

Abstract
Gene expression in the eukaryotic cell is regulated at a number of levels including transcription of genomic DNA into mRNA, nucleocytoplamic export of mRNA, and translation of the exported mRNA into proteins in the cytoplasm by ribosomes. The role played by epigenetics and transcription factors associated with the control of gene expression in the developing neutrophil has been well documented and appreciated over the years. A wealth of information on the role played by transcription factors in myeloid biology has contributed to our understanding of both normal and abnormal neutrophil development. However, the regulation of mRNA translation in myeloid cell maturation is much less well-studied. A better understanding of the translational control of myeloid gene expression may provide important insights into both normal and abnormal myeloid maturation. . This review summarizes our current understanding of the regulation of myeloid gene expression at the mRNA translational level.
Overview
Translation of eukaryotic mRNAs into proteins is a highly regulated and complex process (rev in[1]). Following transcription, mRNA is spliced, capped at the 5′ end, and polyadenylated at the 3′end. It then undergoes export from the nucleus to the cytoplasm to allow the initiation of translation to begin. Translation can be divided into three major sequential steps: initiation, elongation and termination. Since translation initiation has been shown to be a critical rate limiting step in mediating cellular protein synthesis, the current review will focus on that phase of translation. Translation initiation in eukaryotes involves approximately ten initiation factors designated eIFs (eukaryotic initiation factors). Translation initiation begins when the factors eIF-1, eIF-1A, and eIF-3 bind to the 40S ribosomal subunit. EIF-2 (in a complex with GTP) associates with the initiator methionyl tRNA. The eIF-4 family of factors then brings the mRNA to be translated into contact with the 40S ribosomal subunit. The 40S ribosomal subunit, now loaded with the bound methionyl initiator tRNA and eIFs, scans the mRNA to locate the AUG initiation codon. When the AUG codon is reached, eIF-5 triggers the hydrolysis of GTP bound to eIF-2. All the initiation factors are then released, and a 60S subunit binds to the 40S subunit to form the 80S initiation complex; elongation and termination then proceed, resulting in the synthesis of a new polypeptide chain. Little is known about the specific roles played by many of these factors during myeloid maturation. However, it is known that the delicate balance that controls translation initiation is often lost in malignancy. For example, overexpression of the eIF4-E factor resulting in increased proliferation has been observed in the bone marrow of patients in the blast crisis phase of chronic myelogenous leukemia and also in patients with acute myelogenous leukemia. Thus, the critical role of translation initiation in regulating protein synthesis and cell proliferation makes translation initiation factors potential therapeutic targets for the treatment of myeloid malignancies.
Translational initiation control of protein synthesis: general principles
The recruitment of the 40S ribosomal subunit to the 5′ end of mRNA is a crucial, complex and rate-limiting step during 5′cap-dependent translation. Cap-binding protein eukaryotic initiation factor 4E (eIF4E) recognizes and binds to the m7GpppN cap (where m is a methyl group and N any nucleotide) structure at the 5′end of the mRNA. Under basal conditions, eIF4E remains bound to 4E-binding proteins (4E-BPs), blocking translation. However, phosphorylation of the 4E-BPs by means of signal transduction pathways regulated by growth factors and nutrient status, releases the 4E-BPs from eIF4E (Figure 1A). This in turn allows the competing eIF4G scaffold protein to bind to eIF4E. eIF4G then recruits the ATP-dependent RNA helicase eIF4A (eIF4E, 4G and 4A are collectively referred to as eIF4F in the literature), the ubiquitously expressed cofactor eIF4B as well as eIf3, a multisubunit initiation factor, which binds to the 40S ribosomal subunit (rev in[2]) (Figure 1B). Thus, through its ability to bind mRNA in a sequence non-specific manner and its interaction with eIF3, eIF4G brings together the mRNA and the 40S ribosomal subunit. The 40S ribosomal subunit in turn must be loaded with the initiator methionine-tRNA (tRNAiMet). This process is facilitated by yet another factor, eIF2. EIF2 binds both the tRNAiMet as well as GTP giving rise to what is commonly referred to as the “ternary complex” (TC, in Figure 2). Once the ternary complex associates with the 40S ribosomal subunit, eIF3 and eIF1A, it is referred to as the 43S pre-initiation complex (see Figure 1B).
Figure 1. Eukaryotic Cap-dependent translation initiation.
