

Abstract
Bacteriophage RB69 DNA polymerase (RB69 pol) has served as a model for investigating how B family polymerases achieve a high level of fidelity during DNA replication. We report here the structure of an RB69 pol ternary complex at 1.8 Å resolution, extending the resolution from our previously reported structure at 2.6 Å [Franklin, M. C., et al. (2001) Cell 105, 657−667]. In the structure presented here, a network of five highly ordered, buried water molecules can be seen to interact with the N3 and O2 atoms in the minor groove of the DNA duplex. This structure reveals how the formation of the closed ternary complex eliminates two ordered water molecules, which are responsible for a kink in helix P in the apo structure. In addition, three pairs of polar−nonpolar interactions have been observed between (i) the Cα hydrogen of G568 and the N3 atom of the dG templating base, (ii) the O5′ and C5 atoms of the incoming dCTP, and (iii) the OH group of S565 and the aromatic face of the dG templating base. These interactions are optimized in the dehydrated environment that envelops Watson−Crick nascent base pairs and serve to enhance base selectivity in wild-type RB69 pol.
DNA polymerases (pols)1 are essential components of replicases, which are multiprotein complexes responsible for copying genomes of all organisms with high fidelity (for reviews, see refs (1−3)). The mechanistic details of how these pols accomplish this task with such remarkable accuracy are not fully understood. RB69 DNA polymerase (RB69 pol) is the founding member of the B family of pols (4), as it was the first pol to have its structure determined in the apo form and then in a ternary complex (5,6). RB69 pol also shares sequence similarities with a number of eukaryotic pols, including human pols α, δ, and ε (4). Structural studies of all these pols have provided tantalizing clues that continue to be explored as the resolution of ternary complex structures has improved.
One of the hallmarks of DNA pol ternary complex structures is that residues near the active site interact directly with the incoming dNTP as well as with the templating base in the minor groove of a DNA duplex, forming a nascent base pair-binding pocket (NBP) that is essential for recognition of Watson−Crick (W−C) base pairs. Occasionally, an incorrect incoming dNTP is mispaired with the templating base and incorporated onto the 3′ end of the growing primer strand. Even when this occurs, extension beyond this mismatched nucleotide is seldom observed, and the mismatched nucleotide residue is nearly always removed by an exonuclease activity that may be present either in a separate domain of the pol or as a separate subunit of the replicase. However, most of the base discrimination occurs when an incoming dNTP enters the binary complex. The correct incoming dNTP triggers Fingers closing and stabilizes the closed ternary complex, allowing proper alignment of the substrates prior to nucleotidyl transfer. When an incorrect incoming dNTP triggers the open-to-closed transition, the resulting closed ternary complex is unstable. As a consequence, the rate of reopening is much faster than the rate of nucleotidyl transfer and the mismatched dNTP is released (3,7).
Earlier studies have attempted to determine the differences between W−C and non-W−C base pairs in DNA duplexes (8,9). These studies have shown that mispairs distort the geometry of the duplex so that the cross-strand C1′−C1′ distances and the orientation of the glycosidic bonds as well as the positions of common N3 and O2 hydrogen bond acceptors in the minor groove of the duplex are altered relative to those of W−C base pairs. For example, the dT-dG mispair prefers a wobble geometry with the base of dG displaced toward the minor groove of the duplex. When DNA interacts with a pol, the minor groove of the bound DNA is widened near the primer/template (P/T) junction (10), which may also change preferences for mispaired geometries. Thus, depending on the specific interactions between the DNA and the pol at different locations in the P/T duplex, a dT-dG mispair can adopt either wobble or inverted wobble geometry with dG being displaced toward the minor or major groove (11). Because both wobble and inverted wobble forms of a dT-dG mispair have been observed, the energetic and steric differences for N3 and O2 hydrogen bond acceptors between a dT-dG mispair and a dC-dG pair are likely to be relatively small and unable to account for the large disparity in the kinetics exhibited by pols for insertion of dTMP versus dCMP opposite a templating dG. Thus, changes in the orientation of the glycosidic bonds as well as in the C1′−C1′ distances between mispaired and W−C base paired incoming dNTPs may play a greater role in base selectivity than the precise positioning of the N3 and O2 hydrogen bond acceptors.
To identify the structural features of nascent base pairs that allow a pol to discriminate between correct and incorrect dNTPs, structures of ternary complexes are needed with both correct and incorrect nascent base pairs. Structures of a number of ternary complexes with correct dNTPs have been obtained (6,12−14). However, it has been a challenge to capture a pol ternary complex with an incorrect dNTP in the insertion site. With W−C base pairs, the structures of binary and ternary complexes have provided clues about the mechanism of base recognition. For example, the structure of a Taq pol binary complex shows that residues in the NBP directly recognize N3 and O2 hydrogen acceptors in a widened minor groove of the DNA duplex (10). Minor groove widening has also been observed in complexes of other pols such as T7 pol and pol β (12,13,15), implying a common base recognition mechanism. The proper positioning of the N3 and O2 hydrogen bond acceptors in the minor groove of the DNA duplex is one of the features that distinguish W−C from non-W−C base pairs (8,9). In contrast, the same N3 and O2 recognition pattern of nascent base pairs is not observed in RB69 pol ternary complexes (6), suggesting that B family pols recognize the N3 and O2 hydrogen bond acceptors in a different manner. Nevertheless, a common feature among different families of pols is that the Fingers domain undergoes a conformational change after encountering correct dNTPs (6,13,14). This transition has been proposed as a checkpoint for discriminating against incorrect incoming dNTPs, although how this happens remains poorly understood. To address the base selectivity issue for RB69 pol, we have determined the crystal structure of its ternary complex with dCTP opposite a templating dG at 1.8 Å resolution, which is significantly higher than our previous resolution of 2.6 Å for a structure that had dTTP opposite dA (6). With the improved resolution, we have observed structural features that could not be seen previously and have confirmed many important inferences that were based on the 2.6 Å structure, particularly the importance of three unusual nonpolar−polar interactions in the pol active site and the role of the newly observed hydration network in the incorporation of correct dNMPs. We have compared this 1.8 Å resolution structure with the apo form of RB69 pol and provide additional insights into an open-to-closed conformational change of RB69 pol and its effects on the conformation of the incoming dCTP when it is part of the ternary complex. Finally, we have extended our comparison of the RB69 pol structure to the binary, ternary, and apo structures of ϕ29 DNA polymerase (ϕ29 pol) as well as to the ternary complex of yeast pol δ (16,17), in an attempt to identify the common features among the B family pols that are important for base selectivity.
Experimental Procedures
Chemicals
All chemicals were of the highest quality available; dCTP was purchased from Roche (Roche Applied Science, Indianapolis, IN).
