase activity prevents smooth muscle cell anoikis, a potential new antiatherogenic property. FASEB J. 2009;23:3129–3139. doi: 10.1096/fj.08-127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frias MA, et al. Native and reconstituted HDL protect cardiomyocytes from doxorubicin-induced apoptosis. Cardiovasc Res. 2010;85:118–126. doi: 10.1093/cvr/cvp289. [DOI] [PubMed] [Google Scholar]
- 51.Theilmeier G, et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. 2006;114:1403–1409. doi: 10.1161/CIRCULATIONAHA.105.607135. [DOI] [PubMed] [Google Scholar]
- 52.Kontush A, et al. Preferential sphingosine-1-phosphate enrichment and sphingomyelin depletion are key features of small dense HDL3 particles: relevance to antiapoptotic and antioxidative activities. Arterioscler Thromb Vasc Biol. 2007;27:1843–1849. doi: 10.1161/ATVBAHA.107.145672. [DOI] [PubMed] [Google Scholar]
- 53.Rutti S, et al. Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology. 2009;150:4521–4530. doi: 10.1210/en.2009-0252. [DOI] [PubMed] [Google Scholar]
- 54.Terasaka N, et al. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via ABCG1. Proc Natl Acad Sci U S A. 2007;104:15093–15098. doi: 10.1073/pnas.0704602104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yvan-Charvet L, et al. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ Res. 2010;106:1861–1869. doi: 10.1161/CIRCRESAHA.110.217281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radojkovic C, et al. Stimulation of cell surface F1-ATPase activity by apolipoprotein A-I inhibits endothelial cell apoptosis and promotes proliferation. Arterioscler Thromb Vasc Biol. 2009;29:1125–1130. doi: 10.1161/ATVBAHA.109.187997. [DOI] [PubMed] [Google Scholar]
- 57.Brodeur MR, et al. HDL3 reduces the association and modulates the metabolism of oxidized LDL by osteoblastic cells: a protection against cell death. J Cell Biochem. 2008;105:1374–1385. doi: 10.1002/jcb.21938. [DOI] [PubMed] [Google Scholar]
- 58.de Souza JA, et al. Metabolic syndrome features small, apolipoprotein A-I-poor, triglyceride-rich HDL3 particles with defective anti-apoptotic activity. Atherosclerosis. 2008;197:84–94. doi: 10.1016/j.atherosclerosis.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 59.Mythreye K, et al. ApoA-I induced CD31 in bone marrow-derived vascular progenitor cells increases adhesion: implications for vascular repair. Biochim Biophys Acta. 2008;1781:703–709. doi: 10.1016/j.bbalip.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yvan-Charvet L, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fujimoto VY, et al. High-density lipoprotein metabolism and the human embryo. Hum Reprod Update. 2010;16:20–38. doi: 10.1093/humupd/dmp029. [DOI] [PubMed] [Google Scholar]
- 62.Browne RW, et al. Follicular fluid high density lipoprotein-associated micronutrient levels are associated with embryo fragmentation during IVF. J Assist Reprod Genet. 2009;26:557–560. doi: 10.1007/s10815-009-9367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yesilaltay A, et al. Effects of hepatic expression of the high-density lipoprotein receptor SR-BI on lipoprotein metabolism and female fertility. Endocrinology. 2006;147:1577–1588. doi: 10.1210/en.2005-1286. [DOI] [PubMed] [Google Scholar]
- 64.Nofer JR, et al. High-density lipoproteins, platelets and the pathogenesis of atherosclerosis. Clin Exp Pharmacol Physiol. 2010;37:726–735. doi: 10.1111/j.1440-1681.2010.05377.x. [DOI] [PubMed] [Google Scholar]
- 65.Dole VS, et al. Thrombocytopenia and platelet abnormalities in high-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1111–1116. doi: 10.1161/ATVBAHA.108.162347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calkin AC, et al. Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation. 2009;120:2095–2104. doi: 10.1161/CIRCULATIONAHA.109.870709. [DOI] [PubMed] [Google Scholar]
- 67.Gordon SM, et al. Proteomic characterization of human plasma high density lipoprotein fractionated by gel filtration chromatography. J Proteome Res. 2010;9:5239–5249. doi: 10.1021/pr100520x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gordon S, et al. High-density lipoprotein proteomics: Identifying new drug targets and biomarkers by understanding functionality. Curr Cardio Risk Rep. 2010;4:1–8. doi: 10.1007/s12170-009-0069-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heinecke JW. The HDL proteome: a marker–and perhaps mediator–of coronary artery disease. J Lipid Res. 2009;50(Suppl):S167–171. doi: 10.1194/jlr.R800097-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Courtney HS, et al. Serum opacity factor, a streptococcal virulence factor that binds to apolipoproteins A-I and A-II and disrupts high density lipoprotein structure. J Biol Chem. 2006;281:5515–5521. doi: 10.1074/jbc.M512538200. [DOI] [PubMed] [Google Scholar]
- 71.Genovese G, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang C, et al. Janus kinase 2 modulates the apolipoprotein interactions with ABCA1 required for removing cellular cholesterol. J Biol Chem. 2004;279:7622–7628. doi: 10.1074/jbc.M312571200. [DOI] [PubMed] [Google Scholar]
- 73.Tang C, et al. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–32343. doi: 10.1074/jbc.M109.047472. [DOI] [PMC free article] [PubMed] [Google Scholar]