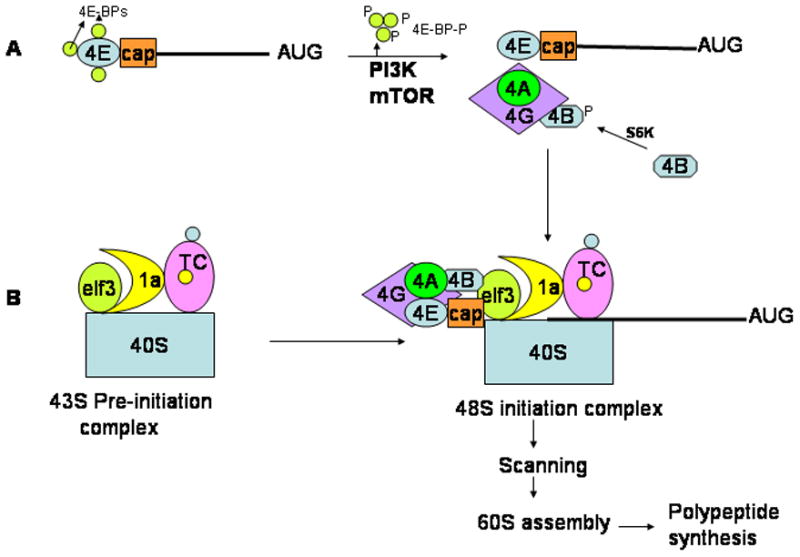
A. Under basal conditions, the initiation factor eIF4E (4E) is sequestered at the 5′Cap site of the mRNA by the 4E-BPs. Following phosphorylation by PI3K or mTOR, the 4E-BPs dissociate from eIF4E allowing it to be incorporated into the eIF4F (includes eIF4A, eIf4G and eIf4B) complex. B. The 40S ribosomal subunit bound to eIF3 associates with the ternary complex (TC) via eIF1A to give rise to the 43S pre-initiation complex. This complex is recruited to the mRNA via the eIF4G complex. The so called 48S initiation complex now scans the mRNA in an ATP-dependent manner and upon identification of the initiator AUG codon, recruits 60S ribosomal subunit whereupon polypeptide synthesis ensues.
Figure 2. Formation and regulation of the ternary complex in eukaryotic translation.

The ternary complex is composed of eIf2, GTP and the initiator methionyl tRNA (iMet). The activity of this complex is modulated by the GTP-exchange factor eIF2B. Stress, amino acid deficiency and heme deficiency result in the activation of the eIF2 kinases which phosphorylate the α-subunit of eIF2. Phosphorylated eIF2a binds eIF2B with high affinity thereby preventing GTP/GDP exchange. Since levels of eIF2B are limiting in the cell, the net result is a reduction in translation initiation via lowered levels of the ternary complex.
The pre-initiation complex then associates with mRNA by way of eIF3 and eIF4G and the resulting complex is termed the 48S initiation complex. Following hydrolysis of the GTP bound to eIF2 to GDP, eIF2 is released and reloaded with another GTP molecule with the aid of the GTP exchange factor eIF2B for another round of initiation (Figure 2). The 40S subunit now scans the mRNA in a 5′ to 3′ direction in search of the first in-frame translation initiation AUG codon. Once this is encountered and a codon-anticodon interaction established, the initiation factors dissociate from the 40S ribosomal subunit allowing for the binding of the large ribosomal 60S subunit and polypeptide synthesis ensues (Figure 1B). In addition to eIF4F, other factors are also involved in the stabilization of the ribosome-mRNA interactions. These include the polyA-binding protein (PABP), which binds at the 3′ end of mRNA and promotes mRNA-ribosome stabilization through its loop-back interaction with eIF4G. In addition, eIF4B and eIF4H are RNA chaperones that assist in mRNA secondary structure unwinding activity of eIF4A [3]
Regulation of Translation initiation
Translation control occurs largely by the regulation of the activity and integrity of the cap-dependent translation initiation complex (rev in [1]). In general, translation initiation is regulated by two major mechanisms. The first, illustrated in Figure 1A, involves a group of proteins termed 4E-BPs (eIF4E binding proteins) which compete with eIF4G to bind to the 5′ CAP associated eIF4E protein. However, the 4E-BPs are phosphorylated upon activation of the phosphoinositol-3-kinase (PI3K) pathway and its downstream target mTOR (mechanistic or mammalian target of rapamycin)[4]. The second mechanism involved in the control of translation initiation is mediated by the phosphorylation of the eIF2α subunit on serine 51 by the four known eIF2α kinases, resulting in global translational arrest. Both these mechanisms are detailed below.
The mTOR pathway
The mTOR pathway is an evolutionarily conserved pathway that is critical for cellular responses to environmental cues. mTOR (mechanistic target of rapamycin) is a serine/threonine protein kinase belonging to the PIKK family of protein kinases (rev in[5]). Mammalian TOR is a functional component of two distinct multiprotein complexes, mTORC1: in which mTOR is complexed with raptor (regulatory protein of mTOR) and LST8 (also called GβL), and mTORC2, harboring both LST8 and rictor. mTORC1 has been shown to be responsive to the inhibitory effects of the antibiotic rapamycin, while mTORC2 is not[6]. MTORC1 functions as a multimeric protein complex that regulates protein synthetic pathways responsive to nutritional, environmental and growth factor mediated signals. The phosphotransferase activity of mTORC1 is modulated by the activation of RHEB (Ras homolog enriched in brain; a GTP-GDP exchange protein) which in turn is regulated by a heterodimeric tumor suppressor containing the proteins tuberous sclerosis 1 (TSC1) and TCS2. The latter is a GTPase activating protein (GAP) which can modulate the activity of RHEB by converting it to its GDP-bound inactive form[7] The two major targets of mTOR are the 4E-BPs and the 40S ribosomal protein S6 kinases (S6K), both important components of the translational machinery. Upon activation, mTOR regulates the phosphorylation/activation of p70 S6 kinase (S6K) and the phosphorylation/deactivation of 4E-BP1[8]. Activation of S6 kinase is thought to modulate ribosome biogenesis through the activation of ribosomal protein S6 (rpS6)[9] (Figure 3). In addition, S6K also phosphorylates eIF2B, SKAR (S6K1 Aly/REF-like target) and eukaryotic elongation factor 2 kinase, thus affecting both the initiation and elongation stages of mRNA translation.