Enzyme
Wild-type RB69 pol in an exonuclease-deficient background (D222A and D327A) was overexpressed in Escherichia coli, purified, and stored as previously described (18).
P/T DNA Substrates
All oligonucleotides were synthesized by W. M. Keck Foundation Biotechnology Resource Laboratory (Yale University). The sequence of the template strand used for cocrystallization was 5′-TCAGGTAAGCAGTCCGCG-3′; the sequence of the primer stand was 5′-GCGGACTGCTTACdd-3′ with a 3′ dideoxy terminus. The P/T was annealed by being heated to 85 °C and then gradually cooled to 25 °C. Sequences of other oligonucleotides for kinetic assays were altered one nucleotide at a time for formation of mismatch-containing DNA duplexes as described below. The 5′ end of each primer was labeled with Cy3 for kinetic studies.
Crystallization of the RB69 pol Ternary Complexes
We prepared the ternary complex by mixing an equimolar ratio of wild-type RB69 pol (exo−) with a freshly annealed P/T duplex and dCTP. After the solution had been mixed, the final concentration of the complex was ∼12 mg/mL and the dCTP concentration was 2.5 mM. Crystals of the ternary complex were grown under oil in a microbatch procedure by mixing equal volumes of the ternary complex with a solution containing 100 mM sodium cacodylate buffer (pH 6.5), 160 mM CaCl2, and 8% (w/v) polyethylene glycol 350 monomethyl ether (PEG350 MME). Rod-shaped crystals typically grew in 3−6 days at 20 °C to a size of 0.2 mm × 0.1 mm × 0.1 mm. The crystals were stabilized and cryoprotected by being transferred first to a stabilization solution containing 100 mM sodium cacodylate buffer (pH 6.5), 20% (w/v) PEG350 MME, and 100 mM CaCl2. Prior to being frozen in liquid nitrogen, the crystals were transformed into another stabilization solution where the concentration of PEG350 MME was increased to 30% (v/v) for cryoprotection.
X-ray Diffraction Data Collection, Structure Determination, and Refinement
X-ray diffraction data were collected at a wavelength of 0.9095 Å and at 110 K at NECAT, beamline 24ID-E (Advanced Photon Source, Argonne National Laboratory, Argonne, IL). The data were processed using the HKL2000 program suite (19), and the data statistics are summarized in Table 1. The crystals belonged to orthorhombic space group P212121 with unit cell dimensions different from those previously published for the 2.6 Å ternary complex of RB69 pol (6).
Table 1. Crystallographic Statistics for Data Collection and Refinement.
| space group | P212121 |
| unit cell dimensions (Å) | a = 75.50, b = 119.40, c = 129.76 |
| resolution (Å)a | 50−1.80 (1.86−1.80) |
| wavelength (Å) | 0.9095 |
| no. of unique reflections | 104627 |
| redundancy | 3.4 (1.9) |
| completeness (%) | 94.9 (74.0) |
| Rmerge (%)b | 7.7 (81.3) |
| I/σ | 13.7 (0.86) |
| refinement statistics | |
| no. of reflections | 99370 |
| Rwork (%)c | 17.5 |
| Rfree (%)d | 20.1 |
| final model | |
| no. of non-hydrogen atoms | 9599 |
| no. of waters | 1373 |
| no. of Ca2+ ions | 3 |
| no. of template nucleotides | 18 |
| no. of primer nucleotides | 13 |
| no. of dCTPs | 1 |
| average B factor (Å2) | |
| protein (w/TLS) | 20.5 |
| waters | 33.0 |
| rmsde | |
| bond lengths (Å) | 0.006 |
| bond angles (deg) | 1.005 |
| PDB entry | 3NCI |
The highest-resolution shell statistics are in parentheses.
Rmerge = ⟨∑hkl∑j|Ij(hkl) − ⟨I(hkl)⟩|⟩/⟨I(hkl)⟩, merging statistics for all symmetry mates.
Rwork = ∑hkl|Fobs(hkl) − Fcalc(hkl)|/∑hkl|Fobs(hkl)| (crystallographic R factor).
Rfree is the cross-validation R factor for ∼5% of the total unique reflections that have been randomly selected.
Root-mean-square deviation from ideal values.
The structure was determined by the automated molecular replacement method Phaser as implemented in CCP4 (20), starting with the wild-type (wt) RB69 pol structure of the ternary complex [PDB entry 1IG9(6)]. The P/T DNA duplex and the incoming dCTP were built into residual electron density maps, which were phased with the partially refined polymerase model, using COOT (21). The structure was refined using Refmac5 (22), and refinement statistics are listed in Table 1. Using the criteria of Wang and Boisvert (23), the resolution of the structure was estimated to be 1.8 Å. All figures were made using Ribbons (24).
PDB Entry
Coordinates and structure factors for the wild-type ternary complex structure have been deposited in the Protein Data Bank as entry 3NCI.
Chemical Quench Experiments
Experiments were performed at 23 °C with a buffer solution of 66 mM Tris-HCl (pH 7.4) as previously described (18). Rapid chemical quenching experiments were performed using a KinTek model RQF-3 instrument (KinTek Co., University Park, PA). For kpol and Kd,app determinations, experiments were performed under single-turnover conditions, with a 10-fold excess of RB69 pol over P/T. Briefly, enzyme and P/T from one syringe were rapidly mixed with Mg2+ and various dNTP concentrations from the other syringe for times ranging from as little as 3 ms to as much as 15 s. The final concentrations after mixing were as follows: 1 μM enzyme, 100 nM P/T, and 10 mM Mg2+. Reactions were quenched with 0.5 M EDTA (pH 8.0). Substrates and products were separated by 19:1% (w/v) PAGE (polyacrylamide/bisacrylamide gels) containing 8 M urea, visualized using a FUJIFILM imager scanner (FLA-5100), and quantified using Multi Gauge version 3.0 (FUJIFILM) and GraphPad Prism 4 (Life Sciences Co., Stamford, CT).
Analysis of Kinetic Data
The amount of product formed versus time was plotted for each dNTP concentration and fitted by nonlinear regression to the general form of eq 1 to obtain observed rates of product formation, kobs (or ki if multiple phases of turnovers exist),
| 1 |
where Y is the concentration of the DNA product formed during the reaction, C is the offset constant, Ai is the observed amplitude of product formation, and kobs or ki (with i = 1 for single-phase turnover) is the observed rate constant. The kinetic parameters kpol (the rate of phosphoryl transfer) and Kd,app (defined as the dNTP concentration at which the rate of phosphoryl transfer reaches 1/2kpol) were obtained by plotting kobs versus dNTP concentration with eq 2:
| 2 |
where kobs represents the observed rate at a given dNTP concentration. Note that the Kd,app values are not necessarily ground-state dissociation constants of dNTP binding, because the observed dNTP concentration dependence of the rates of product formation is affected by “hidden” steps that occur subsequent to dNTP binding but prior to phosphoryl transfer.
