

Abstract
We report the synthesis and evaluation of a series of iodinated celecoxib analogues as cyclooxygenase-2 (COX-2)-targeted single photon emission computerized tomography (SPECT) imaging agents for the detection of inflammation. The structure−activity relationship identified 5-(4-iodophenyl)-1-{4-(methylsulfonyl)phenyl}-3-(trifluoromethyl)-1H-pyrazole (8) as a promising compound with IC50 values of 0.05 μM against purified COX-2 and 0.03 μM against COX-2 in activated macrophages. The arylstannane of 8 undergoes facile radio-[123I]-iodination upon treatment with Na123I/NaI and chloramine T using an EtOAc/H2O two-phase system. The [123I]-8 was produced in a radiochemical yield of 85% and a radiochemical purity of 99%. In vivo SPECT imaging demonstrated that the radiotracer was taken up by inflamed rat paws with an average 1.7-fold enrichment over contralateral noninflamed paws. This study suggests that conversion of celecoxib into its isomeric iodo-[123I]-analogues is a useful approach for generating novel and efficacious agents for COX-2-targeted SPECT imaging of inflammation.
Keywords: Iodine-123, celecoxib, iododestannylation, two-phase reaction, cyclooxygenase-2 (COX-2), in vivo SPECT imaging
Prostaglandins are important biological mediators of inflammation, originating from the biotransformation of arachidonic acid catalyzed by cyclooxygenases (COX).1 Constitutively expressed COX-1 is found in most normal tissues, where it modulates homeostatic functions, such as hemostasis, vascular tone, and cyctoprotection of the gastric mucosa.2 Inducible COX-2 is expressed in inflammatory lesions, where it modulates pain, fever, and edema, and in proliferative lesions where it promotes growth, angiogenesis and enhances the metastatic potential of tumor cells.3 The expression of COX-2 is an early event in tumorigenesis that plays a role in tumor progression.4 COX-2 mRNA and protein are detectable in a significant percentage of precursor lesions (e.g., colon polyps and Barrett's esophagus)5,6 and an even higher percentage of malignant tumors (e.g., colon adenocarcinoma and esophageal adenocarcinoma).7,8 Recent work has shown that selective COX-2 inhibitors are useful in the treatment of certain human cancers.9,10 Therefore, COX-2 is an attractive molecular target for detection of cancers by imaging with radiolabeled COX-2 inhibitors. In fact, syntheses of 18F- and 123I-labeled COX-2 inhibitors as potential imaging agents have been reported.11,12 Preliminary characterization of these compounds indicated that they accumulated in COX-2-expressing macrophages,13−15 but the in vivo uptake in tumors of most of the agents did not correlate to the presence of COX-2.16 We recently reported that COX-2-targeted fluorescent imaging agents can be selectively delivered into inflammatory tissues and COX-2-expressing tumors in vivo.17 Because an enormous amount of medicinal chemistry has been conducted to create COX-2-specific inhibitors, there are numerous classes of potential building blocks that are available for the preparation of single photon emission computerized tomography (SPECT) imaging agents for COX-2.18,19 For the development of COX-2-targeted SPECT imaging agents, we synthesized a series of derivatives of celecoxib. Here, we report the synthesis, evaluation, and radiolabeling of [123I]-celecoxib analogues as selective COX-2 tracers for SPECT imaging and describe the in vivo delivery of a tracer that selectively accumulates in the COX-2-expressing carrageenan-induced rat paw model of acute inflammation.
The Claisen reaction of iodoacetophenone (A, R1 = 3-I or 4-I) with methyltrifluoroacetate in the presence of sodium methoxide gave the expected β-diketones B (R1 = 3-I or 4-I, and R2 = CF3) in 80−88% yield. An ultrasonication-assisted condensation of A (R1 = 3-I or 4-I) with either dimethyl- or diethyloxalate afforded compounds B (R1 = 3-I or 4-I, and R2 = CO2Me or CO2Et) in 65−70% yield. Alternatively, the reaction of A (R1 = 3-I or 4-I) with succinic anhydride in the presence of lithium diisopropylamide (LDA) proceeded smoothly to afford B (R1 = 3-I or 4-I, and R2 = C2H4CO2H) in 68−72% yield. Condensation of compounds B (R1 = 3-I or 4-I, and R2 = C2H4CO2H) with R3-Ph-NHNH2·HCl (R3 = SO2NH2 or SO2Me) afforded the respective pyrazole products, 1−10, in 76−84% yield (Scheme 1). The 1,5-regioisomers were generated almost exclusively by carrying out the reaction in the presence of the hydrochloride salt of the substituted phenylhydrazine in refluxing ethanol.18 However, when required, the 1,5-diarylpyrazoles were separated from the minor 1,3-diarylpyrazole isomers by flash chromatography.