Figure 3. The mTOR signaling network.
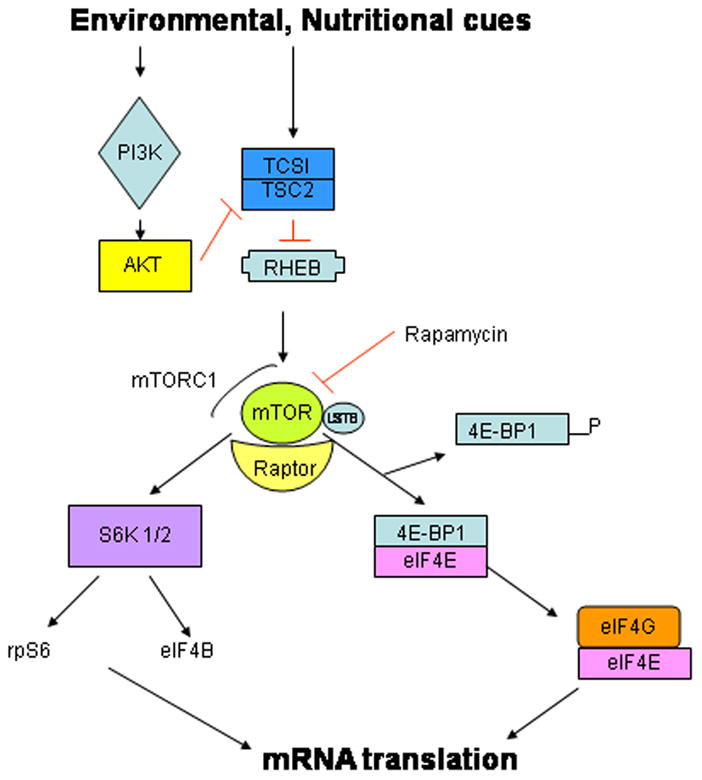
The mammalian target of rapamycin C1 (mTORC1) is activated by the GTP-bound form of RHEB, which in turn is regulated by the tuberous sclerosis1 and 2 tumor suppressor complex. The mTORC1 kinase is a master regulator of protein synthesis as it responds to both environmental and nutritional cues by directly phosphorylating the 4E-binding proteins (4E-BPs) thereby releasing the eIF4E to participate in cap-dependent translation (see Figure 1). Additionally, mTORC1 also promotes the activation of S6kinase which in turn phosphorylates a number of translation initiation factors (eIF4B) and ribosomal protein S6. See text for abbreviations.
Hypophosphorylation of the 4E-BPs is thought to increase their affinity for eIF4E, thus blocking binding of the eIF4G and eIF4A and hampering translation initiation of mRNA from proceeding (Figure 1A). However, phosphorylation of the 4E-BPs by the PI3K-mTOR signaling pathway (Figure 3) results in lowered affinity of these proteins for eIF4E, thereby allowing for the formation of the competing eIF4E-eIF4G-eIF4A (eIF4F)- mRNA complex that permits mRNA translation to proceed[4]. Thus inhibition of mTOR activity results in the down regulation of the activity of several components of the translational machinery resulting in a block in cell proliferation and eventually to cell death.
A recent study in which the 4E-BP1 and 4E-BP2 genes were ablated in mice surprisingly demonstrated an impairment of myelopoiesis with no apparent effect on thymocyte maturation[10]. These mice exhibited an increase in the number of immature granulocytic precursors and a concomitant decrease in the numbers of mature granulocytes compared to their wild type littermates. It has been previously shown in in vitro cell culture studies that expression of the 4E-BPs is markedly increased during granulopoiesis[11]. Based on these observations, it was concluded that 4E-BP1 and 4E-BP2 play a pivotal role in the early phases of granulo-monocytic differentiation thus underscoring the role of translation initiation during granulopoiesis. In this context, 4E-BP1 has been shown to be constitutively phosphorylated in both chronic myeloid leukemia (CML) and in acute myeloid leukemia (AML) as a result of constitutive activation of mTOR and Bcr-Abl in CML[12] and PI3K-Akt in AML[13].