Steady-State Assay
Primer extension assays for mismatch-containing DNA duplex were performed with wild-type RB69 pol (exo−) and various P/Ts (see Table 2 for details of sequences) with four dNTPs at room temperature for 15 s and 2 min. After the solutions had been mixed, the final concentrations were as follows: 40 nM RB69 pol, 200 nM P/T, 10 mM MgSO4, and 66 mM Tris-HCl (pH 7.4).
Table 2. Kinetic Parameters for Primer Extension for Mismatched DNA Duplexes.
| P/Ta | kpol (s−1) | Kd,app (μM) | kpol/Kd,app (s−1 μM−1) |
|---|---|---|---|
| P/T1 | 169 | 70 | 2.4 |
| P/T2b | ND | ND | 4.8 × 10−4 |
| P/T3b | ND | ND | 1.3 × 10−3 |
| P/T4b | ND | ND | 1.5 × 10−2 |
| P/T5 | 85 | 77 | 1.1 |
| P/T6 | 163 | 62 | 2.6 |
Primer/Template (P/T) sequences are shown below: The mismatched base pairs are highlighted in red.
The mismatched base pairs are highlighted in red.
Because of nonsaturating conditions for incoming dNTPs, only the incorporation efficiency of the kcat/KM ratios was determined. Individual kpol and Kd,app values cannot be determined (ND).
Results and Discussion
Overall Structure of the 1.8 Å Resolution Ternary Complex of RB69 pol
The crystal structure of the RB69 pol ternary complex was refined at 1.8 Å resolution to yield crystallographic and free R factors of 18 and 20%, respectively (Table 1). The overall structure of this complex is identical to that previously reported at 2.6 Å resolution with a root-mean-square Cα deviation of 0.54 Å after exclusion of a flexible loop (residues 250−263) in the exonuclease domain and the flexible C-terminal tail (residues 891−901) (6). However, this 1.8 Å resolution structure contains a nascent dG/dCTP base pair instead of a dA/dTTP base pair and has 13 bp rather than 14 bp in the P/T duplex. In both structures, the enzyme binds to the first 10 bp of the P/T, leaving 3 bp in this complex and 4 bp in the previous complex without direct contact with the enzyme. The two ternary complexes also have a different number of nucleotide residues in the 5′ template overhang, four in this complex and three in the 2.6 Å resolution structure. These template overhangs have different sequences and interact with different residues in RB69 pol. When the two structures were superimposed, the phosphodiester backbones of the two P/Ts are also superimposable, a feature that is the hallmark of non-sequence specific binding of DNA to the polymerase.
Recognition of the N3 and O2 Hydrogen Bond Acceptors in the Minor Groove of the P/T Duplex
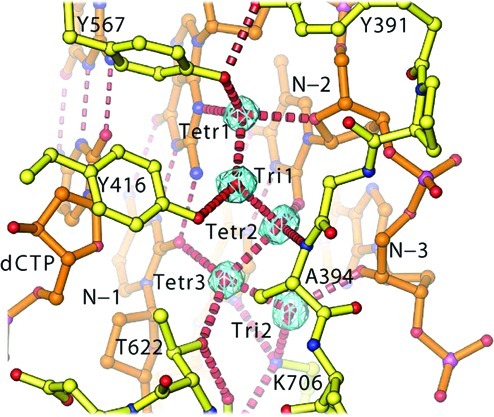
In our 1.8 Å resolution structure, five ordered water molecules serve as extensions of conserved pol residues that recognize the N3 and O2 hydrogen bond acceptors in the minor groove of the P/T duplex (Figure 1 and Figure S1 of the Supporting Information). This is particularly evident for the first 2 bp upstream from the P/T junction. These water molecules are buried at the interface between the DNA duplex and the enzyme. Three of these five water molecules, Tetr1, Tetr2, and Tetr3, have tetrahedral geometry, and the other two, Tri1 and Tri2, have unusual planar trigonal geometry (Figure 1B,C). All of these water molecules are polarized so that they serve as extensions of hydrogen bond donors for the side chains of K706, Y567, Y416, and T622 and an extension of the hydrogen bond acceptor for the main chain carbonyl of A394. The first water molecule, Tetr1, interacts with the N3 and O2 hydrogen bond acceptors of the 3′ nucleotide residue of the primer and is in perfect tetrahedral coordination (Figure 1). Its hydrogen atom is oriented directly by the side chain of Y567 and indirectly by the side chain of Y416 through a trigonally coordinated water molecule, Tri1. The second tetrahedrally coordinated water molecule, Tetr2, interacts with N3 and O2 of the second 3′ nucleotide residue of the primer. The third tetrahedrally coordinated water molecule, Tetr3, interacts with N3 and O2 of the third nucleotide residue of the primer (N − 3rd nucleotide residue in Figure 1). Its hydrogen atom is oriented by the side chain of T622 and another trigonally coordinated water molecule, Tri2, which forms a hydrogen bond with K706. Finally, the ε-amino group of K706 interacts with N3 and O2 of the third nucleotide residue of the template from the P/T junction in the duplex (Figure 1). This is the only side chain directly interacting with the N3 and O2 hydrogen bond acceptors in the P/T duplex that does not involve a water molecule. Surprisingly, neither do the residues of RB69 pol serve as hydrogen bond donors, nor do the water molecules interact with the N3/O2 atom of the incoming dCTP in the RB69 pol ternary complex. The overall water-mediated mechanism by which RB69 pol recognizes N3 and O2 in the minor groove observed here is also found in the ternary complex of ϕ29 pol, another B family pol (Figure S1 of the Supporting Information). This pattern is different from that found in members of the A family pols, where the side chains of conserved pol residues bind directly to the N3 atoms of purines (Pu) or the O2 atoms of pyrimidines (Py) once the minor groove of the duplex has been widened (10,12,13,15). Nevertheless, all pol−DNA interactions, like those of many nonspecific DNA-binding proteins, occur mainly with the minor groove (25). It is well-known from the crystal structure of the Trp repressor complex with DNA that ordered water molecules can serve as extensions of protein side chains to mediate sequence specific protein−DNA interactions (26), a mechanism that has now been extended to non-sequence specific pol−DNA interactions.
Figure 1.