Scheme 1. Synthesis of Isomeric Iodo Compounds 1−10.

Reagents and conditions: (a) 25% NaOMe/MeOH, methyl t-butyl ether, 25 °C, 48 h, or 25% NaOMe/MeOH, ultrasound, 45 °C, 16 h, or LDA, succinic anhydride, THF, −78 °C. (b) R3-Ph-NHNH2·HCl, MeOH, reflux 16 h, or R3-Ph-NHNH2·HCl, TEA, MeOH, reflux 16 h.
The IC50 values for inhibition of purified human COX-2 or ovine COX-1 by test compounds were determined by a thin-layer chromatography (TLC) assay.20 Hematin-reconstituted COX-2 (66 nM) or COX-1 (44 nM) in 100 mM Tris-HCl, pH 8.0, containing 500 μM phenol was treated with several concentrations of inhibitors (0−66 μM) at 25 °C for 17 min and 37 °C for 3 min followed by metabolism of 14C-arachidonic acid (50 μM) for 30 s at 37 °C. Table 1 displays the in vitro COX-1 and COX-2 inhibition data. We found that the iodo-celecoxib derivative, 1, which contains an iodo group at the meta-position of the 5-phenyl substituent, is a potent and selective COX-2 inhibitor with a COX-2 IC50 value of 0.08 μM (COX-1 IC50 > 66 μM). When the CF3 group of 1 was replaced by a CO2Me substituent, the COX-2 inhibitory potency was significantly decreased (2, COX-2 IC50 = 3.8 μM). Interestingly, the para-iodo isomer, 3, showed better potency against purified COX-2 (IC50 = 0.56 μM). A further increase in COX-2 potency was observed when the CO2Me group of 3 was replaced with a CO2Et group (4, COX-2 IC50 = 0.26 μM). Replacement of the CO2Me group of 3 or 4 with a propionic acid (R2 = C2H4CO2H) moiety afforded inactive compounds, 5 and 6. However, isomeric iodination or replacement of the SO2NH2 group of compound 1 with a SO2Me group increased the COX-2 inhibitory potency and selectivity as exhibited by compound 8 (R3 = SO2Me, COX-2 IC50 = 0.05 μM). A complete loss of poor inhibition was observed when the CF3 group of 7 or 8 was replaced with a CO2Me group (compounds 9 and 10).
Table 1. In Vitro Purified COX-1 and COX-2 and Lipopolysaccaride (LPS)-Activated Macrophage-Like (RAW254.7 Cell) Cellular COX-2 Enzyme Inhibition Assay Data of Isomeric Iodo Compounds 1−10.
| IC50(μM)a |
|||
|---|---|---|---|
| compd no. | purified COX-1 | purified COX-2 | RAW 264.7 cell COX-2 |
| 1 | >4 | 0.08 | 0.04 |
| 2 | >4 | 3.80 | NT |
| 3 | >4 | 0.56 | NT |
| 4 | >4 | 0.26 | 2.70 |
| 5 | >4 | >4 | NT |
| 6 | >4 | >4 | NT |
| 7 | >4 | 0.32 | NT |
| 8 | >4 | 0.05 | 0.03 |
| 9 | >4 | >4 | NT |
| 10 | >4 | 3.00 | NT |
| celecoxib | >4 | 0.03 | NT |
IC50 values were determined by incubating several concentrations of inhibitors in DMSO with purified murine COX-2 (66 nM) and ovine COX-1 (44 nM) for 20 min followed by treatment with 1-14C-AA (50 mM) at 37 °C for 30 s. Assays were run in duplicate. NT, not tested.
