

Abstract
Composite tumors of the adrenal medulla, containing pheochromocytoma and ganglioneuroma, are rare. A 27-year-old male presented to us with dyspnea and was found to have labile hypertension. A left suprarenal mass was detected on computed tomography. The patient was operated under the cover of alpha anti-adrenergic drugs. The histopathological examination demonstrated that the tumor consisted of pheochromocytoma and ganglioneuroma elements, and hence, a diagnosis of composite adrenal medullary tumor (CAMT) was made. To the best of our knowledge, this is the first case of CAMT reported from India.
Keywords: Composite adrenal medullary tumor, ganglioneuroma, pheochromocytoma
INTRODUCTION
Composite adrenal medullary tumors (CAMTs) are rare. They are either composed of pheochromocytoma and ganglioneuroma or pheochromocytoma and ganglioneuroblastoma. This type of pheochromocytoma is called “composite” or “mixed” depending on whether pheochromocytoma or non-pheochromocytoma components are of the same or different embryonic origin. The non-pheochromocytoma components reported in the “composite” pheochromocytoma include ganglioneuroma,[1,2] ganglioneuroblastoma,[3,4] neuroblastoma, malignant schwannoma and neuroendocrine carcinoma.[5] Their association with adrenal cortical tumors, multiple neuroendocrine neoplasia (MEN-2A) syndrome and neurofibromatosis type 1 (NF-1) has also been described.[1–6] They usually present with signs and symptoms of excess catecholamine production in the form of palpitations, breathlessness and labile hypertension. However, sometimes they may be found incidentally at autopsy or may even masquerade as acute pancreatitis.[7] We report an unusual case of composite pheochromocytoma with ganglioneuroma with normal vanyl mandelic acid (VMA) levels in a young adult who presented with hypertension.
CASE REPORT
A 27-year-old male presented with history of breathlessness and sensation of tightness of chest for the last 3 months. On general physical examination, he was found to have labile hypertension with the systolic blood pressure ranging from 170 to 200 and diastolic values fluctuating from 100 to 130. The urinary VMA level was 5.4 mg/day (normal 1.5-7.5 mg/day). Computer tomography (CT) scan of the abdomen revealed a well-defined, lobulated, solid, homogenously enhancing mass in the left suprarenal region. Other organs including contralateral adrenal gland were normal in size and appearance. The left adrenal mass was excised surgically under the cover of two alpha anti-adrenergic drugs. Intraoperatively, the blood pressure was stable and the postoperative period was uneventful. On gross examination, the capsulated left adrenal gland specimen measured 7.5Χ6.5Χ3.5 cm and weighed 455 g. The cut surface showed a firm, tan-colored tumor with cyst-like areas. Normal adrenal gland tissue was not evident on the gross examination. Microscopically, the normal adrenal cortex was seen to be peripherally compressed by a well-circumscribed tumor comprising pheochromocytoma and ganglioneuroma [Figure 1]. The ganglioneuromatous component predominated, accounting for over 80-90% of the total mass. It consisted of bundles of spindle cells (Schwann cells) admixed with mature ganglion cells [Figure 2]. The pheochromocytoma component (accounting for 10% of the mass) consisted of large polygonal cells arranged in nests and cords (Zellballen pattern), bordered by spindle-shaped sustentacular cells with delicate fibrovascular stroma [Figure 3]. No mitoses or confluent necrosis were found. Amyloid and intracytoplasmic hyaline inclusions were not seen. Neuroblastic component was avidly searched for but was found to be absent.
Figure 1.
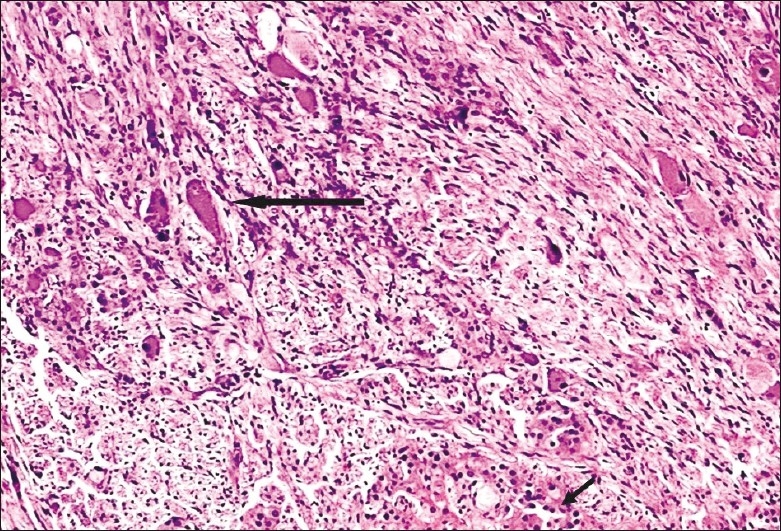
CAMT: Areas of ganglioneuroma (thick arrow) are seen admixed with areas of pheochromocytoma (thin arrow) (original magnification ×20)
Figure 2.