Five buried water molecules. (A−C) Standard and close-up views of stereodiagrams for five buried water molecules (contoured at 0.7σ and edited to remove nonsolvent density for the sake of clarity), including two planar triangularly coordinated water molecules (B and C).
In our 1.8 Å resolution structure, we noted three pairs of unusual nonpolar−polar interactions within the hydrophobic environment of the nascent base pair-binding pocket between (i) the Cα hydrogen of G568 and the N3 hydrogen acceptor of the templating dG, (ii) the C6 hydrogen of dCTP and the O5′ hydrogen acceptor of its ribosyl moiety, and (iii) the aromatic face of the templating dG base and the OH group of S565. Interactions between nonpolar and polar groups occur rarely, because of the free energy cost of desolvating a hydrogen bond donor or acceptor without a compensating hydrogen bond, yet nonpolar−polar interactions can provide some specificity among interacting partners; on the other hand, classic hydrophobic interactions cannot, where two interacting partners can readily slide relative to one other.
We suggest that in the unusual nonpolar−polar interaction between the Cα hydrogen of G568 and the N3 hydrogen acceptor of the templating dG, a hydrogen bond forms because one of the two G568 Cα hydrogen atoms is perfectly positioned to do so in a hydrophobic environment (Figure 2 and Figure S2 of the Supporting Information). There is a well-documented example of this type of hydrogen bond in a lipid bilayer that involves a glycine residue (27). This putative hydrogen bond could tightly anchor the templating base within the NBP and would be applicable to all four possible templating bases because there is always a hydrogen bond acceptor at the N3 or O2 position. It is interesting to note that there is no room for a water molecule near G568 in the ternary complex, but there are two ordered water molecules that interrupt the backbone hydrogen bonding pattern of the P helix near G568 in the apo structure, causing a significant kink in the α-helix (see below).
Figure 2.
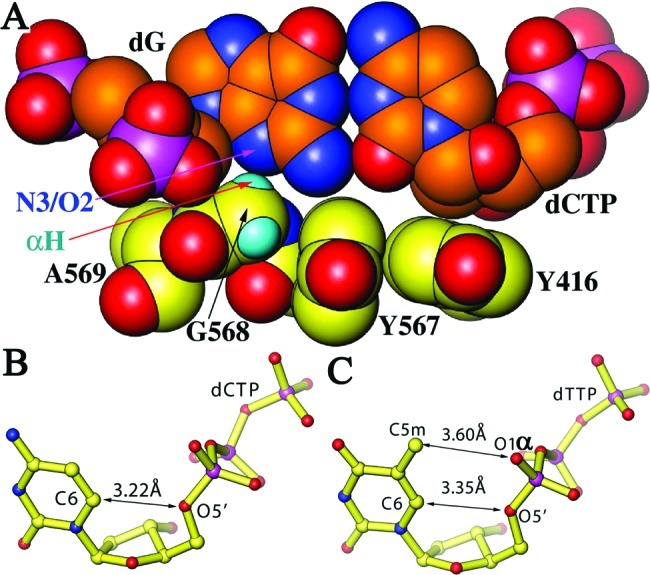
Eclipsed conformation of the incoming dCTP in the nascent base pair. (A) Recognition of N3 and O2 hydrogen bond acceptors by the G568 Cα hydrogen (small cyan spheres). (B) Eclipsed conformation of dCTP in this ternary complex. (C) Eclipsed conformation of dTTP in the previously reported RB69 pol ternary complex (6).
Formation of an Internal C6−O5′ Hydrogen Bond in dCTP and Its Potential Role in Catalysis
We propose that a hydrogen bond forms between the C6 and O5′ atoms of the incoming dCTP, when it becomes enmeshed in the closed ternary complex (Figure 2), because the observed interatomic distance between these two atoms is 3.22 Å in our structure (Figure 2B) rather than a usual longer separation, for example, 4.5 Å for the templating dG residue or 4.8 Å for the priming ddC residue in the structure. Moreover, the predicted hydrogen atom on C6 is an optimal orientation for the formation of a hydrogen bond. This is another example of an unusual hydrogen bond, because (i) the O5′ atom is normally a weak hydrogen acceptor and the hydrogen on C6 of dCTP is a poor donor, (ii) such short C6−O5′ interatomic distances are not found in other nucleotide residues within P/T duplexes, and (iii) in solution, rNTPs (presumably dNTPs as well) assume an extended conformation with the polar O5′ atom positioned as far away as possible from the nonpolar Py’s C6 or Pu’s C8 atom (28,29). We refer to the conformation of the incoming dCTP in our structure as being in an eclipsed conformation where the orientation of the O5′−C5′ bond of the ribosyl moiety is nearly coplanar with the base such that the O5′ atom eclipses the Py’s C6−H6 moiety (Figure 2B,C). Pseudotorsion angles from the O5′−C5′ bond to the C1′−N1 bond and to the C1′···C6 bond of the dCTP in the ternary complex are 5.2° and 3.4°, respectively, which are smaller than corresponding angles of most ribonucleotide residues in the structure of the 50S ribosome (Figure S3 of the Supporting Information). Thus, incoming dNTPs undergo an extended-to-eclipsed transition when they become a part of the ternary complex.
We also suggest that this putative Py’s hydrogen bond between C6 and O5′ could provide additional stabilization of the transition state in which the O5′ atom is one of three equatorial oxygen atoms in the pentacovalent phosphoryl intermediate. This oxygen atom does not have direct interaction with any pol residues. The other two equatorial oxygen atoms are stabilized by the ε-amino group of K560 and divalent metal ions in the ternary complex. In the transition state, the C6−O5′ distance should be further reduced as the Pα−O5′ distance increases, and the electronegativity of the O5′ atom should increase as the positive partial charge develops at the Pα center. All these features could further strengthen the proposed C6−O5′ hydrogen bond.
The observed eclipsed geometry of dCTP and its C6−O5′ internal interaction could provide a direct link between correct W−C base pairing geometry and catalysis through transition-state stabilization as suggested above, because the C6−O5′ interaction is likely to be absent in non-W−C base paired incoming dNTPs. On the basis of structures of mispaired bases embedded in DNA duplexes (8,9), non-W−C base pairs can be classified into three groups according to their distinct C1′−C1′ distance relative to W−C base pairs: (i) large Pu-Pu mis pairs, (ii) similar size of Pu-Py mispairs, and (iii) small Py-Py mispairs. In our ternary complex, the base of dCTP fits snugly into the NBP and is sandwiched between the dG templating base and its own O5′ moiety. This space cannot accommodate the base of Pu dNTPs in Pu-Pu mispairs. Thus, any Pu-Pu mispaired dNTPs would prevent the ternary complex from being fully closed. While the dG templating base in our ternary complex could be computationally replaced with smaller Py bases without steric clashes for Py-Py mispairs, the incoming dCTP may not adopt an eclipsed geometry without a tightly enclosed templating base. Consequently, the putative C6−O5′ hydrogen bond could not form.