The ability of promising compounds to inhibit COX-2 in intact cells was assayed in the RAW264.7 murine macrophage-like cell line.21 The RAW264.7 cells were treated with lipopolysaccharide (200 ng/mL) and γ-interferon (10 U/mL) for 7 h to induce COX-2 expression and then treated with vehicle or the test compounds at several concentrations. The IC50 values for inhibition of COX-2 by compounds 1 and 8 were 0.04 and 0.03 μM, respectively (Table 1). Among these compounds, 8 showed the most potent inhibitory activity against COX-2 in cultured inflammatory cells without inhibition of COX-1 at concentrations up to 5 μM. Thus, compound 8 was selected for radio-123I-iodination. The remaining isomeric iodo compounds that have low to moderate COX-2 inhibitory potency and selectivity in the purified COX enzyme assay were not tested for their inhibitory activity against COX-2 enzyme in the activated intact RAW264.7 cell line assay.
We recently reported an efficient two-phase radioiodination method that was used in the present case to radiolabel compound 8.22 The aryltributylstannane 11 of the stable iodo compound 8 was generated by a palladium-catalyzed deiodostannylation reaction using hexabutylditin and tetrakis-(triphenylphosphine)palladium(0) in refluxing 1,4-dioxane. The radioiodination of tributylstannyl derivative 11 was conducted by electrophilic 123I-iododestannylation in an EtOAc/H2O binary-phase system using Na123I/NaI, chloramine T, and aqueous 1 N HCl (Scheme 2). The radiolabeled product was isolated by collecting the organic layer from the two-phase system and evaporating the solvent under a flow of argon. The electrophilic 123I species is generated in the water layer at pH 3.5 and rapidly extracted into the EtOAc layer for subsequent electrophilic aromatic substitution reaction. The final product stays in the EtOAc layer, which lacks the polar byproducts of the reaction to give the high radiochemical yield and purity of the product. This phase transfer technology does not require any further chromatographic purification. In a typical experiment, we reacted the precursor 11 (20 μg in 1.5 mL of EtOAc) with the radioactive Na123I (17.3 mCi in 500 μL of 0.01 N NaOH) in the presence of carrier NaI (4.5 μg in 300 μL of H2O), chloramine-T trihydrate (8.4 μg in 300 μL of H2O), and aqueous 1 N HCl (300 μL). After the reaction mixture was stirred for 5 min at room temperature, water (1 mL) followed by EtOAc (1 mL) was added. The organic layer was collected, washed with water, and evaporated under argon flow to afford [123I]-8 (product, 14.7 mCi; radiochemical yield, 85%; and radiochemical purity, 99%). The radiochemical purity was determined by a radio-TLC scan. The radiotracer [123I]-8 coeluted with an unlabeled standard. The specific activity of [123I]-8 was 491 Ci/mmol, which was calculated based on the final radiotracer obtained from the organic layer of the iodination reaction as compared with the added carrier in the reaction (specific activity = 0.01473 Ci/0.00003 mmol).
Scheme 2. Radiosynthesis of [123I]-8.

Reagents and conditions: (a) tetrakis-(Triphenylphosphine)palladium(0), bis-tributyltin, 1,4-dioxane, reflux 16 h. (b) Na123I/NaI, chloramine T, EtOAc, H2O, 1 N HCl, 25 °C, 4 min.
The rat paw model is well-documented for the role of COX-2-derived prostaglandins as a major driving force for the acute edema that results after carrageenan injection into the paw.23 One of the significant advantages of this animal model of inflammation is the ability to image the inflamed paw in comparison to the noninflamed contralateral footpad, which does not express COX-2. We injected 100 μL of 1% carrageenan in the rear right footpad of Sprague−Dawley rats (325−350 g) and waited 3 h for inflammation to develop. Then, we tail vein injected [123I]-8 dissolved in a formulation solvent consisting of dimethyl sulfoxide (10%), ethanol (40%), and sterile saline (50%) (300 μL, 600—800 μCi) into the anesthetized animals. Three hours later, the rats were reanesthetized with 2% isoflurane and placed in a BioScan NanoSPECT/CT, and a 30 min acquisition (24 projections × 60 s per projection) was initiated. The images were reconstructed with a resolution of 170 × 170 × 184 at 0.4 mm × 0.4 mm × 0.4 mm. The SPECT images were analyzed using AMIDE.24 Compound [123I]-8 targeted the swollen footpad selectively over the contralateral control footpad (Figure 1). The rats were sacrificed at various time points postinjection by isoflurane overdose. The hind feet were removed and weighed, and radioactivity associated with each footpad was counted with a well γ-counter. Figure 2 displays the relative uptake of [123I]-8 in the carrageenan-injected footpad over the control footpad obtained from measurements of individual footpad radioactivity after removal at two different time points (3 and 6 h; p = 0.005 at 3 h). The uptake of the radiotracer in the inflamed paw was 23.5 ± 5% of the injected dose/g and was 11.5 ± 1% in the noninflamed paw.
