

Abstract
Objectives
As an initial step in developing novel antibacterials against Burkholderia pseudomallei, we have characterized the FabI enoyl-ACP reductase homologues in the type II fatty acid biosynthesis pathway from this organism and performed an initial enzyme inhibition study.
Methods
A BLAST analysis identified two FabI enoyl-ACP reductase homologues, bpmFabI-1 and bpmFabI-2, in the B. pseudomallei genome, which were cloned, overexpressed in Escherichia coli and purified. Steady-state kinetics was used to determine the reaction mechanism and the sensitivity of bpmFabI-1 to four diphenyl ether FabI inhibitors. The antibacterial activity of the inhibitors was assessed using a wild-type strain of Burkholderia thailandensis (E264) and an efflux pump mutant (Bt38).
Results
Consistent with its annotation as an enoyl-ACP reductase, bpmFabI-1 catalysed the NADH-dependent reduction of 2-trans-dodecenoyl-CoA via a sequential Bi Bi mechanism. In contrast, bpmFabI-2 was inactive with all substrates tested and only bpmfabI-1 was transcriptionally active under the growth conditions employed. The sensitivity of bpmFabI-1 to four diphenyl ethers was evaluated and in each case the compounds were slow-onset inhibitors with Ki values of 0.5–2 nM. In addition, triclosan and PT01 had MIC values of 30 and 70 mg/L for B. pseudomallei as well as a wild-type strain of B. thailandensis (E264), but MIC values of <1 mg/L for the efflux pump mutant Bt38. A reduction in MIC values was also observed for the pump mutant strain with the other diphenyl ethers.
Conclusions
Provided that efflux can be circumvented, bpmFabI-1 is a suitable target for drug discovery.
Keywords: B. pseudomallei, slow-onset inhibition, efflux pumps
Introduction
Burkholderia pseudomallei is classified as a category B bioterrorism pathogen by the US National Institute of Allergy and Infectious Diseases.1,2 This organism causes the disease melioidosis, which is primarily found in South-East Asia and Northern Australia. Although only a few cases of the disease are reported each year, it is thought that the lack of research and medical facilities in the areas of incidence may have resulted in an underestimate of the numbers of individuals that are affected.3 Currently, there is no vaccine to prevent melioidosis and mortality is still very high, even with treatment using the first-line agents ceftazidime or imipenem, while relapse is often observed.4
Fatty acid biosynthesis (FAS) is used to synthesize the metabolic precursors for membrane phospholipids in the cell wall. In eukaryotes, fatty acid biosynthesis is catalysed by a type I fatty acid synthesis (FAS-I), in which the different enzyme activities are encoded by domains of a large polypeptide. In contrast, fatty acids are synthesized in prokaryotes by a type II pathway (FAS-II) in which each reaction is catalysed by individually encoded enzymes (Figure 1).5 Due to the essential role that fatty acids play in bacterial cell survival and the low degree of sequence homology with the mammalian FAS-I synthase, the FAS-II pathway is thought to be an attractive antibacterial drug target.6,10 In particular, the FAS-II enoyl-acyl carrier protein (ACP) reductase, which catalyses the final step in the elongation cycle, is thought to be a key regulator of fatty acid biosynthesis and to be essential for the viability of bacteria.7 Although a recent report concluded that the FAS-II pathway in Streptococcus agalactiae and, by extension, other Gram-positive bacteria is not essential for growth in the presence of fatty acids,8 the generality of this conclusion, at least with regard to the important nosocomial pathogen Staphylococcus aureus, has since been questioned.9 Importantly, there is no evidence that Gram-negative bacteria, such as B. pseudomallei, can sequester fatty acids from their environment to counter the impact of FAS-II inhibition, and an overall goal of our programme is to validate the FAS-II pathway in B. pseudomallei and other pathogenic bacteria as a novel target for drug discovery.
Figure 1.

The E. coli fatty acid biosynthesis pathway.
Although there are currently four subtypes of enoyl-ACP reductases (FabI, FabK, FabL and FabV), most drug discovery efforts have focused on organisms that contain only the FabI homologue.10 Triclosan is the paradigm FabI inhibitor,10–12 with picomolar binding affinity for the enzymes from Escherichia coli (ecFabI), S. aureus (saFabI) and Francisella tularensis (ftuFabI).10,13–16 In addition, the antitubercular drug isoniazid is a potent inhibitor of the FabI enzyme in Mycobacterium tuberculosis (mtFabI and InhA).17 Our group has reported the synthesis of several diphenyl ethers with subnanomolar affinity for saFabI, ftuFabI and mtFabI, where the lowest MIC values of these compounds for the respective organisms are <0.1–1 mg/L.14,16,18,19 However, organisms that encode alternative and/or additional enoyl-ACP reductases, such as Streptococcus pneumoniae that has the flavin-dependent FabK reductase, are less susceptible to triclosan.20
In this work, we have studied the mechanism of the FabI enzyme from B. pseudomallei. There are two fabI gene homologues, one on each of the two chromosomes.21 Both of the two fabIs have been cloned and expressed in E. coli. While a detailed mechanistic analysis has been performed for bpmFabI-1 (chromosome 1), the FabI homologue encoded on chromosome 2 (bpmFabI-2) was not transcriptionally active and displayed no activity with any of the enoyl reductase substrates tested. Due to the interest in developing chemotherapeutics against B. pseudomallei, we have studied the inhibition of bpmFabI-1 by triclosan and three other lead diphenyl ethers, and also evaluated the whole cell activity of these compounds against B. pseudomallei and Burkholderia thailandensis.
Materials and methods
Substrate synthesis
Trans-2-octenoyl-CoA (Oct-CoA), trans-2-decenoyl-CoA (Dec-CoA) and trans-2-dodecenoyl-CoA (DD-CoA) were synthesized from trans-2-ocetonoic acid, trans-2-decenoic acid and trans-2-dodecenoic acid, respectively, using the mixed anhydride method.22 Crotonyl-CoA (Cr-CoA) was prepared by coupling crotonic anhydride with CoA, as described previously.23 Crotonyl-ACP was prepared using the ACP from F. tularensis (NCBI reference sequence: YP_170325), which is 68% identical and 79% similar to the ACP from B. pseudomallei.24 All products were characterized by electrospray ionization mass spectrometry.