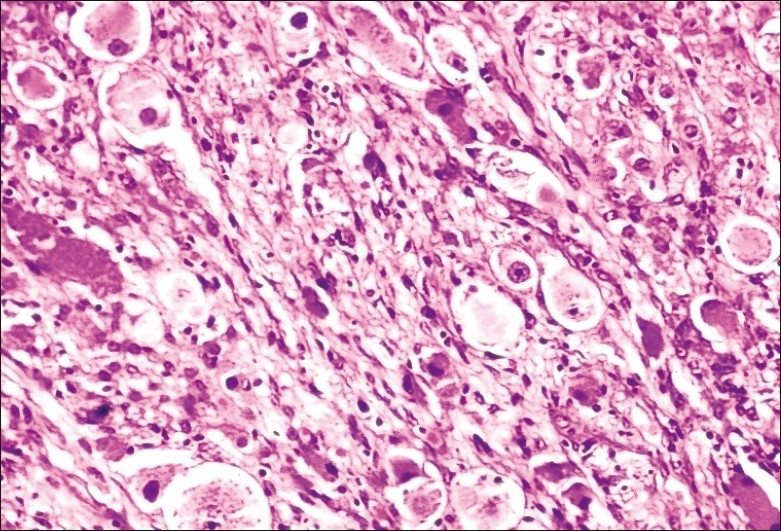
Ganglioneuroma component of CAMT with many ganglion cells intricately associated with spindle schwannian stroma (original magnification ×40)
Figure 3.
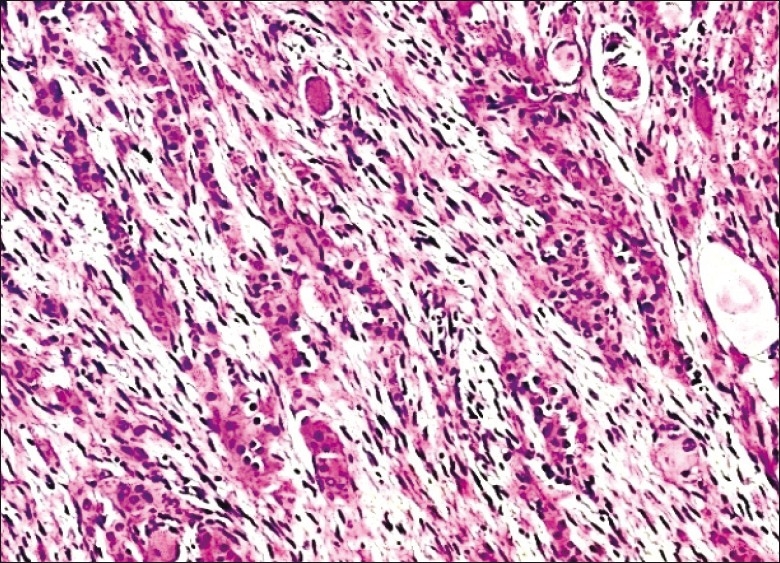
Pheochromocytoma component of CAMT with polygonal cells arranged in cords and trabeculae within loose Schwannian matrix (original magnification ×20)
Pheochromocytoma cells were highlighted by chromogranin [Figure 4], synaptophysin and neuron specific enolase (NSE). The sustentacular cells and Schwann cells stained with S-100 protein. The ganglion cells were decorated by synaptophysin.
Figure 4.