Figure 1.
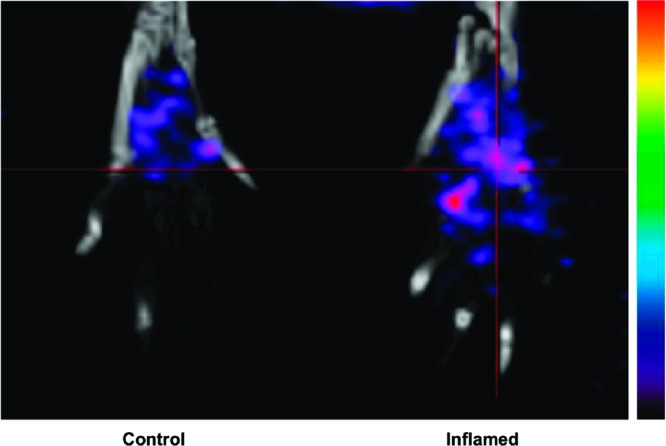
In vivo SPECT/CT image of a Sprague−Dawley rat footpad at 3 h post tail vein administration of [123I]-8.
Figure 2.
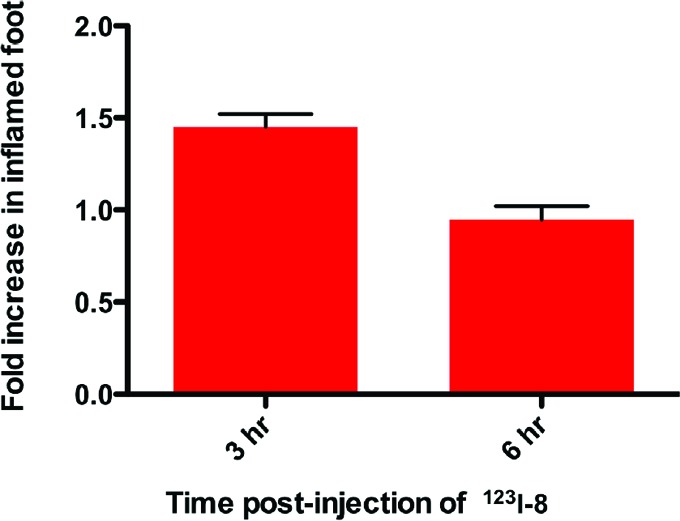
Relative uptake of [123I]-8 in inflamed vs noninflamed rat footpads at 3 and 6 h postinjection of [123I]-8.
The importance of COX-2 in the uptake of isotope into the inflamed footpad was probed by blocking the COX-2 active site with an excess of unlabeled 8. We administered the nonradioactive compound 8 at a dose of 55 mg/kg (ip) at 2 h postcarrageenan and waited 1 h for absorption and blockage of the COX-2 active site prior to dosing with [123I]-8 (∼1 mCi, tail vein). At 3 h postinjection of [123I]-8, we euthanized the animals, removed the hind paws, and measured the radioactivity of the individual paws in the well counter. There was no increase of radiotracer in the inflamed footpad as compared to the noninflamed control footpad (calculated fold increase = 1.0 ± 0.2).
In summary, isomeric iodo analogues of celecoxib were generated and radioiodinated such that they retain the ability of the parent celecoxib to inhibit COX-2 selectively in purified enzyme as well as in live inflammatory cells (e.g., compound 1 or 8). A striking observation from this study is that replacement of the p-tolyl ring with a p-iodophenyl ring, as well as substitution of the p-SO2NH2 group of celecoxib with a p-SO2Me group, generates compounds like 1 or 8 that are highly potent and selective COX-2 inhibitors. It is likely that the [123I]-substituent in compound 8 and the Me group in celecoxib are bioisoteric. This observation, coupled with the structural flexibility revealed in the present study, suggests that isomeric iodinated analogues of celecoxib are efficacious COX-2 inhibitors, and these compounds can be efficiently labeled with [123I] for use in COX-2-targeted SPECT imaging.