Cloning, expression and purification of bpmFabI-1 and bpmFabI-2
Since bpmFabI-1 (BURPS1710b_2636, chromosome 1: 2917100–891) and bpmFabI-2 (BURPS1710b_A2297, chromosome 2: 2788921–9682) from B. pseudomallei are 100% identical to bmfabI-1 (BMA1608, chromosome 1: 1671734–2525) and bmfabI-2 (BMAA1403, chromosome 2: 1510367–1128) from Burkholderia mallei, genomic DNA from B. mallei ATCC 23344 (NCBI reference sequence: YP_102617.1) was used for cloning. Amplification was performed using puReTaq Ready-To-Go PCR Beads (Amersham Biosciences) and the following primers (Integrated DNA Technologies): bmFabI-1 5′-GGAATTCCATATGGGCTTTCTCGACGGTAAAC-3′ (forward) and 5′-CCCAAGCTTTTCCTCGAGGCCGGCCATC-3′ (reverse); and bmFabI-2 5′-GGAATTCCATATGCGACTTCAGCACAAGC-3′ (forward) and 5′-CCCAAGCTTGCCGACGACGTGATAG-3′ (reverse). Both PCR products were digested with NdeI and HindIII, and then inserted into the pET23b plasmid (Novagen) so that a His-tag was encoded at the C terminus of the coding sequence for each protein. In addition, in order to provide a bpmFabI-2 construct with a cleavable N-terminal His-tag, bmfabI-2 was amplified using the primers 5′-GGAATTCCATATGCGACTTCAGCACAAGC-3′ (forward) and 5′- CGCGGATCCTCAGCCGACGACGTGATAG-3′ (reverse), digested with NdeI and BamHI, and then inserted into the pET15b plasmid. The correct sequence of each plasmid was confirmed by DNA sequencing (DNA Sequencing Facility, Health Science Center, Stony Brook University).
Protein expression and purification were performed as described previously using E. coli BL21(DE3) pLysS cells, and the N-terminal His-tag on bpmFabI-2 was cleaved by treatment with thrombin.16 The purity of each protein was verified by 12% SDS–PAGE, which gave an apparent molecular weight of ∼28 kDa in each case. The proteins were concentrated using centriplus YM-30 concentrators (Amicon), and protein concentrations were determined by measuring the absorption at 280 nm and using an extinction coefficient (ε) of 13 490 M−1 cm−1 for bpmFabI-1 and 16 170 M−1 cm−1 for bpmFabI-2, calculated in each case from the primary sequence. The bpmFabI-1 protein was stored at 4°C at a concentration of ∼350 μM.
Circular dichroism (CD) spectroscopy
CD spectra of bpmFabI-1 and bpmFabI-2 (15 µM) were recorded at room temperature on an Aviv model 202SF CD spectrometer (Lakewood, NJ, USA), using a 1 mm path length cell, 1 nm bandwidth, 1 nm resolution, 0.5 s response time and a scan speed of 50 nm/min. The secondary structure content of each protein was subsequently estimated using CDNN Deconvolution software (version 2; Bioinformatik.biochemtech.uni-halle.de/cdnn).
Steady-state kinetic assays
All kinetic experiments were performed on a Cary 300 Bio (Varian) spectrometer at 25°C in 30 mM PIPES buffer, pH 6.8, containing 150 mM NaCl and 1.0 mM EDTA. Initial velocities were measured by monitoring the oxidation of NADH to NAD+ at 340 nm (ε = 6300 M−1 cm−1) and kinetic parameters (kcat and kcat/Km) were determined as previously described.24 Initial characterization of the enzyme mechanism was performed in reaction mixtures containing 30 nM bpmFabI-1 and by measuring initial velocities at several fixed concentrations of NADH (50, 120, 190 and 250 µM) and by varying the concentration of Oct-CoA (16–128 µM), or at a fixed concentration of Oct-CoA (16, 32, 64 and 128 µM) and by varying the concentrations of NADH (50–250 µM). Double reciprocal plots were then used to differentiate between ping-pong or ternary-complex mechanisms.
To further investigate the binding order of the two substrates, product inhibition studies were performed in which the concentration of each substrate (NADH and Oct-CoA) was varied in the presence of the NAD+ product (0, 200 and 2000 µM). Lineweaver–Burk plots were subsequently used to determine whether enzyme inhibition was competitive, uncompetitive or non-competitive.
Fluorescence titrations
Equilibrium fluorescence titrations were conducted using a Quanto MasterTM-4/2005 spectrometer (Photon Technologies International). Binding experiments were performed at 25°C using the same buffer as that used for kinetic studies. Microlitre aliquots of NADH stock solution (1 mM) were titrated into a 1 mL solution of bpmFabI-1 or bpmFabI-2 (1 µM), and the fluorescence of NADH was monitored using 350 nm excitation and 455 nm emission with excitation and emission slit widths of 5.0 and 1.0 nm, respectively. A control experiment was also conducted in which there was no enzyme in the cuvette. The dilution of the protein concentration was kept to a minimum (<1%) and Kd values were calculated as described previously.16
Progress curve analysis
Progress curve analysis was performed to identify slow-onset inhibitors of bpmFabI-1. Reactions were performed by adding enzyme (5 nM) to assay mixtures containing glycerol (8%, v/v), BSA (0.1 mg/mL), DMSO (2%, v/v), Oct-CoA (300 µM), NADH (250 µM), NAD+ (200 µM) and inhibitor (0–1000 nM). Reactions were monitored until the progress curve became linear, indicating that the steady-state had been reached. In this protocol, the low enzyme concentration and high substrate concentration ensure that substrate depletion was minimized so that the progress curves in the absence of inhibitors were approximately linear over a period of 30 min.14,31 Because triclosan and the diphenyl ether inhibitors bind in presence of NAD+, 200 µM NAD+ was added so that the NAD+ concentration was constant during progress curve data collection. Subsequently, the data were fitted to the integrated rate equation (equation 1):
| (1) |
where At and A0 are the absorbances at time t and time 0, vi and vs are the initial velocity and steady-state velocity from the progress curve, respectively, t is time, γ = [E] × (1 − vs/vi)2/[I] and kobs is the observed rate constant for each curve. The kobs values were then analysed using equation 2, which describes a two-step inhibition mechanism in which the initial rapid binding of the inhibitor to the enzyme is followed by a second slow step that leads to the formation of the final enzyme–inhibitor complex (E-I*).