Acute Myeloid Leukemia and deregulation of translation
Upregulation of eIF4E is sufficient to drive protein translation and to transform cells. Previous work has demonstrated an overexpression of eIF4E in bone marrow mononuclear cells from patients with AML. Upregulation of a subclass of proto-oncogenes that have been referred to as eIF4E-sensitive including c-myc, cyclin D1 and Bcl-xl [14], is thought to contribute to the underlying pathology of AML.
The mTORC1 axis plays an important role in controlling factors associated with cellular processes that have commonly gone awry in human cancers including acute myeloid leukemia (rev in[15]). Additionally both upstream (eg AKT) and downstream (eg eIF4E) mediators within the mTORC1 pathway have been shown to be mutated in hematopoietic and other malignancies. Constitutive activation of mTORC1 has been reported in all cases of AML while only 50% of AML patients have mutation in the PI3K/AKT pathway. The mechanism underlying the activation of mTORC1 is thus not fully understood[16]. This pathway is therefore viewed as an important potential target for therapeutic strategies for leukemia, lymphoma and multiple myeloma. In this context, the use of rapamycin (mTORC1 inhibitor, see Fig 3), its analogs (rapalogs) and second generation derivatives are at different stages of pre-clinical investigation or are in clinical trials(rev in [15]). In a recent study, Tambirini et al showed that rapamycin did not consistently inhibit protein translation in AML cells. The authors did show however, that the 4E-BP1 memetic 4EGI-1 did inhibit translation of the eIF4E-sensitive oncogenes including c-myc and cyclin D1 and induced cell death of AML cells in culture. This study demonstrated that protein translation in AML cells is regulated by an mTORC1-independent, 4E-BP1/eIF4E-dependent pathway[17]. A better understanding of these pathways is clearly warranted and will likely provide a more objective roadmap to the rational development of improved therapies for AML.
EIf2α phosphorylation pathway
Regulation of tRNA i met binding step during mRNA translation, a second rate limiting step in translation initiation, occurs independently of the mTOR pathway (Figure 2). Phosphorylation of the alpha subunit of eIF2 on Serine 51 by stress-induced enzymatic activity of four known kinases: heme-regulated inhibitor (HRI), Protein kinase R (PKR), PKR-like endoplasmic reticulum-associated eIF2a kinase (PERK) and mammalian general control non-derepressing 2 (mGCN2), transforms eIF2 from a substrate for the guanine exchange factor eIF2B to a competitive inhibitor[18]. This is because phosphorylated eIF2α has a higher affinity for eIF2B than upphosphorylated eIF2α. This results in the sequestering of eIF2B thus preventing it from functioning as a guanine nucleotide exchange factor (Figure 2). The resultant reduction in levels of the ternary complex causes global inhibition of translation. Limiting levels of eIF2B in the cell make this a critical regulatory factor in contributing to global translational regulation within the cell[19]
Role of uORFs in regulating myeloid transcription factors
Approximately 10% of vertebrate mRNAs possess specialized cis-regulatory elements that make them specifically responsive to translational control. This feature allows for a subgroup of mRNAs to be translated in the wake of global translational arrest in response to environmental stress. mRNAs endowed with these cis-elements which include upstream initiation codons, upstream open-reading frames (uORFs) and internal ribosome entry sites (IRESs), include key regulatory proteins, cytokines, growth factors and components of the cell cycle, the expression of which are necessary to return the cell to homeostasis. Examples of such mRNAs include cyclin D1[20], thrombopoietin (TPO)[21], BCL-2 [22] the myeloid master transcriptional regulator C/EBPα and its family member C/EBPβ, [23] among others.
Translational control of C/EBPα expression
C/EBPα is the founding member of a family of basic region/leucine zipper (bzip) transcription factors and has been shown to be a master regulator of granulopoiesis. (Rev in[24–26][27]). It is expressed at high levels throughout myeloid differentiation and has been shown to bind to the promoters of multiple myeloid-specific genes at different stages of myeloid maturation [24,25].[26]. Profound hematopoietic abnormalities have been reported for mice nullizygous for C/EBPα[28]. Although C/EBPα−/− mice die perinatally due to defects in gluconeogenesis which result in fatal hypoglycemia [29], they also have a selective early block in the differentiation of granulocytes. The C/EBPα intronless and generates two isoforms as a result the differential utilization of alternate translation start codons. The resultant p42kD (full length) and p30kD (truncated) C/EBPα proteins differ from each other at the N-terminus, which is abbreviated in the p30kD protein (see Figure 4). Translational control of C/EBPα-isoform expression is orchestrated by a conserved cis-regulatory uORF in the 5′UTR (untranslated region) that is out of frame with the coding region of C/EBPα and is thought to be responsive to the activities of both eIF4E and eIF2 (Figure 4). Thus, an increase in the activity of eIF2 or eIF4E, as may be expected during neoplastic cell proliferation, results in the increase in expression of the shorter p30 isoform (rev in[23]). As is indicated in Figure 4, a small upstream open reading frame (uORF) monitors the site of translation initiation by sensing the activity of the translation initiation factors eIF2 and eIF4E. When levels of these factors are high, the out-of-frame uORF is translated, but termination of its translation very close to the AUG for p42 is thought to prevent reinitiation at AUG1. Instead, ribosomes continue to scan and reinitiate at AUG2, resulting in the expression of C/EBPα p30. In contrast, under basal conditions, when levels of the initiation factors are relatively low, most ribosomes do not initiate translation at the uORF but instead initiate translation at AUG1 by a process involving “leaky ribosome scanning”, resulting in translation of the full length C/EBPα p42 isoform[23]. This mechanism of translational control appears to be conserved among key regulatory proteins which govern differentiation and proliferation. It has been hypothesized that the expression of mRNAs encoding these key regulatory proteins that determine cell fate are translated only at permissive levels of the translation initiation factors eIF2 and eIF4E, which in turn are responsive to environmental and other cues. Under these conditions, via an uORF- mechanism, the ratios of the different isoforms of these key regulatory proteins is adjusted to allow for either proliferation or differentiation[23].