Acknowledgments
We are grateful to Jeffrey A. Clanton of the Department of Radiology and Radiological Sciences for assistance with radiochemical syntheses. We are also grateful to Dr. Carol A. Rouzer of the Vanderbilt Institute for Chemical Biology for critical reading of this manuscript.
Supporting Information Available
Full synthetic procedures and analytical and spectral characterization data of the synthesized compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
This work has been supported by grants from the National Institutes of Health (CA128323 and CA89450).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Vane J. R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol. 1971, 231, 232–235. [DOI] [PubMed] [Google Scholar]
- Smith W. L.; Garavito R. M.; DeWitt D. L. Prostaglandin Endoperoxide H Synthases (Cyclooxygenases)-1 and -2. J. Biol. Chem. 1996, 271, 33157–33160. [DOI] [PubMed] [Google Scholar]
- Li G.; Yang T.; Yan J. Cyclooxygenase-2 increased the angiogenic and metastatic potential of tumor cells. Biochem. Biophys. Res. Commun. 2002, 299, 886–890. [DOI] [PubMed] [Google Scholar]
- Taketo M. M. Cyclooxygenase-2 Inhibitors in Tumorigenesis (Part II). J. Natl. Cancer Inst. 1998, 90, 1609–1620. [DOI] [PubMed] [Google Scholar]
- Taketo M. M. COX-2 and colon cancer. Inflammation Res. 1998, 47, 112–116. [DOI] [PubMed] [Google Scholar]
- Abdalla S. I.; Lao-Sirieix P.; Novelli M. R.; Lovat L. B.; Sanderson I. R.; Fitzgerald R. C. Astrin-Induced Cyclooxygenase-2 Expression in Barrett's Carcinogenesis. Clin. Cancer Res. 2004, 10, 4784–4792. [DOI] [PubMed] [Google Scholar]
- Sano H.; Kawahito Y.; Wilder R. L.; Hashiramoto A.; Mukai S.; Asai K.; Kimura S.; Kato H.; Kondo M.; Hla T. Expression of Cyclooxygenase-1 and -2 in Human Colorectal Cancer. Cancer Res. 1995, 55, 3785–3789. [PubMed] [Google Scholar]
- Shirvani V. N.; Ouatu-Lascar R.; Kaur B. S.; Omary M. B.; Triadafilopoulos G. Cyclooxygenase 2 expression in Barrett’s esophagus and adenocarcinoma: Ex vivo induction by bile salts and acid exposure. Gastroenterology 2000, 118, 487–496. [DOI] [PubMed] [Google Scholar]
- Gupta R. A.; DuBois N. Cyclooxygenase-2 Inhibitor Therapy for the Prevention of Esophageal Adenocarcinoma in Barrett's Esophagus. J. Natl. Cancer Inst. 2002, 94, 406–407. [DOI] [PubMed] [Google Scholar]
- Maier T. J.; Schilling K.; Schmidt R.; Geisslinger G.; Grosch S. Cyclooxygenase-2 (COX-2)-dependent and -independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. Biochem. Pharmacol. 2004, 67, 1469–1474. [DOI] [PubMed] [Google Scholar]
- Kuge Y.; Katada Y.; Shimonaka S.; Temma T.; Kimura H.; Kiyono Y.; Yokota C.; Minematsu K.; Seki K.; Tamaki N.; Kazue Ohkura K.; Saji H. Synthesis and evaluation of radioiodinated cyclooxygenase-2 inhibitors as potential SPECT tracers for cyclooxygenase-2 expression. Nucl. Med. Biol. 2006, 33, 21–27. [DOI] [PubMed] [Google Scholar]
- Kabalka G. W.; Mereddy A. R.; Schuller H. M. Synthesis of iodine-123-labeled celecoxib analgue: a potential SPECT agent. J. Labelled Compd. Radiopharm. 2005, 48, 295–300. [Google Scholar]
- Vries E. F. J. D.; Waarde A. V.; Buursma A. R.; Vaalburg W. Synthesis and In Vivo Evaluation of 18F-Desbromo-DuP-697 as a PET Tracer for Cyclooxygenase-2 Expression. J. Nucl. Med. 2003, 44, 1700–1706. [PubMed] [Google Scholar]