| (2) |
In equation 2, k2 and k−2 are the association and dissociation rates for the second step, respectively, [I] is inhibitor concentration and 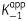 is the apparent dissociation constant for the initial enzyme–inhibitor complex (E-I).
is the apparent dissociation constant for the initial enzyme–inhibitor complex (E-I).
Inhibition of bpmFabI-1 by triclosan and select diphenyl ether inhibitors
Pre-incubation assays were performed to obtain the true inhibition constants and to determine the preference of slow-onset inhibitors for the different cofactor-bound forms of bpmFabI-1. Enzyme (5 nM) was pre-incubated in the presence of a fixed concentration of DMSO (2% v/v), NAD+ (10–200 µM), NADH (250 µM) and inhibitors (0–1000 nM) for 5 h at 4°C. After warming to 25°C, assays were initiated by the addition of Oct-CoA (30 µM). The inhibition constant K1 was calculated as previously described.13,15
Quantitative real-time PCR (RT-PCR)
RT-PCR was used to assess the transcriptional activity of bpmfabI-1 and bpmfabI-2 during growth of B. pseudomallei. RT-PCR was performed as described previously.26,27 Briefly, PCR amplification was performed using gene-specific primers in the presence of SYBR-green dye. The relative number of transcripts of each gene was determined based on linear regression analysis of genomic DNA for each primer pair, where data represent independent biological analyses of bacteria grown to mid-logarithmic phase growth.
MIC measurements
MICs required to reduce growth by 90% were determined using the microplate Alamar blue assay, as described previously.13,14,27 Briefly, B. pseudomallei and B. thailandensis were grown in Luria Broth (BD Difco) to mid-log phase and diluted to 2 × 103 cfu/mL in Mueller–Hinton broth (BD Difco). Compounds were diluted in Mueller–Hinton broth 1:2 from 250 mg/L to 0.2 mg/L in 96-well microtitre plates. Bacteria were added to compounds 1:2 and plates incubated for 18 h at 37°C. Alamar blue (Invitrogen) was then added to each well and incubated for 4 h at 37°C, and Alamar blue reduction was measured spectrophotometrically at 570 and 600 nm. MIC90 values were determined by plotting the percentage inhibition calculated from spectrophotometric readings over the drug concentration series. All MIC90 values were verified by assessing growth.
Results
Enzymatic characterization of bpmFabI-1 and bpmFabI-2
In order to identify putative FabI homologues in B. pseudomallei, the sequences of the FabI proteins from E. coli (ecFabI) and from F. tularensis (ftuFabI) were used as the query sequence for a BLAST analysis of proteins encoded in the B. pseudomallei genome. This analysis revealed that there are two FabI homologues, one on each of the chromosomes, with bpmFabI-1 encoded on chromosome 1 having 63.9% identity over 252 residues to ecFabI and 61.6% identity over 258 residues to ftuFabI, and bpmFabI-2 encoded on chromosome 2 having 42.4% identity over 252 residues to ecFabI and 40.9% identity over 252 residues to ftuFabI. In addition, this analysis also revealed 41.4% identity over 251 residues between bpmFabI-1 and bpmFabI-2, in which active site residues28 were conserved between both enzymes (Figure 2). Following overexpression in E. coli, the two FabI homologues were purified to homogeneity using His-tag affinity chromatography, following which the N-terminal His-tag was removed from bpmFabI-2 using thrombin. SDS–PAGE demonstrated that each protein was >95% pure and provided apparent molecular weights of 28 kDa in each case, consistent with the predicted molecular weights of 27 770 Da and 27 823 Da for bpmFabI-1 and bpmFabI-2, respectively.
Figure 2.

Sequence alignment of the FabI enzymes from E. coli, F. tularensis and B. pseudomallei. Residues in the rectangle contribute to the substrate binding loop. The three active site residues are highlighted by a diamond. The sequence alignment was performed using Clustal W45 and the figure was made using Jalview.46
The secondary structure of bpmFabI-1 and bpmFabI-2 were estimated by CD spectroscopy. Data analysis indicated that the α-helical content of bpmFabI-1 and bpmFabI-2 were 39.2% and 45.0%, respectively, while the β-sheet contents were 15.8% and 14.9%, respectively.
The catalytic activity of the two bpmFabI enzymes was subsequently characterized using CoA-based substrate analogues carrying crotonyl (C4), octenoyl (C8), decenoyl (C10) and dodecenoyl (C12) fatty acids. The steady-state kinetic parameters for bpmFabI-1 are summarized in Table 1, where it can be seen that this FabI homologue is able to catalyse the reduction of all the CoA substrates. In contrast, bpmFabI-2 was not active with any substrate tested, a result that was unaffected by removal of the His-tag purification sequence from this protein. Crotonyl-ACP was also synthesized in order to evaluate whether the lack of activity detected with bpmFabI-2 was simply the result of a complete inability to accept CoA-based substrates. However, bpmFabI-2 was also inactive with crotonyl-ACP, whereas bpmFabI-1 efficiently reduced this substrate with a kcat/Km value that was 9-fold larger than the corresponding value for crotonyl-CoA. In the bpmFabI-1 assays, substrate inhibition was observed at high concentrations of decenoyl-CoA and dodecenoyl-CoA, possibly as a result of binding of these compounds to the NADH binding site. Thus, the observed variation in kcat/Km through the enoyl-CoA substrate series may actually be an underestimate. Due to substrate inhibition observed with the longer acyl chain substrate, octenoyl-CoA was used for the progress curve analysis where high substrate concentrations are required.