Although Pu-Py mismatches and W−C base pairs are similar in size, the orientation of the glycosidic bonds between them is very different. For example, the orientation of the glycosidic bonds of the dT-dG mispair within a non-cDNA duplex differs by 15° relative to that observed with W−C base pairs (8,9). Any change in the orientation of the glycosidic bond in dNTPs would prevent them from adopting an eclipsed geometry, because the orientation of the glycosidic bond is restricted so that it is nearly parallel to the C4′−C5′ bond in the dCTP observed in our structure. Like in Py-Py mispairs discussed above, Pu-Py mispairs without an eclipsed geometry would not have a favorable C6−O5′ (for Py dNTPs) or C8−O5′ (for Pu dNTPs) internal interaction.
Anchoring the dG Templating Base by the Hydroxyl Side Chain of S565
The last nonpolar−polar interaction in our structure is between the aromatic face of the dG templating base and the hydroxyl side chain of S565 (Figure 3). This side chain sits atop the templating dG with an interatomic distance of ∼3.33 Å between it and all five atoms of the dG’s five-membered ring (Figure 3A,B). The hydrogen atom of this OH group is oriented so that it can form a hydrogen bond with the backbone carbonyl oxygen of L561. Given such an orientation, the lone pair electrons of the polar oxygen atom contact the aromatic face of the dG templating base. The same interaction of S565 with the dA templating base was also observed in the previous 2.6 Å resolution structure (6), and it is reasonable to assume that it can also occur with Py templating bases. With this interaction, the templating base is tightly anchored within the NBP and cannot slide relative to other NBP residues. This differs from classic hydrophobic interactions without any specificity. This polar−nonpolar interaction is conserved among all B family pols for which structures have been determined.
Figure 3.

Rigidity of the templating nucleotide-binding pocket. (A) Interactions with interatomic distances (in angstroms) indicated for the base of the dG (in cyan) observed in this ternary complex. Essential residues for the binding pocket are colored yellow, and nonessential residues are colored silver. The nucleotide 3′ to the dG is colored gold. (B) Interactions of the ribosyl moiety of dG with G568.
Additionally, there are other interactions that help to immobilize the templating base within the NBP, including (i) extensive van der Waals contacts between its ribosyl moiety and the G568-A569 peptide bond and (ii) hydrogen bonds with its flanking 5′ and 3′ phosphodiester linkages by the side chains of S360 and N572, respectively (Figure S4 of the Supporting Information). These hydrogen donors in turn are defined by other hydrogen donors from backbone amides of K363 and W574. pol residues that interact with the templating base but not directly with the incoming dNTPs such as S565, Y567, and L561 serve only to firmly anchor the templating base within the NBP and would not be expected to have a significant influence on pre-steady-state kinetic parameters for incorporation of the correct dNMPs (30).
New Insights into the Open-to-Closed Transition of the Fingers Domain
The open-to-closed transition of the Fingers domain involves many complex motions that help the incoming dCTP undergo an extended-to-eclipsed conformational change. In this structure, the guanidinium group of R482 in helix N makes electrostatic interactions with the γ-phosphate group of the incoming dCTP; K562 in helix P binds to the α-phosphate, and N564 also in helix P binds to the β-phosphate through a water molecule. These three residues are located in the Fingers domain (Figure 4), which moves during an open-to-closed transition. The importance of these interactions has been established by the functional consequences of their replacement with Ala (31). As a part of the open-to-closed conformational transition, the backbone Cα atoms of the three Fingers residues shift toward the Palm domain (where the dG templating base binds) as follows: R482 by 5.0 Å, K560 by 4.3 Å, and N564 by 3.7 Å (Figure 4C). During the reversal of this process, these three residues move away from the Palm domain, and the distance between two anchoring points for the incoming dCTP (one for its triphosphate moiety by the three residues in the Fingers domain and the other for its base by residues in the Palm domain as well as the dG templating base) increases so that dCTP relaxes from an eclipsed to an extended conformation. The opening and closing motions of the Fingers domain in pols are known to be rapid and non-rate-limiting steps (32). We suggest that dCTP alternates between the extended and eclipsed conformations in response to the opening and closing motions of the Fingers domain. When the base of dCTP is tethered to the dG templating base through W−C hydrogen bonds, the putative C6−O5′ hydrogen bond should stabilize the eclipsed conformation and suppress the opening process. Without W−C hydrogen bonds, dCTP would be released whenever the Fingers domain opens. It is tempting to speculate that if the tethering interbase hydrogen bonds are solely responsible for retaining dCTP in the pol active site whenever the Fingers domain opens, and thus for confining dCTP in an eclipsed geometry when RB69 pol is in the closed ternary complex, the actual energy from interbase hydrogen bonds may no longer be reflected in the apparent binding affinity of correct dNTPs. This structural feature would explain the observation that the binding affinity for correct dNTPs is independent of the number of interbase hydrogen bonds, three for dG-dC and two for dA-dT pairs (18).
Figure 4.

Formation of the NBP upon Fingers domain closing. (A) Superposition of the ternary complex (yellow and silver) with the apo structure (magenta and silver) using main atoms of Y577, G568, and A569 shows the effects of the kinked-to-straight helical transition on interactions with the dG. Large movements of key residues are indicated. (B) Same as panel A, but using Cα atoms of the C-terminus of helix P for superposition. (C) Same as panel A, but using Cα atoms of the Palm domain. (D) Contribution of K560, N564, and R482 (gold) to kinetic parameters for the correct dNTP in the ternary complex is indicated next to each residue as the KD(mutant)/KD(wt) ratio and the kpol(wt)/kpol(wt) ratio when each residue was substituted by an Ala residue (31).
In our structure, we are able to provide additional information about the open-to-closed transition by observing specific hydrogen bonding patterns in the P helix backbone (Figure 5) and comparing them with the patterns in the apo structure (5,6). In the structure presented here, helix P is nearly straight, although it deviates slightly from an idealized α-helix with canonical backbone hydrogen bonds between residues i and i + 4. This helix includes many nonstandard hydrogen bonds (Figure 5B). In contrast, helix P in the apo structure has a kink near Y567 and G568 (5,6). This kink results from indirect hydrogen bonds that are mediated through two ordered water molecules that are inserted between the N564 O atom and the G568 N atom and between the I563 O atom and the Y567 N atom, causing the helix to bend by ∼23° toward S565 (Figure 5). These two ordered water molecules are part of a large hydration network surrounding the P and N helices of the Fingers domain. Thus, the open-to-closed change of the Fingers domain includes the kinked-to-straight transition of helix P and the dehydration of helices of the Fingers domain.