Figure 4. Schematic of translational control of the C/EBPα mRNA.
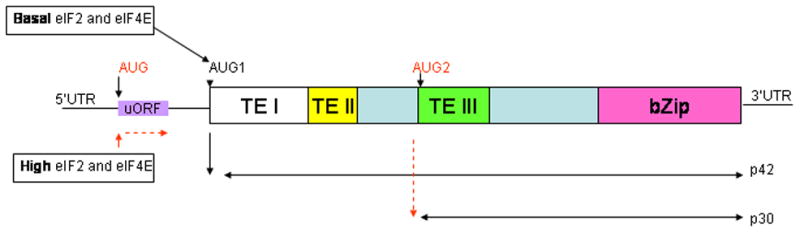
The upstream open reading frame (uORF) directs translation of the two isoforms of C/EBPα p42 (wildtype) and p30 (truncated) depending on the availability of translation initiation factors eIF2 and eIf4E. High levels of these factors result in the ribosomes translating the uORF followed by reinitiation at the downstream AUG (AUG2) giving rise to the p30 isoform. Under basal conditions, on the other hand, ribosomes skip translation initiation at the uORF, initiating translation instead at the first AUG (AUG1) resulting in the formation of the p42 isoform.
TEI, II ανδ III αρε τρανσχατιϖατιoν δoμαινσ ιν τηε X/EBΠα protein, bZip is the DNA binding and dimerization basic Leucine zipper domain.
It has been demonstrated that the p30 C/EBPα protein not only interferes with the DNA binding ability of p42 C/EBPα, thus inhibiting transactivation of key granulocytic target genes in a dominant-negative manner[30], but also binds to the promoters of a distinctive set of target genes to alter their transcription[31] Additionally, modification of the mouse locus to express only the p30 isoform led to the formation of granulocyte-macrophage progenitors. However, deficiency of p42 in these mice led to the development of AML with complete penetrance[32] Thus changes in the ratio of p42:p30 isoforms of C/EBPα play a critical role in contributing to AML[33].
Suppression of C/EBPα translation has also been shown to occur in blasts from patients with CML in blast crisis. This is brought about via an RNA binding protein, hnRNP-E2, which binds to the uORF of the C/EBPα mRNA, thereby blocking translation. Expression of hnRNP-E2 is thought to be upregulated by the activity of the oncogenic BCR-ABL fusion protein in CML patients, and downregulation of hnRNP-E2 by the BCR-ABL inhibitor imatinib results in restoration of C/EBPα protein expression and granulocytic differentiation of the CML blasts[34].
Role of microRNAs in controlling translation in myelopoiesis
Examples of regulatory elements within mRNAs that alter translation include microRNA binding elements usually located at the 3′UTR of the mRNA. MicroRNAs (miRNAs) are 18-24 nucleotides long non-coding RNAs that regulate eukaryotic gene expression. miRNAs are encoded in the genome and are initially transcribed by RNA polymerase II as long primary transcripts referred to as primary miRNAs (pri-miRNAs). These transcripts are recognized and processed by a ribonuclease called Drosha into 60–80 nucleotide intermediates called precursor miRNAs (pre-miRNAs) which are then exported to the cytoplasm where a second ribonuclease termed Dicer cleaves pre-miRNAs to generate double stranded 18-24 nucleotide long miRNAs. The miRNAs are then incorporated into the RNA-induced silencing complex or RISC, a large protein complex that also contains the Argonaute or mRNA cleaving proteins. The miRNA guides the RISC complex to target complementary regions in the 3′UTRs of mRNAs, leading to repression of translation or destabilization of the mRNA by deadenylation. (rev in[35]).
Increasing evidence has implicated miRNAs as components of oncogene and tumor suppressor pathways. Alterations in miRNA expression or structure have been documented in a variety of malignancies,[36] and several miRNA families have now been functionally implicated as having tumor-suppressive and oncogenic potential.[37,38].