Table 1.
Steady-state kinetic parameters for bpmFabI-1
| Substrate | kcat (min−1) | Km (μM) | kcat/Km (μM−1 min−1) |
|---|---|---|---|
| Crotonyl-CoA | 215 ± 8 | 188 ± 15 | 1.2 ± 0.1 |
| Crotonyl-ACP | 242 ± 22 | 27 ± 6 | 9 ± 2 |
| Trans-2-octenoyl-CoA | 1700 ± 132 | 160 ± 22 | 11 ± 1 |
| Trans-2-decenoyl-CoA | 335 ± 10 | 5.6 ± 0.7 | 60 ± 8 |
| Trans-2-dodecenoyl-CoA | 504 ± 6 | 1.7 ± 0.1 | 300 ± 17 |
As expected for an enoyl-ACP reductase, double-reciprocal plots of the kinetic data displayed intersecting lines to the left of the y-axis, consistent with a ternary-complex mechanism (Figure 3).29,30 To further determine whether the reaction proceeded via an ordered Bi Bi mechanism or a random Bi Bi mechanism, product inhibition studies were conducted (Figure 4), which showed that NAD+ is a competitive inhibitor with respect to NADH and a mixed-type competitive inhibitor with respect to octenoyl-CoA. This inhibition pattern is consistent with an ordered Bi Bi mechanism in which NADH binds first to the enzyme.29,30 Consistent with this result, fluorescence titration experiments demonstrated that NADH was able to bind to the free enzyme with a Kd value of 1.02 ± 0.02 μM (Figure 5).
Figure 3.

Two-substrate steady-state kinetics. Initial velocity patterns: (a) 1/v versus 1/[Oct-CoA] double-reciprocal plot in which [NADH] was fixed; and (b) 1/v versus 1/[NADH] double-reciprocal plot in which [Oct-CoA] was fixed.
Figure 4.

Product inhibition studies to determine the order of substrate binding. Assays were performed by varying the concentration of one substrate at a fixed concentration of the second substrate and in the presence of NAD+. Initial velocity patterns: (a) 1/v versus 1/[Oct-CoA] double-reciprocal plot in which [NAD+] was fixed; and (b) 1/v versus 1/[NADH] double-reciprocal plot in which [NAD+] was fixed.
Figure 5.

Fluorescence titration of bpmFabI-1 and bpmFabI-2 with NADH. The plot shows the change in fluorescence when 1 µM bpmFabI-1 (open circles) or bpmFabI-2 (open triangles) is titrated with NADH. The continuous lines are the best fits of the data to the Scatchard equation with Kd = 1.02 ± 0.02 µM and Kd = 1.07 ± 0.02 µM, respectively.
Antimicrobial activity of the diphenyl ether bpmFabI-1 inhibitors
As the first step towards the development of bpmFabI inhibitors, we evaluated the ability of the diphenyl ether class of compounds to inhibit this enzyme. Progress curve analysis was used to demonstrate that all four diphenyl ethers tested were slow-onset inhibitors of bpmFabI-1.14,31 Representative data are shown in Figure 6 for the inhibition of bpmFabI-1 by triclosan. In these experiments, sufficient substrate is used so that the reaction rate is linear for a significant period of time (30 min) in the absence of inhibitor and so that the observation of curvature in the presence of inhibitor can be used as a diagnostic for slow-onset inhibition. In Figure 6(a), it can be seen that in the presence of triclosan the rate decreased exponentially with time, from an initial velocity (vi) to a steady-state velocity (vs). In addition, both vi and vs decreased with increasing inhibitor concentration, while kobs increased and the time required to reach vs decreased (Figure 6a). This behaviour is a classic example of slow-onset inhibition in which the rapid formation of the initial E-I complex is followed by a second slow step leading to the formation of the final E-I* complex (Figure 7a). Fitting the data to equation 1 provided values for vi, vs and kobs. The hyperbolic dependence of kobs on the concentration of inhibitor was then fitted to equation 2 (Figure 6b), allowing the calculation of the kinetic constants for the interconversion of E-I and E-I*, and also providing a value for 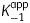 , the dissociation constant of E-I (Table 2). Assuming that the rate of dissociation of the inhibitor from the enzyme (koff) can be approximated by k−2, the residence time of triclosan on bpmFabI-1 (1/k−2) is 35 min, which is similar to that determined previously for the inhibition of ftuFabI by triclosan (Table 2).14
, the dissociation constant of E-I (Table 2). Assuming that the rate of dissociation of the inhibitor from the enzyme (koff) can be approximated by k−2, the residence time of triclosan on bpmFabI-1 (1/k−2) is 35 min, which is similar to that determined previously for the inhibition of ftuFabI by triclosan (Table 2).14
Figure 6.

Progress curve and pre-incubation analysis for the inhibition of bpmFabI-1 by triclosan and PT01. (a) Progress curves were obtained for triclosan concentrations ranging from 0 to 1000 nM. The continuous lines are the best fits of the data to equation 1. (b) kobs from (a) plotted as a hyperbolic function of [triclosan] using equation 2. (c) Effect of NAD+ on the apparent inhibition constant of triclosan. The continuous, dashed and dotted lines are the fits of the data to equations in which the inhibitor binds to both E-NADH and E-NAD+ (continuous line), only E-NAD+ (dashed line) or only E-NADH (dotted line).14 The best fit (continuous line) is obtained using 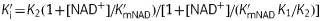 , with K1 = 1.57 ± 0.13 nM and K2 = 1096 ± 74 nM. (d) The effect of NAD+ on the apparent inhibition constant of PT01. The continuous, dashed and dotted lines are as described in (c), and the best fit (continuous line) is again obtained with the equation in which PT01 binds to both E-NAD+ and E-NADH with K1 = 0.51 ± 0.04 nM and K2 = 910 ± 101 nM.