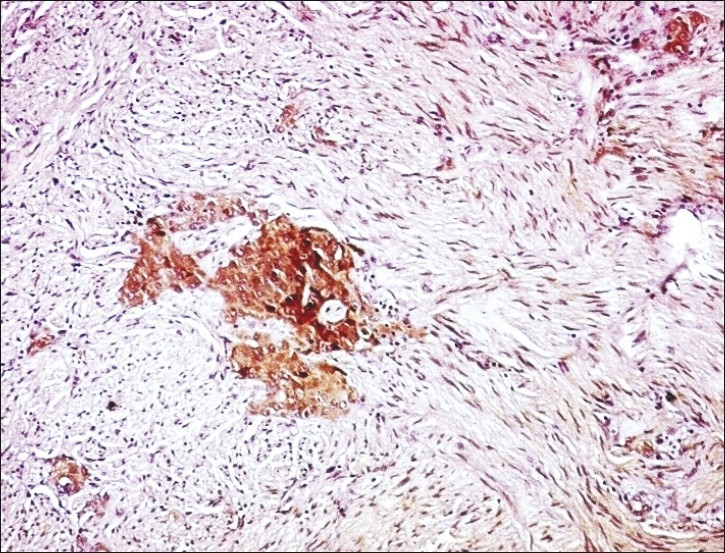
Immunostaining with chromogranin highlights the pheochromocytoma element of CAMT (original magnification ×10
DISCUSSION
The occurrence of pheochromocytoma and other tumor types in a single adrenal gland has been occasionally reported. The designation “composite” pheochromocytoma has been used to describe tumors in which the components theoretically arise from a common embryologic progenitor. Rare cases of adrenal tumors in which two different histological elements, with no common embryologic ancestry, occur are designated as mixed adrenal tumors. The components found in the “mixed” pheochromocytoma include adrenal cortical neoplasms[1] and spindle cell sarcoma.[5]
CAMTs composed of pheochromocytoma and ganglioneuroma or ganglioneuroblastoma are rare.[1–4] These neoplasms reflect phenotypic plasticity shown by primitive sympathetic cells and mature chromaffin cells in vitro.[8] The incidence of these tumors among sympathoadrenal pheochromocytoma, in reported case studies, ranges from 3 to 8.7%.[9,10] Most of the cases have been reported to have occurred in individuals above 30 years of age. Majority of these were found after fifth decade. Males and females are equally affected.[9,10] Our case is one of the youngest patients documented with CAMT. It is the first case from India to the best of our knowledge. These tumors present with signs and symptoms attributable to catecholamine and/or neuropeptide production. Most of the cases in literature were clinically suspected in view of increased catecholamine secretion and, rarely, vasoactive intestinal polypeptide secretion. In the present case, the patient's urinary VMA levels were normal, which is in contrast with most of the cases reported in literature. The normal VMA level could be due to pheochromocytoma being only a minor component in this composite tumor. In a study by Aiba et al.,[1] it was postulated that the ganglioneuromatous component may play a significant role in modifying the effects of hormone secreted by the pheochromocytoma component, by metabolizing the catecholamines.[1] This effect may also have contributed to the lowered VMA level in our patient.
The association of CAMTs with NF-1 (von Recklinghausen's disease), MEN-2A and adrenal cortical tumors including bilateral involvement of adrenals has been documented.[1–5] Recently, a case of composite pheochromocytoma associated with MEN-2B syndrome has also been reported.[11] No features of either NF-1 or MEN-2A or adrenocortical tumor were noted in our case.
The differential diagnosis in such a situation would include other adrenal tumors like adrenocortical adenoma or hyperplasia, adrenocortical carcinoma and a pheochromocytoma. Hence, a thorough pathological examination is essential to delineate the components of this tumor. Microscopically, the predominant component was a ganglioneuroma and the pheochromocytoma component was found only on extensive and meticulous search. The chromaffin cells of pheochromocytoma were more readily highlighted with chromogranin staining. In a study of 46 cases of pheochromocytoma, Lam et al.[10] found four cases of composite tumors with ganglioneuromatous component. These cases had heterogenous radiologic, gross and microscopic features with varying admixtures of ganglioneuroma and pheochromocytoma components, although mostly the ganglioneuroma component predominated.
CAMTs are considered as benign in nature even in the presence of a neuroblastic component. However, one of the case studies has reported a single case of pheochromocytoma-ganglioneuroma with distant metastasis.[10] Our patient was disease and symptom free at 6 months, and his hypertension was controlled without any medication.