An increasing body of evidence implicates miRNA activity in mediating both normal and abmormal myelopoiesis (rev in [39]). MiRNAs have in particular been shown to activate or be activated by myeloid specific transcription factors such as C/EBPα and Gfi-1. For example, mir-223 is thought to be a direct target of C/EBPα and its expression increases during granulopoiesis. Ablating mir-223 in mice results in the expansion of granulocyte precursor cells resulting from a cell autonomous increase in the number of granulocytic progenitors[40]. Additionally, overexpression of mir-223 in acute promyelocytic leukemia (APL) cells results in an enhanced capacity for granulocytic differentiation[41]. Mir-223 is thus thought to be a positive regulator of granulopoietic differentiation. More recently it has been shown that mir-223 targets E2F1, a master cell cycle regulator, by inhibiting translation of its mRNA. Thus, granulopoiesis appears to be regulated by a C/EBPα–miR-223–E2F1 axis, wherein miR-223 functions as a key regulator of myeloid cell proliferation associated with E2F1 in a mutual negative feedback loop[42]. In a paradigm shifting study, Eiring et al demonstrated a new role for miRNAs. They demonstrated that mir-328 is down regulated in CML patients in blast crisis. Restoration of mir-328 expression however restores differentiation by simultaneous interaction with the C/EBPα translational regulator hnRNP-E2 (see above) as well as with the mRNA for PIM1, a survival factor. Interestingly, the interaction with hnRNP-E2 occurs independently of the microRNAs seed sequence leading to the release of C/EBPα mRNA from hnRNA-E2-mediated translational inhibition. Thus mir-328 appears to control cell fate by its ability to base pair with the 3′UTR of target mRNAs (PIM1) as well as by acting as a decoy for hnRNP binding thus interfering with cell fate by releasing C/EBPα from translational inhibition[43].
The role of mir-27 in granulopoiesis has also been documented. This miR has been shown to target the myeloid transcription factor AML1, whose expression decreases during granulocytic differentiation in a mir-27 dependent manner. Anti-mir-27 treatment of immature myeloid progenitors resulted in an increase in the expression of AML1 and impaired granulocytic differentiation[44]
Numerous studies have analyzed the expression of miRNAs in acute myeloid leukemias and the resulting miR signatures generated have proved to be helpful in classifying sub-types of AML and hence the choice of treatment options to be used, as well as in determining the efficacy of targeted therapies against AML. These studies have been detailed elsewhere[39].
The GAIT system: regulation of inflammation by translation repression in myeloid cells
Neutrophils, monocytes and macrophages provide the first line of defense against invading organisms or following cellular injury. When activated, these cells secrete antimicrobial agents and cytokines which promote the elimination of the invading organisms. However, to prevent overexpression of these toxic agents once the cellular insult has abated, which would otherwise result in a chronic state of inflammation, the cells have developed mechanisms both at the transcriptional and translational levels to turn off cytokine production [45].
Recent studies have demonstrated that the formation of the so called GAIT (gamma interferon inhibitor of translation) complex at the 3′UTR (untranslated region) of target genes involved in the inflammatory response occurs in myeloid cells (Rev in[46]). In response to IFN-γ signaling, the tetrameric GAIT complex composed of glutamyl propyl tRNA synthetase (EPRS), NS1-associated protein (NSAP1), ribosomal protein L13a and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) binds defined 3′UTR cis elements within a family of inflammatory mRNAs and suppresses their translation. 3′UTR GAIT elements have been identified in ceruloplasmin (Cp, nt 78–106), vascular endothelial growth factor-A (VEGF-A, nt 358–386), death-associated protein kinase (DAPK, nt 1141–1169), zipper-interacting protein kinase (ZIPK, nt 174–206) and chemokine C-C motif ligand 22 (CCL22, nt 433–462) mRNAs, all associated with the inflammatory response (rev in [46]).
Two distinct steps are involved in the assembly of the GAIT complex following IFN-γ treatment of myeloid cells. First, approximately two hours post IFN-γ stimulation, EPRS is released from the tRNA multisynthetase complex (MSC) where it resides and forms a non-functional pre-GAIT complex together with NSAP1. Later ribosomal protein L13a which is released from the 60S ribosomal subunit by an unclear mechanism is joined by GAPDH to form a functional GAIT complex at the 3′UTR of the target gene. EPRS is thought to recognize and bind to target mRNAs, NSAP1 negatively regulates RNA binding, and L13a binds to and inhibits translation initiation by interacting with eIF4G by competing with eIF3 and the 40S subunit containing 43S pre-initiation complex [47](see Figure 1B). The dismantling and subsequent reformation of the GAIT complex thus renders the 3′UTR GAIT-regulated mRNAs susceptible to rapid expression in response to cellular insult and later to silencing once the threat has passed. The mechanism underlying the dynamics and release of the four components of the GAIT complex remains under investigation. It is however evident that repression of a post-transcriptional regulon by the GAIT system contributes to the prevention of chronic inflammation.