, with K1 = 1.57 ± 0.13 nM and K2 = 1096 ± 74 nM. (d) The effect of NAD+ on the apparent inhibition constant of PT01. The continuous, dashed and dotted lines are as described in (c), and the best fit (continuous line) is again obtained with the equation in which PT01 binds to both E-NAD+ and E-NADH with K1 = 0.51 ± 0.04 nM and K2 = 910 ± 101 nM.
Figure 7.
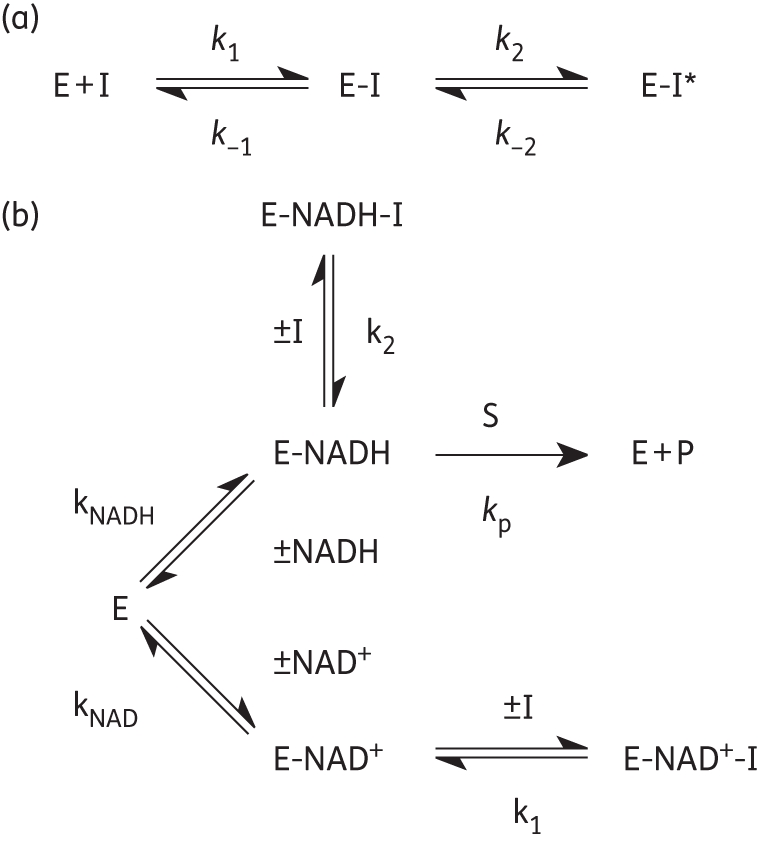
Kinetic schemes for the inhibition of bpmFabI-1. (a) Slow-onset inhibition in which formation of the final E-I* inhibitor complex occurs in two steps. (b) Kinetic scheme for the interaction of inhibitors with E-NAD+ and E-NADH.
Table 2.
Inhibition of ftuFabI and bpmFabI-1 by triclosan
| Enzyme inhibitor pair | k2 (min−1) | k−2 (min−1) | t1/2 (min) |
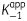 (nM) (nM) |
K1 (nM) |
|---|---|---|---|---|---|
| ftuFabI-triclosana | 0.56 ± 0.04 | 0.025 ± 0.003 | 28 ± 2 | 407 ± 48 | 0.051 ± 0.003 |
| bpmFabI-1-triclosan | 0.87 ± 0.03 | 0.020 ± 0.006 | 35 ± 8 | 647 ± 50 | 1.57 ± 0.13 |
aData taken from Lu et al.14
Since most diphenyl ethers preferentially bind to the FabI-NAD+ product complex and only occasionally prefer the FabI-NADH form of the enzyme,10,28,32,38 we performed pre-incubation experiments with the diphenyl ether inhibitors and bpmFabI-1.14 This enables the dependence of enzyme inhibition on NADH or NAD+ to be evaluated, and also provides the true thermodynamic affinity of the inhibitor for E-NAD+ (K1) and E-NADH (K2) (Figure 7b). Following pre-incubation of bpmFabI-1 with inhibitor in the presence of saturating NADH and various concentrations of NAD+ for 5 h, the reaction was initiated by adding octenoyl-CoA to obtain the apparent inhibition constant 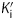 at each [NAD+]. Subsequently, the dependence of
at each [NAD+]. Subsequently, the dependence of 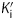 on [NAD+] was examined. For triclosan, the dependence of
on [NAD+] was examined. For triclosan, the dependence of 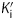 on [NAD+] was best described by the equation in which the inhibitor binds to both E-NAD+ and E-NADH forms of bpmFabI-1 (Figure 6c), albeit with ∼1000-fold preference for E-NAD+. The K1 and K2 values determined using this method were 1.57 nM and 1.10 µM, respectively (Table 3). The three other diphenyl ethers behaved similarly to triclosan (Table 3), with representative data shown in Figure 6(d) for PT01. Thus, all four compounds tested are slow-onset inhibitors of bpmFabI-1 with nanomolar affinity for the E-NAD+ form of this enzyme.
on [NAD+] was best described by the equation in which the inhibitor binds to both E-NAD+ and E-NADH forms of bpmFabI-1 (Figure 6c), albeit with ∼1000-fold preference for E-NAD+. The K1 and K2 values determined using this method were 1.57 nM and 1.10 µM, respectively (Table 3). The three other diphenyl ethers behaved similarly to triclosan (Table 3), with representative data shown in Figure 6(d) for PT01. Thus, all four compounds tested are slow-onset inhibitors of bpmFabI-1 with nanomolar affinity for the E-NAD+ form of this enzyme.
Table 3.