Figure 5.

Helices of the Fingers domain. (A) Final 2Fo − Fc map contoured at 1.4σ superimposed with the refined model of helix P. (B) Hydrogen atoms are generated for explicit hydrogen bonds in the backbone of helix P. Nonstandard backbone hydrogen bonds are colored yellow. (C) Superposition of helices of the Fingers domain between the structure of the current complex (yellow) and the previous apo structure (cyan and yellow) of RB69 pol using the Cα atoms of H485−T554 (5,6). Large displacements for G569 and V573 between the two structures are indicated. (D) Same as panel C, but the Cα atoms of L562−L566 were used for superposition. (E) Same as panel C, but the Cα atoms of G568−N572 were used for superposition. (F) Helix P of the apo structure with two water molecules inserted into its backbone. (G) Superposition of helix P (yellow) of our ternary complex with its equivalent helix (cyan) of the ternary complex of ϕ29 pol (16). (H) Superposition of helix P (yellow) of the apo structure of RB69 pol with its equivalent helix (cyan) of the apo form of ϕ29 pol (16).
Fingers domain closing has generally been portrayed as a rotation with a single hinge point at the Palm−Fingers junction (6). From this structure, it is clear that this involves complex changes involving many moving parts. Some of these became apparent when different segments of the Fingers domain in the ternary complex were superimposed onto corresponding segments of the apo structure (Figure 5C−E). This comparison showed that the ends of the P and N helices of the Fingers domain were displaced to different extents, one end by 9 Å (G496 of helix N) and the other by 13 Å (V573 of helix P). For example, helix N does not undergo a kinked-to-straight transition but twists and curves around helix P. A consequence of these complex motions is the simultaneous maintenance of the interaction of residues such as R482 and K560 with the triphosphate moiety of dCTP via rotation of the triphosphate moiety into alignment with the other half of the binding pocket, which is made up mainly of the two conserved residues, D411 and D623, in the Palm domain. These two acidic residues interact with the triphosphate tail of dCTP through two divalent metal ions that are essential for catalysis (31).
Single-Stranded 5′ Template Overhang and Other pol−DNA Interactions
One extra nucleotide in this structure compared to the previous structure (6) on the 5′ template strand has resulted in a well-defined structure for its three 5′-overhanging nucleotide residues (N + 1 to N + 3) in this complex (Figure 6). In this complex, bases N + 1 and N + 2 are stacked on each other and against F359 on one side and I253 on the other side, but not on the bases of their N and N + 3 neighbors. The phosphodiester groups of nucleotides N + 1 and N + 2 are hydrogen bonded to the guanidinium group of R260 and O5′ of nucleotide N + 3, respectively. Because these interactions would not be expected to be sequence specific, they should occur with any single-stranded template overhang. In addition, N786 forms two hydrogen bonds with the N1 and O2 atoms of nucleotide residue N + 3 (dT), and E219 forms a hydrogen bond with the N2 atom of nucleotide N + 2 (dC) (Figure 6). The new structure of the single-stranded 5′ template overhang suggests that it might contribute substantially to the pol−P/T binding with specificity for properly positioning the P/T junction at the pol active site. This interaction differs from other interactions with the duplex region, which also contributes to additional binding affinity but cannot properly position the P/T junction at the pol active site for catalysis. Interestingly, a similar 5′ template overhang structure has also been observed in the ternary complex of yeast pol δ (17), suggesting that this structure might also have functional significance. For example, the single-stranded 5′ template exists only before termination of Okazaki fragment synthesis. After completion, the single-stranded DNA−pol interactions disappear and the P/T rapidly dissociates from the polymerase (33−36). In addition, the phosphodiester backbone conformation in the 5′ template overhang differs from one residue to the next. This would likely prevent nonspecific sliding of the P/T during translocation by locking the P/T junction at the pol active site while waiting for next correct incorrect dNTP to enter (33−36). Except for the interactions just mentioned, all other pol contacts with DNA are mediated through the phosphodiester linkages, where the common hydration pattern in the minor groove largely remains unaltered upon binding of the DNA duplex to the pol (Figure S5 of the Supporting Information).
Figure 6.
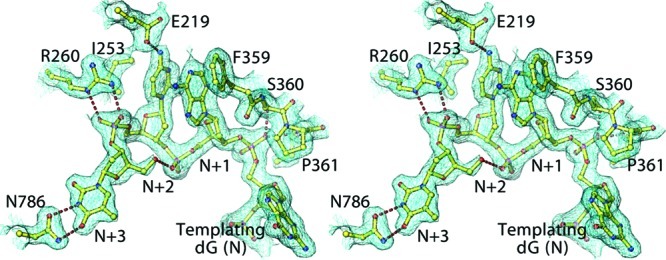
Structure of the overhanging 5′ template in stereodiagram superimposed onto the final 2Fo − Fc map contoured at 0.5σ.
Comparison with Other B Family pols
Since our initial reports on the crystal structures of the apo form and the ternary complex of RB69 pol (5,6), structures of a large number of B family pols have been determined, including the catalytic subunit of yeast pol δ (16,17,37−43). Among structures of all B family pols, ϕ29 pol and RB69 pol have been determined with the highest resolution (6,16). The structures of the apo form as well as binary and ternary complexes of ϕ29 pol have been determined. It is of particular interest that binding of the P/T to ϕ29 pol does not cause a major conformational change (16), so that residues involved in the open-to-closed conformational change can be identified by comparison between the apo form and the ternary complex as well as between the binary and ternary complexes.
Comparison of RB69 pol with ϕ29 pol (16) has shown that all important structural features observed in our 1.8 Å resolution structure are also found in the ϕ29 pol structure, including the open-to-closed conformational change of the Fingers domain upon binding of correct dNTPs, a kinked-to-straight helical transition in the Fingers domain (Figure 5G,H), a putative extended-to-eclipsed transition of the incoming dNTPs, three unusual nonpolar−polar interactions, and five buried water molecules (Figures 1−3). Nearly all of these structural features are also found in yeast pol δ; however, pol δ has an expanded recognition pattern for the N3 and O2 atoms that extends beyond the first three base pairs of the P/T junction at the minor groove (17). Moreover, comparison of RB69 pol with members of different pol families has shown that correct incoming dNTPs in the ternary complexes always adopt the same eclipsed conformation as the dCTP observed in our 1.8 Å structure. This includes the dNTPs bound in the ternary complexes of pol β, T7 pol, and Dpo4 (12,13,44), suggesting that it might have a universal role in base selectivity and catalysis.