Conclusions and perspectives
Control of gene expression at the mRNA translational level has been a particularly neglected area of investigation, especially in myeloid cells. However, a recent surge in the interest in this pathway as a result of the realization that cellular pathways commonly deregulated in AML including cell cycle progression, proliferation and differentiation are mechanistically tied to translation. For example, several upstream (AKT, TSC1/2) and downstream (eIF4E) mediators of the mTORC1 pathway are either mutated or activated in AML. Although there has been an intense search for therapeutic strategies targeting the mTOR pathway in myeloid cells, much work is yet to be done to gain a fundamental understanding of the role of the players that contribute to translation initiation and control in normal myeloid cells.
Acknowledgments
The author wishes to thank her mentor Dr Nancy Berliner and members of the Berliner lab for helpful discussions, valuable advice and support. This work has been supported by NIH PO1-HL63357.
Footnotes
Conflict of interest disclosure
No financial interest/relationships with financial interest relating to the topic of this article have been declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Van Der Kelen K, Beyaert R, Inzé D, De Veylder L. Translational control of eukaryotic gene expression. Crit Rev Biochem Mol Biol. 2009;44:143–168. doi: 10.1080/10409230902882090. [DOI] [PubMed] [Google Scholar]
- 2.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonenberg N, Dever TE. Eukaryotic translation initiation factors and regulators. Curr Opin Struct Biol. 2003;13:56–63. doi: 10.1016/s0959-440x(03)00009-5. [DOI] [PubMed] [Google Scholar]
- 4.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Gene Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 5.Ma X, Blenis J. Molecular mechanisms of mTOR mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 6.Wullschleger S, Loewith R, Hall MN. TOR signalling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 7.Martin KA, Blenis J. Coordinate regulation of translation by the PI3-kinase and mTOR pathways. Adv Cancer Res. 2002;86:1–39. doi: 10.1016/s0065-230x(02)86001-8. [DOI] [PubMed] [Google Scholar]
- 8.Platanias LC. Mechanisms of type-1 and type-II interferon-mediated signaling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 9.Lee-Fruman KK, Kuo CJ, Lippincott J, Terada N, Blenis J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene. 1999;18:5108–5114. doi: 10.1038/sj.onc.1202894. [DOI] [PubMed] [Google Scholar]
- 10.Olson K, Booth GC, Poulin F, Sonenberg N, Beretta L. Impaired myelopoiesis in mice lacking the repressors of translation initiation, 4E-BP1 and 4E-BP2. Immunology. 2009;128:e376–384. doi: 10.1111/j.1365-2567.2008.02981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grolleau A, Sonenberg N, Wietzerbin J, Beretta L. Differential regulation of 4E-BP1 and 4E-BP2, two repressors of translation initiation, during human myeloid cell differentiation. J Immunol. 1999;162:3491–3497. [PubMed] [Google Scholar]
- 12.Ly C, Arechiga AF, Melo JV, Walsh CM, Ong ST. Bcr-Abl kinase modulates the translation regulators ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia via the mammalian target of rapamycin. Cancer Res. 2003;63:5716–5722. [PubMed] [Google Scholar]
- 13.Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of Acute Myelod Leukemia cells requires PI3 kinase activation. Blood. 2003;102:972–980. doi: 10.1182/blood-2002-11-3429. [DOI] [PubMed] [Google Scholar]
- 14.Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KL. eIF4E is a central node of an RNA regulon that governs cellular proliferation. J Biol Chem. 2006;175:415–426. doi: 10.1083/jcb.200607020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagner P, Schneider A, Gartenhaus RB. Targeting the translational machinery as a novel treatment strategy for hematologic malignancies. Blood. 2010;115:2127–2135. doi: 10.1182/blood-2009-09-220020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park S, Chapiuis N, Tamburini J, et al. Role of the PI3K/AKT and mTOR pathways in acute myeloid leukemia. Haematologica. 2010;95:819–828. doi: 10.3324/haematol.2009.013797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamburini J, Green AS, Bardet V, et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood. 2009;114:1618–1627. doi: 10.1182/blood-2008-10-184515. [DOI] [PubMed] [Google Scholar]
- 18.Hinnebusch A. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. In: Sonnenberg N, Hershey JWB, Mathews MB, editors. CSH Monographs Volume 39: Translational Control of Gene Expression. Cold Spring Harbor NY: Cold Spring harbor Laboratory Press; 2000. pp. 185–243. [Google Scholar]
- 19.Oldfield S, Jones J, Tanton D, Proud C. Use of monoclonal antibody to study the structure and function of eukaryotic protein synthesis initiation factor eIF2B. Eur J Biochem. 1994;221:399–410. doi: 10.1111/j.1432-1033.1994.tb18752.x. [DOI] [PubMed] [Google Scholar]