Antibacterial activity and inhibition of bpmFabI-1 by triclosan and the diphenyl ethers
|
B. thailandensis |
||||||
|---|---|---|---|---|---|---|
| Inhibitor | K1 (nM) | K2 (nM) | B. pseudomallei MIC (mg/L) | E264 MIC (mg/L) | Bt38 MIC (mg/L) | |
| Triclosan | 1.57 ± 0.13 | 1096 ± 74 | 30 | 30 | 0.2–0.5 | |
| PT01 | 0.51 ± 0.04 | 910 ± 101 | 70 | 70 | 0.2–0.5 | |
| PT02 | 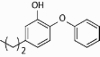 |
1.30 ± 0.10 | 361 ± 14 | >250 | >250 | 1.2 |
| PT03 |  |
1.80 ± 0.20 | 428 ± 20 | >250 | >250 | 33 |
The ability of the diphenyl ethers to inhibit bacterial growth was also evaluated. MIC values for B. pseudomallei varied from 30 and 70 mg/L for triclosan and PT01, respectively, to >250 mg/L for PT02 and PT03 (Table 3). Since efflux is a major mechanism of drug resistance in Burkholderia spp.,33 we also evaluated antibacterial activity using a B. thailandensis strain (Bt38) in which the bpeAB-oprB and amrAB efflux pumps have been disabled.34 FabI-1 and FabI-2 in B. pseudomallei and B. thailandensis are very similar, with 98% and 97% identity, respectively, so it is likely that the FabI-1 enzyme in B. thailandensis is also very sensitive to the compounds used in this study. As a control, we first determined MIC values of the diphenyl ethers for a wild-type strain of B. thailandensis (E264) and observed the same values as those determined for B. pseudomallei (Table 3). However, MICs for pump mutant strain Bt38 were dramatically reduced, with values of 0.2–0.5 mg/L for triclosan and PT01, 1.2 mg/L for PT02, and 33 mg/L for PT03 (Table 3).
Discussion
While the natural substrates for the FabI enzymes are fatty acid thioesters of ACP, all FabIs evaluated to date are able to accept enoyl substrates based on CoA or other artificial carrier molecules.10,13–16,35 Consequently, enoyl-CoA substrates are normally used to assay these enzymes, since enoyl-CoAs are significantly easier to synthesize and purify compared with the corresponding enoyl-ACPs. The kcat/Km values vary by a factor of 300, from crotonyl-CoA to dodecenoyl-CoA, indicating that the enzyme catalyses the reduction of long-chain fatty acids most efficiently. The increase in kcat/Km from crotonyl-CoA to crotonyl-ACP is due primarily to a reduction in Km for the ACP substrate, consistent with the expectation that ACP is the preferred substrate carrier for this class of enzyme.36
Currently, it is not known why bpmFabI-2 is inactive. However, either the enzyme is not folded correctly or we have so far not presented the enzyme with the correct substrate. In order to ascertain whether activity was lost during purification, we evaluated the enoyl-ACP reductase activity of the E. coli cell lysate following overexpression of bpmFabI-2. However, no activity, above that assigned to the endogenous E. coli enzymes, could be detected. In addition, bpmFabI-2 is soluble, suggesting that if the protein is incorrectly folded it is not grossly so, while CD spectroscopy revealed that bpmFabI-2 has similar secondary structure content to bpmFabI-1. Further perusal of the sequence data reveals a minor change in the alignment of the catalytic residues between bpmFabI-2 and the other FabI enzymes. Similar to other FabI short-chain dehydrogenase reductases (SDRs), bpmFabI-1 has the typical Y-Y-K catalytic triad, specifically Y146-Y156-K163.37 However, in bpmFabI-2, the residue equivalent to Y146 is displaced by one residue to position 147, so that a phenylalanine occupies position 146. In ecFabI, ftuFabI and bpmFabI-1, the sequence around Y146 is L-S/T-Y-L-G-A-E-R/K, whereas in bpmFabI-2 it is L-T-F-Y-G-A-E-R. Experiments with ecFabI have shown that Y146 plays a key role in catalysis36 and it is plausible that the position of Y146 in bpmFabI-2 has been altered to accommodate an alternative substrate. Interestingly, fluorescence titration reveals that bpmFabI-2 binds NADH with a Kd value of 1.07 ± 0.02 μM. Thus, we currently believe that while bpmFabI-2 is an NAD-dependent enzyme, it does not catalyse the reduction of fatty acid substrates. In this regard, we note that the SDR family proteins catalyse the oxidation/reduction of a wide range of substrates.37
To provide additional information on the role of bpmFabI-2, the transcriptional activity of both FabI homologues was evaluated using RT-PCR. This analysis demonstrated that bpmFabI-1 is strongly transcribed, while the transcriptional level of bpmFabI-2 is ≥1000-fold less than that for bpmFabI-1. These data support the importance of bpmFabI-1 in the fatty acid biosynthesis pathway and indicate that bpmFabI-2, if indeed it is an enoyl-ACP reductase, is not required for fatty acid biosynthesis under the growth conditions employed. In this regard, it has previously been observed that chromosome 1 encodes many of the core functions associated with the central metabolism and cell growth of B. pseudomallei, whereas chromosome 2 encodes accessory functions associated with adaptation and survival in atypical conditions, possibly accounting for the lack of bpmFabI-2 expression observed here.21 Thus, we cannot rule out the possibility that bpmFabI-2 assumes one or more important functions under alternative growth conditions, e.g. when the organism replicates in vivo.
Triclosan is a potent inhibitor of the FabI enzymes from organisms such as E. coli,13,15 F. tularensis14 and S. aureus.16 This molecule has been used as a starting point for developing long residence time diphenyl ether inhibitors of ftuFabI,14 in addition to the FabI from M. tuberculosis (mtFabI and InhA),18,19 which is relatively insensitive to triclosan.38 Slow-onset inhibition of the FabI enzymes is coupled to ordering of a loop of amino acids close to the active site10,14,19,28 and the long residence time of the slow-onset inhibitors is thought to be critical for in vivo drug activity,10,39–43 as demonstrated directly by us for a series of inhibitors of ftuFabI.14 Consequently, as a prelude to rational inhibitor discovery, we were interested in assessing the ability of the diphenyl ether class of compounds to inhibit this enzyme and to determine the ability of these compounds to inhibit the growth of B. pseudomallei. Triclosan and three other diphenyl ethers are slow-onset inhibitors of bpmFabI-1, binding preferentially to the E-NAD+ product complex with K1 values of ∼1 nM, and progress curve analysis revealed that triclosan has a residence time of 35 min on the enzyme target.