- Ishikawa T.; Jain N. K.; Taketo M. M.; Herschman H. R. Imaging Cyclooxygenase-2 (Cox-2) Gene Expression in Living Animals with a Luciferase Knock-in Reporter Gene. Mol. Imaging Biol. 2006, 8, 171–187. [DOI] [PubMed] [Google Scholar]
- McCarthy T. J.; Sheriff A. U.; Graneto M. J.; Talley J. J.; Welch M. J. Radiosynthesis, In Vitro Validation, and In Vivo Evaluation of 18F-Labeled COX-1 and COX-2 Inhibitors. J. Nucl. Med. 2002, 43, 117–124. [PubMed] [Google Scholar]
- Schuller H. M.; Kabalka G.; Smith G.; Meredy A.; Akula M.; Cekanova M. Detection of Overexpressed COX-2 in Precancerous Lesions of Hamster Pancreas and Lungs by Molecular Imaging: Implications for Early Diagnosis and Prevention. ChemMedChem 2006, 1, 603–610. [DOI] [PubMed] [Google Scholar]
- Uddin M. J.; Crews B. C.; Blobaum A. L.; Kingsley P. J.; Gorden D. L.; McIntyre J. O.; Matrisian L. M.; Subbaramaiah K.; Dannenberg A. J.; Piston D. W.; Marnett L. J. Selective Visualization of Cyclooxygenase-2 in Inflammation and Cancer by Targeted Fluorescent Imaging Agents. Cancer Res. 2010, 70, 3618–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning T. D.; Talley J. J.; Bertenshaw S. R.; Carter J. S.; Collins P. W.; Docter S.; Graneto M. J.; Lee L. F.; Malecha J. W.; Miyashiro J. M.; Rogers R. S.; Rogier D. J.; Yu S. S.; Anderson G. D.; Burton E. G.; Cogburn J. N.; Gregory S. A.; Koboldt C. M.; Perkins W. E.; Seibert K.; Veenhuizen A. W.; Zhang Y. Y.; Isakson P. C. Synthesis and Biological Evaluation of the 1,5-Diarylpyrazole Class of Cyclooxygenase-2 Inhibitors: Identification of 4-[5-(4-Methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib). J. Med. Chem. 1997, 40, 1347–1375. [DOI] [PubMed] [Google Scholar]
- Prasit P.; Wang Z.; Brideau C.; Chan C. C.; Charleson S.; Cromlish W.; Ethier D.; Evans J. F.; Ford-Hutchinson A. W.; Gauthier J. Y.; Gordon R.; Guay J.; Gresser M.; Kargman S.; Kennedy B.; Leblanc Y.; Leger S.; Mancini J.; O'Neill G. P.; Ouellet M.; Percival M. D.; Perrier H.; Riendeau D.; Rodger I.; Zamboni R.; Boyce S.; Rupniak N.; Forrest M.; Visco D.; Patrick D. The discovery of rofecoxib, [MK 966, VIOXX, 4-(4′-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone], an orally active cyclooxygenase-2 inhibitor. Bioorg. Med. Chem. Lett. 1999, 9, 1773–1778. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Crews B. C.; Rowlinson S. W.; Marnett A. B.; Kozak K. R.; Remmel R. P.; Marnett L. J. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: Facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 925–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Kozak K. R.; Crews B. C.; Hochgesang G. P. Jr.; Marnett L. J. Covalent Modification of Cyclooxygenase-2 (COX-2) by 2-Acetoxyphenyl Alkyl Sulfides, a New Class of Selective COX-2 Inactivators. J. Med. Chem. 1998, 41, 4800–4818. [DOI] [PubMed] [Google Scholar]
- Uddin M. J.; Crews B. C.; Blobaum A. L.; Kingslay P. J.; Ghebraselase K.; Saleh S. S.; Clanton J. A.; Baldwin R. M.; Marnett L. J. Synthesis and evaluation of [123I]-indomethacin derivatives as COX-2 targeted imaging agents. J. Labelled Compd. Radiopharm. 2010, 52, 387–393. [Google Scholar]
- Winter C. A.; Risley E. A.; Nuss G. W. Carrageenin-induced edema in hind paw of the rat as an assay for antiinflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [DOI] [PubMed] [Google Scholar]
- Loening A. M.; Gambhir S. S. AMIDE: A free software tool for multimodality medical image analysis. Mol. Imaging 2003, 2, 131–137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.