- 20.Rousseau D, Kaspar R, Rosenwald I, Gehrke L, Sonenberg N. Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proc Natl Acad Sci USA. 1996;93:1065–1070. doi: 10.1073/pnas.93.3.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghilardi N, Wiestner A, Skoda RC. Thrombopoietin production is inhibited by a translational mechanism. Blood. 1998;92:4023–4030. [PubMed] [Google Scholar]
- 22.Harigai M, Miyashita T, Hanada M, Reed JC. A cis-acting element in the BCL-2 gene controls expression through translational mechanisms. Oncogene. 1996;12:1369–1374. [PubMed] [Google Scholar]
- 23.Calkhoven C, Müller C, Leutz A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Gene Dev. 2000;14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- 24.Mueller BU, Pabst T. C/EBPα and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol. 2006;13:7–14. doi: 10.1097/01.moh.0000190110.08156.96. [DOI] [PubMed] [Google Scholar]
- 25.Schuster M, Porse B. C/EBPa: a tumour suppressor in multiple tissues? Biochim. Biophys Acta. 2006;1766:88–103. doi: 10.1016/j.bbcan.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Fuchs O. Growth-inhibiting activity of transcription factor C/EBPa, its role in hematopoiesis and its tumour suppressor or oncogenic properties in leukaemias. Folia Biologica (Praha) 2007;53:97–108. doi: 10.14712/fb2007053030097. [DOI] [PubMed] [Google Scholar]
- 27.Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol. 2009;27:619–628. doi: 10.1200/JCO.2008.17.9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang P, Iwama A, Datta MW, Darlington GJ, Link DC, Tenen DG. Granulocyte development in C/EBP-alpha−/−mice: The role of expression of the IL-6 and G-CSF receptors. 39th Annual Meeting of the American Society of Hematology; San Diego, California, USA. December 1997; p. 90. [Google Scholar]
- 29.Wang ND, Finegold MJ, Bradley A, et al. Impaired energy homeostasis in C/EBP-alpha knockout mice. Science. 1995;269:1108–1112. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- 30.Pabst T, Muller B, Zhang P. Dominant negative mutations of CEBPA encodong CCAAT/enhancer binding protein-α (C/EBPα), in acute myeloid leukemia. Nat Genet. 2001;27:263–270. doi: 10.1038/85820. [DOI] [PubMed] [Google Scholar]
- 31.Geletu M, Balkhi MY, Peer Zada AA, et al. Target proteins of C/EBPalphap30 in AML: C/EBPalphap30 enhances sumoylation of C/EBPalphap42 via up-regulation of Ubc9. Blood. 2007;110:3301–3309. doi: 10.1182/blood-2007-01-071035. [DOI] [PubMed] [Google Scholar]
- 32.Kirstetter P, Schuster MB, Bereshchenko O, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13:299–310. doi: 10.1016/j.ccr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 33.Fu CT, Zhu KY, Mi JQ, et al. An evolutionarily conserved PTEN-C/EBPα-CTNNA1 axis controls myeloid development and transformation. Blood ePublication. 2010;115:4715–4724. doi: 10.1182/blood-2009-11-255778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perrotti D, Cesi V, Trotta R, et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30:48–58. doi: 10.1038/ng791. [DOI] [PubMed] [Google Scholar]
- 35.Manikandan J, Aarthi JJ, Kumar SD, Pushparaj PN. Oncomirs: The potential role of non-coding microRNAs in understanding cancer. Bioinformation. 2008;8:330–334. doi: 10.6026/97320630002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 37.Calin G, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pelosi E, Labbaye C, Testa U. MicroRNAs in normal and malignant myelopoiesis. Lukemia Res. 2009;33:1584–1593. doi: 10.1016/j.leukres.2009.04.039. [DOI] [PubMed] [Google Scholar]
- 40.Johnnidis J, Harris MH, Wheeler RT, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 41.Fazi F, Rosa A, Fatica A, et al. Aminicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005;123:819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 42.Pulikkan J, Dengler V, Peramangalam PS, et al. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood. 2010;115:1768–1778. doi: 10.1182/blood-2009-08-240101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eiring A, Harb JG, Neviani P, et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell. 2010;140:652–665. doi: 10.1016/j.cell.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng J, Iwama A, Satake M, Kohu K. MicroRNA-27 enhances differentiation of myeloblasts into granulocytes by post-transcriptionally downregulating Runx1. Brit J Haematol. 2009;145:412–423. doi: 10.1111/j.1365-2141.2009.07632.x. [DOI] [PubMed] [Google Scholar]
- 45.Serhan C, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 46.Mukhopadhyay R, Jia J, Arif A, Ray PS, Fox PL. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci. 2009;34:324–331. doi: 10.1016/j.tibs.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kapasi P, Chaudhuri S, Vyas K, et al. L13a Blocks 48S Assembly: Role of a General Initiation Factor in mRNA-Specific Translational Control. Mol Cell. 2007;25:113–126. doi: 10.1016/j.molcel.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]