MIC values of the four compounds for B. pseudomallei and the wild-type strain of the non-pathogenic organism B. thailandensis ranged from 30 mg/L for triclosan and 70 mg/L for PT01, to >250 mg/L for PT02 and PT03. In addition, for the pump mutant the MIC values of triclosan and PT01 are in the range observed for this class of compounds with other susceptible organisms,14,16,18 suggesting that their antibacterial activity is due to FabI-1 inhibition in Bt38. Since all four compounds have similar affinities for bpmFabI, it is presently unclear why the MIC values of PT02 and PT03 differ from those of triclosan and PT01 for the three strains tested. Clearly, all are substrates for the efflux pumps that have been inactivated in Bt38, since the MIC values are lower for this strain; however, it is plausible that PT02 and, especially, PT03 are substrates for additional efflux system(s) or detoxification pathways that are still present in Bt38, or that these compounds have more difficulty in crossing the cell wall.
Finally, it is important to comment on the presence of the FabV enoyl-ACP reductase homologue in B. pseudomallei. In addition to the two FabI homologues, B. pseudomallei also contains a homologue of the recently discovered FabV enoyl-ACP reductase.24,44 It is currently not clear what role FabI and FabV play in Burkholderia spp. However, it is possible that both have to be inhibited in order to fully compromise fatty acid biosynthesis, a view that is supported by recent work on Pseudomonas aeruginosa PAO1.44 Like B. pseudomallei, P. aeruginosa also contains both FabI (paFabI) and FabV (paFabV) homologues, which are 65% and 74% identical, respectively, to the corresponding enzymes in B. pseudomallei. Interestingly, Zhu et al.44 have demonstrated that deletion of the gene for paFabV leads to a >2000-fold increase in the susceptibility of P. aeruginosa to triclosan (MIC > 2000 to 1 mg/L), supporting their conclusion that triclosan resistance in this organism is due to the presence of the ‘triclosan-resistant’ FabV enzyme rather than to efflux. However, in the case of Burkholderia spp., experiments with strain Bt38 indicate that efflux plays an important role in modulating the susceptibility of this organism to triclosan and the other diphenyl ethers. Additionally, the triclosan MIC value for Bt38 of 0.2–0.5 mg/L is similar to that for the P. aeruginosa FabV knockout strain, indicating that in Burkholderia either both enzymes are essential or that this concentration is sufficient to inhibit both bpmFabI-1 and bpmFabV. In this regard, we know that the Ki value of triclosan for bpmFabV is 0.4 µM (0.12 µg/mL),24 which is similar to the MIC value for the efflux pump mutant strain. Thus, even though bpmFabV is ∼250-fold less sensitive to triclosan than bpmFabI-1, the concentration of inhibitor required to prevent bacterial growth is indeed sufficient to inhibit both enoyl-ACP reductases. Currently, we are constructing genetic knockouts of the respective genes in B. thailandensis and B. pseudomallei to provide additional insight into the function of FabI and FabV in Burkholderia spp., and to evaluate the mechanism of action of the enoyl-ACP reductase inhibitors.
Funding
This study was supported by funding from the NIH (AI065357, AI082164, AI044639 and AI070383).
Transparency declarations
None to declare.
References
- 1.Gilad J, Harary I, Dushnitsky T, et al. Burkholderia mallei and Burkholderia pseudomallei as bioterrorism agents: national aspects of emergency preparedness. Isr Med Assoc J. 2007;9:499–503. doi:10.1128/CMR.00061-07. [PubMed] [Google Scholar]
- 2.Harding SV, Sarkar-Tyson M, Smither SJ, et al. The identification of surface proteins of Burkholderia pseudomallei. Vaccine. 2007;25:2664–72. doi: 10.1016/j.vaccine.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Cheng AC, Currie BJ. Melioidosis: epidemiology, pathophysiology, and management. Clin Microbiol Rev. 2005;18:383–416. doi: 10.1128/CMR.18.2.383-416.2005. doi:10.1128/AAC.00866-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaowagul W. Recent advances in the treatment of severe melioidosis. Acta Trop. 2000;74:133–7. doi: 10.1016/s0001-706x(99)00062-5. doi:10.1128/AEM.66.5.1933-1938.2000. [DOI] [PubMed] [Google Scholar]
- 5.White SW, Zheng J, Zhang YM, et al. The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. doi:10.1016/j.micinf.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Baldock C, Rafferty JB, Sedelnikova SE, et al. A mechanism of drug action revealed by structural studies of enoyl reductase. Science. 1996;274:2107–10. doi: 10.1126/science.274.5295.2107. doi:10.1086/507710. [DOI] [PubMed] [Google Scholar]
- 7.Heath RJ, Rock CO. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem. 1995;270:26538–42. doi: 10.1074/jbc.270.44.26538. doi:10.1179/000349803225002516. [DOI] [PubMed] [Google Scholar]
- 8.Brinster S, Lamberet G, Staels B, et al. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature. 2009;458:83–6. doi: 10.1038/nature07772. doi:10.1016/j.drup.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Balemans W, Lounis N, Gilissen R, et al. Essentiality of FASII pathway for Staphylococcus aureus. Nature. 2010;463:E3–4. doi: 10.1038/nature08667. [DOI] [PubMed] [Google Scholar]
- 10.Lu H, Tonge PJ. Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc Chem Res. 2008;41:11–20. doi: 10.1021/ar700156e. [DOI] [PubMed] [Google Scholar]
- 11.Escalada MG, Harwood JL, Maillard JY, et al. Triclosan inhibition of fatty acid synthesis and its effect on growth of Escherichia coli and Pseudomonas aeruginosa. J Antimicrob Chemother. 2005;55:879–82. doi: 10.1093/jac/dki123. [DOI] [PubMed] [Google Scholar]
- 12.Levy CW, Roujeinikova A, Sedelnikova S, et al. Molecular basis of triclosan activity. Nature. 1999;398:383–4. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- 13.Sivaraman S, Zwahlen J, Bell AF, et al. Structure–activity studies of the inhibition of FabI, the enoyl reductase from Escherichia coli, by triclosan: kinetic analysis of mutant. Biochemistry. 2003;42:4406–13. doi: 10.1021/bi0300229. [DOI] [PubMed] [Google Scholar]