Functional Assays for the Observed Hydration Network Mediating Enzyme−DNA Interactions
We have already shown elsewhere that the NBP residues are important for catalysis (18,30,31). However, we were previously unaware of any ordered water molecules mediating polymerase−DNA duplex interactions (6). To determine whether the observed five ordered water molecules buried between the enzyme and DNA duplex next to the P/T junction play an important role in recognition of W−C base pairs of DNA duplexes (other than just filling an empty space between the enzyme and the DNA duplex), we have conducted pre-steady-state and steady-state kinetic studies with non-W−C base pair DNA duplexes. In the steady-state assays, we observed virtually no primer extension for the first 15 s when a dT/dG mismatch was included at the N − 1 or N − 2 positions of DNA duplexes (Figure 7, lanes 3 and 4), whereas the extension beyond a mismatch at the N − 4 and N − 5 positions of the DNA duplex (Figure 7, lanes 6 and 7) was almost as efficient as the W−C base paired DNA duplex (Figure 7, lane 2). In a 2 min assay, we found an accumulation of extension of one nucleotide residue with DNA duplexes containing mismatches at the N − 1 and N − 2 positions (i.e., stalled extension after one nucleotide extension), but not with DNA duplexes containing mismatches at the N − 4 and N − 5 positions, i.e., fully extended as a W−C base pair DNA duplex (Figure 7). Consistent with steady-state kinetic results, we showed that the incorporation efficiency for correct dNMPs to the DNA duplexes containing mismatches at positions N − 4 and N − 5 was similar to that of the W−C base paired DNA duplex, including similar kpol, Kd,app, and kpol/Kd,app values (Table 2). However, when the mismatch was moved from position N − 3 to position N − 2 , and then to position N − 1, the incorporation efficiency was reduced exponentially, approximately 10-fold per nucleotide position (Table 2). When the mismatch was moved from the N − 3 to N − 4 positions (which are outside the network of five ordered water molecules), the incorporation efficiency for the primer extension increased by almost 100-fold to match that of the W−C base paired DNA duplex (Table 2). A mismatch would have to go through four successive steps of mismatch extension before it could be completely buried. During each step, water-mediated interactions slow the extension rates so that the ternary complex is effectively stalled and the mismatch has a high probability for being edited out by the exonuclease. All these results suggest that the five conserved water molecules mediating polymerase−DNA duplex interactions among B family polymerases are indeed important for primer extension because they are specifically involved in recognition of W−C base pairing geometry of DNA duplexes.
Figure 7.

Primer extension assays of mismatched DNA duplexes. Lanes 1 and 8 were no-extension control experiments. Lanes 2−7 were primer extension assays with all four dNTPs and wild-type RB69 pol (exo−) incubated for 15 s and lanes 9−14 for 2 min. Lane 1: P/T1 only, without enzyme or dNTPs. Lane 8: P/T1 and enzyme without dNTPs. Lanes 2−7 and 9−14: P/Ts and enzyme with four dNTPs, where the P/Ts for the six sequential lanes are P/T1, P/T2, P/T3, P/T4, P/T5, and P/T6, respectively. Sequences of P/T1−P/T6 are given in the footnote of Table 2.
Supporting Information Available
Six additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was in part funded by National Institutes of Health (NIH) Grant R01 GM057510-13 to T.A.S., NIH Grants R01 GM063276-9 and 3R01GM063276-08S1 to W.H.K., and the Steitz Center for Structural Biology, Gwangju Institute of Science and Technology, Republic of Korea, to T.A.S., J.W., and Professor S. H. Eom.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: pol, DNA polymerase; RB69 pol, bacteriophage RB69 DNA polymerase; NBP, nascent base pair-binding pocket; W−C, Watson−Crick; P/T, primer/template; Pu, purine; Py, pyrimidine; PEG350 MME, polyethylene glycol 350 monomethyl ether; PDB, Protein Data Bank.
Supplementary Material
References
- Echols H.; Goodman M. F. (1991) Fidelity mechanisms in DNA replication. Annu. Rev. Biochem. 60, 477–511. [DOI] [PubMed] [Google Scholar]
- Joyce C. M.; Steitz T. A. (1994) Function and structure relationship in DNA polymerases. Annu. Rev. Biochem. 63, 777–822. [DOI] [PubMed] [Google Scholar]
- Johnson K. A. (2010) The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta 1804, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite D. K.; Ito J. (1993) Compilation, alignment, and phlogenetic relationships of DNA polymerases. Nucleic Acids Res. 21, 787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Sattar A. K.; Wang C. C.; Karam J. D.; Konigsberg W. H.; Steitz T. A. (1997) Crystal structure of a pol α family replication DNA polymerase from bacteriophage RB69. Cell 89, 1087–1099. [DOI] [PubMed] [Google Scholar]
- Franklin M. C.; Wang J.; Steitz T. A. (2001) Structure of the replicating complex of a pol α family DNA polymerase. Cell 105, 657–667. [DOI] [PubMed] [Google Scholar]
- Tsai Y. C.; Johnson K. A. (2006) A new paradigm for DNA polymerase specificity. Biochemistry 45, 9675–9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennard O.; Hunter W. N. (1989) Oligonucleotide structure: A decade of results from single crystal X-ray diffraction studies. Q. Rev. Biophys. 22, 327–379. [DOI] [PubMed] [Google Scholar]
- Kennard O.; Salisbury S. A. (1993) Oligonucleotide X-ray structures in the study of conformation and interactions of nucleic acids. J. Biol. Chem. 268, 10701–10704. [PubMed] [Google Scholar]
- Eom S. H.; Wang J.; Steitz T. A. (1996) Structure of Taq polymerase with DNA at the polymerase active site. Nature 382, 278–281. [DOI] [PubMed] [Google Scholar]
- Johnson S. J.; Beese L. S. (2004) Structures of mismatch replication errors observed in a DNA polymerase. Cell 116, 803–816. [DOI] [PubMed] [Google Scholar]
- Pelletier H.; Sawaya M. R.; Kumar A.; Wilson S. H.; Kraut J. (1994) Structures of ternary complexes of rat DNA polymerase β, a DNA template-primer, and ddCTP. Science 264, 1891–1903. [PubMed] [Google Scholar]
- Doublie S.; Tabor S.; Long A. M.; Richardson C. C.; Ellenberger T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391, 251–258. [DOI] [PubMed] [Google Scholar]