The current case reinstates the need of considering CAMT as a rare but important cause of hypertension in young individuals. It also highlights the fact that in hypertensives with adrenal CAMT, catecholamines may be within normal limits and that this depends on the complex biochemical interaction and proportion of pheochromocytoma and ganglioneuromatous element. Careful examination of the resected adrenal tumor specimen with adequate sectioning and use of immunohistochemistry to establish the components of a composite tumor is stressed. It is important to investigate young hypertensives thoroughly to rule out a CAMT as a curative cause of secondary hypertension.
Footnotes
Source of Support: Nil
Conflict of Interest: None.
REFERENCES
- 1.Aiba M, Hirayama A, Ito Y, Fujimoto Y, Nakagami Y, Demura H, et al. Compound adrenal medullary tumor (pheochromocytoma and ganglioneuroma) and a cortical adenoma in the ipsilateral adrenal gland.A case report with enzyme histochemical and immunohistochemical studies. Am J Surg Pathol. 1988;12:559–66. doi: 10.1097/00000478-198807000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Brady S, Lechan RM, Schwaitzberg SD, Dayal Y, Ziar J, Tischler AS. Composite pheochromocytoma/ganglioneuroma of the adrenal gland associated with multiple endocrine neoplasia 2A: Case report with immunohistochemical analysis. Am J Surg Pathol. 1997;21:102–8. doi: 10.1097/00000478-199701000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Matias-Guiu X, Garrastazu MT. Composite phaeochromocytoma ganglioneuroblastoma in a patient with multiple endocrine neoplasia type IIA. Histopathology. 1998;32:281–2. doi: 10.1046/j.1365-2559.1998.0372g.x. [DOI] [PubMed] [Google Scholar]
- 4.Nakagawara A, Ikeda K, Tsuneyoshi M, Daimaru Y, Enjoji M. Malignant pheochromocytoma with ganglioneuroblastoma elements in a patient with von Recklinghausen's disease. Cancer. 1985;55:2794–8. doi: 10.1002/1097-0142(19850615)55:12<2794::aid-cncr2820551213>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 5.Juarez D, Brown RW, Ostrowski M, Reardon MJ, Lechago J, Truong LD. Pheochromocytoma associated with neuroendocrine carcinoma.A new type of composite pheochromocytoma. Arch Pathol Lab Med. 1999;123:1274–9. doi: 10.5858/1999-123-1274-PAWNC. [DOI] [PubMed] [Google Scholar]
- 6.Mezitis SG, Geller M, Bocchieri E, Del Pizzo J, Merlin S. Association of pheochromocytoma and ganglioneuroma: Unusual finding in neurofibromatosis type 1. Endocr Pract. 2007;13:647–51. doi: 10.4158/EP.13.6.647. [DOI] [PubMed] [Google Scholar]
- 7.Choi EK, Kim WH, Park KY. A case of a composite adrenal medullary tumor of pheochromocytoma and ganglioneuroma masquerading as acute pancreatitis. Korean J Intern Med. 2006;21:141–5. doi: 10.3904/kjim.2006.21.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tischler AS. Divergent differentiation in neuroendocrine tumors of adrenal gland. Semin Diagn Pathol. 2000;17:120–6. [PubMed] [Google Scholar]
- 9.Linnoila RI, Keiser HR, Steinberg SM, Lack EE. Histopathology of benign versus malignant sympathoadrenal paragangliomas: Clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol. 1990;21:1168–80. doi: 10.1016/0046-8177(90)90155-x. [DOI] [PubMed] [Google Scholar]
- 10.Lam KY, Lo CY. Composite pheochromocytoma-ganglioneuroma of the adrenal gland: An uncommon entity with distinctive clinicopathologic features. Endocr Pathol. 1999;10:343–52. doi: 10.1007/BF02739777. [DOI] [PubMed] [Google Scholar]
- 11.Charfi S, Ayadi L, Ellouze S, Ghorbel R, Khabir A, Gouiaa N, et al. Composite pheochromocytoma associated with multiple endocrine neoplasia type 2B. Ann Pathol. 2008;28:225–8. doi: 10.1016/j.annpat.2008.06.003. [DOI] [PubMed] [Google Scholar]