- 14.Lu H, England K, Ende CA, et al. Slow-onset inhibition of the Fabl enoyl reductase from Francisella tularensis: residence time and in vivo activity. ACS Chem Biol. 2009;4:221–31. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward WHJ, Holdgate GA, Rowsell S, et al. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry. 1999;38:12514–25. doi: 10.1021/bi9907779. [DOI] [PubMed] [Google Scholar]
- 16.Xu H, Sullivan TJ, Sekiguchi JI, et al. Mechanism and inhibition of saFabI, the enoyl reductase from Staphylococcus aureus. Biochemistry. 2008;47:4228–36. doi: 10.1021/bi800023a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci USA. 2003;100:13881–6. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sullivan TJ, Truglio JJ, Boyne ME, et al. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem Biol. 2006;1:43–53. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- 19.Luckner SR, Liu NN, Ende CWA, et al. A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J Biol Chem. 2010;285:14330–7. doi: 10.1074/jbc.M109.090373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heath RJ, Rock CO. Microbiology—a triclosan-resistant bacterial enzyme. Nature. 2000;406:145–6. doi: 10.1038/35018162. [DOI] [PubMed] [Google Scholar]
- 21.Holden MTG, Titball RW, Peacock SJ, et al. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci USA. 2004;101:14240–5. doi: 10.1073/pnas.0403302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parikh S, Moynihan DP, Xiao GP, et al. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry. 1999;38:13623–34. doi: 10.1021/bi990529c. [DOI] [PubMed] [Google Scholar]
- 23.Hofstein HA, Feng YG, Anderson VE, et al. Role of glutamate 144 and glutamate 164 in the catalytic mechanism of enoyl-CoA hydratase. Biochemistry. 1999;38:9508–16. doi: 10.1021/bi990506y. [DOI] [PubMed] [Google Scholar]
- 24.Lu H, Tonge PJ. Mechanism and inhibition of the FabV enoyl-ACP reductase from Burkholderia mallei. Biochemistry. 2010;49:1281–9. doi: 10.1021/bi902001a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reference deleted. [Google Scholar]
- 26.Groathouse NA, Brown SE, Knudson DL, et al. Isothermal amplification and molecular typing of the obligate intracellular pathogen Mycobacterium leprae isolated from tissues of unknown origins. J Clin Microbiol. 2006;44:1502–8. doi: 10.1128/JCM.44.4.1502-1508.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.England K, Ende CA, Lu H, et al. Substituted diphenyl ethers as a broad-spectrum platform for the development of chemotherapeutics for the treatment of tularaemia. J Antimicrob Chemother. 2009;64:1052–61. doi: 10.1093/jac/dkp307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart MJ, Parikh S, Xiao GP, et al. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J Mol Biol. 1999;290:859–65. doi: 10.1006/jmbi.1999.2907. [DOI] [PubMed] [Google Scholar]
- 29.Cleland WW. Kinetics of enzyme-catalyzed reactions with two or more substrates or products. II. Inhibition—nomenclature and theory. Biochim Biophys Acta. 1963;67:173–87. doi: 10.1016/0006-3002(63)91815-8. [DOI] [PubMed] [Google Scholar]
- 30.Segel IH. Enzyme Kinetics. New York: Wiley; 1975. [Google Scholar]
- 31.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 32.Sivaraman S, Sullivan TJ, Johnson F, et al. Inhibition of the bacterial enoyl reductase FabI by triclosan: a structure–reactivity analysis of FabI inhibition by triclosan analogues. J Med Chem. 2004;47:509–18. doi: 10.1021/jm030182i. [DOI] [PubMed] [Google Scholar]
- 33.Moore RA, DeShazer D, Reckseidler S, et al. Efflux-mediated aminoglycoside and macrolide resistance in Burkholderia pseudomallei. Antimicrob Agents Chemother. 1999;43:465–70. doi: 10.1128/aac.43.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan YY, Tan TMC, Ong YA, et al. BpeAB-OpRB, a multidrug efflux pump in Burkholderia pseudomallei. Antimicrob Agents Chemother. 2004;48:1128–35. doi: 10.1128/AAC.48.4.1128-1135.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quemard A, Sacchettini JC, Dessen A, et al. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry. 1995;34:8235–41. doi: 10.1021/bi00026a004. [DOI] [PubMed] [Google Scholar]
- 36.Rafi S, Novichenok P, Kolappan S, et al. Structure of acyl carrier protein bound to FabI, the FASII enoyl reductase from Escherichia coli. J Biol Chem. 2006;281:39285–93. doi: 10.1074/jbc.M608758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jornvall H, Persson B, Krook M, et al. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–13. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 38.Parikh SL, Xiao GP, Tonge PJ. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry. 2000;39:7645–50. doi: 10.1021/bi0008940. [DOI] [PubMed] [Google Scholar]
- 39.Swinney DC. The role of binding kinetics in therapeutically useful drug action. Curr Opin Drug Disc. 2009;12:31–9. [PubMed] [Google Scholar]
- 40.Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov. 2004;3:801–8. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- 41.Tummino PJ, Copeland RA. Residence time of receptor–ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481–92. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- 42.Copeland RA, Pompliano DL, Meek TD. Opinion—drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–9. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 43.Lu H, Tonge PJ. Drug-target residence time: critical information for lead optimization. Curr Opin Chem Biol. 2010;14:467–74. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu L, Lin JS, Ma JC, et al. Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob Agents Chemother. 2010;54:689–98. doi: 10.1128/AAC.01152-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W—improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waterhouse AM, Procter JB, Martin DMA, et al. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–91. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]