- Huang H.; Chopra R.; Verdine G. L.; Harrison S. C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science 282, 1669–1675. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Prasad R.; Wilson S. H.; Kraut J.; Pelletier H. (1997) Crystal structures of human DNA polymerase β complexes with gapped and nicked DNA: Evidence for an induced fit mechanism. Biochemistry 36, 11205–11215. [DOI] [PubMed] [Google Scholar]
- Berman A. J.; Kamtekar S.; Goodman J. L.; Lazaro J. M.; deVega M.; Blanco L.; Salas M.; Steitz T. A. (2007) Structures of ϕ29 DNA polymerase complexed with substrate: The mechanism of tanslocation in B-family polymerases. EMBO J. 26, 3496–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan M. S.; Johnson R. E.; Prakash L.; Prakash S.; Aggarwal A. K. (2007) Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase δ. Nat. Struct. Mol. Biol. 16, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Rhee C.; Bebenek A.; Drake J. W.; Wang J.; Konigsberg W. (2006) The L561A substitution in the nascent base-pair binding pocket of RB69 DNA polymerase reduces base discrimination. Biochemistry 45, 2211–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology (Carter C. W., and Sweet R. M., Eds.) pp 307−326, Academic Press, San Diego. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstev R. W.; Adams P. D.; Win L. C.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 458–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Wang J.; Boisvert D. C. (2003) Structural basis for GroEL-assisted protein folding from the crystal structure of (GroEL-KMgATP)14 at 2.0 Å resolution. J. Mol. Biol. 327, 843–855. [DOI] [PubMed] [Google Scholar]
- Carson M. (1991) Ribbons 2.0. J. Appl. Crystallogr. 24, 958–961. [Google Scholar]
- Steitz T. A. (1990) Structural studies of protein-nucleic acid interaction: The sources of sequence-specific binding. Q. Rev. Biophys. 23, 205–280. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Schevitz R. W.; Zhang R. G.; Lawson C. L.; Joachimiak A.; Marmorstein R. Q.; Luisi B. F.; Sigler P. B. (1988) Crystal structure of trp repressor/operator complex at atomic resolution. Nature 335, 321–329. [DOI] [PubMed] [Google Scholar]
- Senes A.; Ubarretxena-Belandia I.; Engelman D. M. (2001) The Cα-H···O hydrogen bond: A determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. U.S.A. 98, 9056–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanswell P.; Thornton J. M.; Korda A. V.; Williams R. J. P. (1975) Quantitative determination of the conformation of ATP in aqueous solution using the lanthanide cations as nuclear-magnetic-resonance probes. Eur. J. Biochem. 57, 135–145. [DOI] [PubMed] [Google Scholar]
- Saenger W. (1984). Principles of nucleic acid structure, Springer-Verlag, New York. [Google Scholar]
- Zhang H.; Beckman J.; Wang J.; Konigsberg W. (2009) RB69 DNA polymerase mutants with expanded nascent base-pair-binding pockets are highly efficient but reduced base selectivity. Biochemistry 48, 6940–6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Franklin M.; Li. J.; Lin T. C.; Konigsberg W. (2002) Correlation of the kinetics of finger domain mutants in RB69 DNA polymerase with its structure. Biochemistry 41, 2526–2534. [DOI] [PubMed] [Google Scholar]
- Rothwell P. J.; Mitaksov V.; Waksman G. (2005) Motions of the fingers subdomain of klentaq1 are fast and not rate limiting: Implications for the molecular basis of fidelity in DNA polymerases. Mol. Cell 19, 345–355. [DOI] [PubMed] [Google Scholar]
- Hacker K. J.; Alberts B. M. (1994) The slow dissociation of the T4 DNA polymerase holoenzyme when stalled by nucleotide omission. An indication of a highly processive enzyme. J. Biol. Chem. 269, 24209–24220. [PubMed] [Google Scholar]
- Hacker K. J.; Alberts B. M. (1994) The rapid dissociation of the T4 DNA polymerase holoenzyme when stopped by a DNA hairping helix. A model for polymerase release following the termination of each Okazaki fragment. J. Biol. Chem. 269, 24221–24228. [PubMed] [Google Scholar]
- Carver T. E. Jr.; Sexton D. J.; Benkovic S. J. (1997) Dissociation of bacteriophage T4 DNA polymerase and its processivity clamp after completion of Okazaki fragment synthesis. Biochemistry 36, 14409–14417. [DOI] [PubMed] [Google Scholar]
- Yang J.; Nelson S. W.; Benkovic S. J. (2006) The control mechanism for lagging strand polymerase recycling during bacteriophage T4 DNA replication. Mol. Cell 21, 153–164. [DOI] [PubMed] [Google Scholar]
- Hopfner K. P.; Eichinger A.; Engh R. A.; Laue F.; Ankenbauer W.; Huber R.; Angerer B. (1999) Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc. Natl. Acad. Sci. U.S.A. 96, 3600–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Jeruzalmi D.; Moarefi I.; Leighton L.; Lasken R.; Kuriyan J. (1999) Crystal structure of an archaebacterial DNA polymerase. Structure 7, 1189–1199. [DOI] [PubMed] [Google Scholar]
- Rodriguez A. C.; Park H. K.; Mao C.; Beese L. (2000) Crystal structure of pol α family DNA polymerase from the hyperthermophilic archaeon thermococcus sp. 9°N-7. J. Mol. Biol. 299, 447–462. [DOI] [PubMed] [Google Scholar]
- Hashimoto H.; Nishioka M.; Fujiwara S.; Takagi M.; Imanaka T.; Inoue T.; Kai Y. (2001) Crystal structure of DNA polymerase fro hyperthermophilic archaeon Pyrococcus kodakaranesis KOD1. J. Mol. Biol. 306, 469–477. [DOI] [PubMed] [Google Scholar]
- Fogg M. J.; Pearl L. H.; Connolly B. A. (2002) Structural basis for uracil recognition by archaeal family B DNA polymerase. Nat. Struct. Biol. 9, 922–927. [DOI] [PubMed] [Google Scholar]
- Savino C.; Federici L.; Johnson K. A.; Vallone B.; Nastopoulos V.; Rossi M.; Pisani F. M.; Tsernoglou D. (2004) Insights into DNA replication: The crystal structure of DNA polymerase B1 from the archeaon Sulfolobus solfataricus. Structure 12, 2001–2008. [DOI] [PubMed] [Google Scholar]
- Firbank S. J.; Wardle J.; Heslop P.; Lewis R. J.; Connolly B. A. (2008) Uracil recognition in archaeal DNA polymerase captured by X-ray crystallography. J. Mol. Biol. 381, 529–539. [DOI] [PubMed] [Google Scholar]
- Ling H.; Boudsocq F.; Woodgate R.; Yang W. (2001) Crystal structure of a Y-family DNA polymerase in action: A mechanism for error-prone and lesion-bypass replication. Cell 107, 91–102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.