

Abstract
Type I lissencephaly or agyria-pachygyria is a rare developmental disorder which results from a defect of neuronal migration. It is characterized by the absence of gyri and a thickening of the cerebral cortex and can be associated with other brain and visceral anomalies. Since the discovery of the first genetic cause (deletion of chromosome 17p13.3), six additional genes have been found to be responsible for agyria–pachygyria. In this review, we summarize the current knowledge concerning these genetic disorders including clinical, neuropathological and molecular results. Genetic alterations of LIS1, DCX, ARX, TUBA1A, VLDLR, RELN and more recently WDR62 genes cause migrational abnormalities along with more complex and subtle anomalies affecting cell proliferation and differentiation, i.e., neurite outgrowth, axonal pathfinding, axonal transport, connectivity and even myelination. The number and heterogeneity of clinical, neuropathological and radiological defects suggest that type I lissencephaly now includes several forms of cerebral malformations. In vitro experiments and mutant animal studies, along with neuropathological abnormalities in humans are of invaluable interest for the understanding of pathophysiological mechanisms, highlighting the central role of cytoskeletal dynamics required for a proper achievement of cell proliferation, neuronal migration and differentiation.
Keywords: Agyria–pachygyria, Epilepsy, Neuronal migration, Microtubules, Nucleokinesis, Microtubule-associated proteins
Introduction
Neuronal migration disorders account for a substantial number of cortical malformations, including microcephaly, agyria–pachygyria, heterotopia and polymicrogyria. Original classifications made through neuropathological and clinical descriptions are now supplemented by magnetic resonance nuclear imaging (MRI) and molecular genetics. A few years ago, Barkovich et al. [7] proposed a new classification of cortical malformations based on two additional factors, gene abnormality and timing of the earliest stage of development at which cortical development is affected, leading to the recognition of four major groups. The first group comprises cortical malformations resulting from abnormal neuronal and glial proliferation in the ventricular zone (VZ) and subventricular zone (SVZ) giving rise to abnormal brain size; in the second group, abnormal migration of post-mitotic neuroblasts results in lissencephaly and heterotopias; in the third group, abnormal cortical organization and/or late neuronal migration are responsible for polymicrogyria and schizencephaly; the fourth group is associated with either inherited errors of metabolism or diseases still to be classified.
The term “lissencephaly”, or agyria–pachygyria, was introduced by Owen in 1868 to distinguish the flat “lissencephalic” brains of lower mammalian species or fetuses from the folded “gyrencephalic” brains. This term was then applied to human malformed brains lacking or having a substantial reduction in the number of cortical convolutions by homology to the brain of inferior vertebrates [55]. Though agyria is characterized by a total absence of gyri and sulci, most patients rather present with pachygyria, a more patchy malformation in which the medial and ventral gyri are relatively spared, along with other brain regions, depending on the type of gene and mutation involved [70].
General considerations
Lissencephaly, whose overall incidence is estimated around 1.2/100,000 births, has traditionally been separated into two categories: type I (MIM#607432) or classical type, also named agyria–pachygyria, and type II or cobblestone lissencephaly (MIM#236670). The latter is related, in more than 60% of cases, to a defect of O-glycosylation of extracellular matrix proteins resulting in a defect of the integrity of the pial surface and subsequent overmigration of cortical neurons [16]. In classical lissencephaly, the pia is intact but the normal six-layered organization of the cortex is severely impaired. A few other pathologically distinct forms of lissencephaly have been described, but the genes or the cellular mechanisms involved remain to be identified [46, 52, 99]. Depending on associated defects such as cerebellar hypoplasia, corpus callosum, basal ganglia or white matter defects, classical lissencephaly resulting from mutations in LIS1 (also called PAFAH1B1, platelet-activating factor acetylhydrolase isoform1B, alpha subunit; MIM#601545) or DCX (Doublecortin; MIM#300121) is easily differentiated from lissencephaly-variants due to mutations in ARX (Aristaless-related homeobox gene; MIM# #300382), TUBA1A (Tubulin alpha1A; MIM#602529), WDR62 (WD repeat domain 62), RELN (Reelin; MIM#600514) or VLDLR (very low density lipoprotein receptor; MIM#224050) [12, 17, 37, 65, 85, 97, 101, 137]. Although most forms of lissencephaly have a genetic origin, lissencephalic-like syndromes may also result from environmental insults such as hypoxic–ischemic damage, fetal alcohol syndrome, cocaine intoxication during pregnancy or fetal cytomegalovirus infection, as well as from maternal diseases, in particular, maternal diabetes and phenylketonuria [72, 73].
Clinical presentation
Clinical presentation of lissencephalic syndromes may vary depending on the severity and the molecular basis of the agyria–pachygyria. The most severe syndromes, prenatally detected using ultrasound performed during the second trimester of the pregnancy, show an absence of primary sulci, and are confirmed by antenatal cerebral MRI performed from 30 weeks of gestation.
Most patients present with hypotonia or severe neurological distress at birth. In classical lissencephaly due to LIS1 or DCX mutations, seizures occur in 75% of infants and develop during the first year of life [35, 110]. In the Miller–Dieker syndrome (MDS), clinical symptoms at birth also include facial dysmorphism and visceral abnormalities. Other specific phenotypes are observed exclusively in males, as for example, in the severe X-linked lissencephaly associated with abnormal genitalia (XLAG) due to severe ARX mutations [95, 101]. This form of lissencephaly is typically characterized by respiratory distress at birth, neonatal-onset intractable epilepsy, severe hypotonia from birth, hypothalamic dysfunction with deficient temperature regulation, and chronic diarrhea. External genitalia are ambiguous or underdeveloped. Adducted thumbs may also be present. All patients display cranio-facial dysmorphism including congenital microcephaly, psychomotor delay and most of them die before the age of 18 months [42, 95, 101, 126].
Lissencephalies with cerebellar hypoplasia (LCH) encompass heterogeneous disorders which have been related to mutations in LIS1, DCX, RELN and TUBA1A. In the LCH due to RELN or VLDLR mutations, phenotypic features, besides the classical clinical symptoms, include myopia, nystagmus and lymphoedema [17, 18, 24, 85]. More recently reported, WDR62- and TUBA1A-patients present with congenital microcephaly, neurological disabilities and epilepsy. Additional features such as strabismus and facial diplegia with transient oropharyngoglossal dysfunction responsible for neonatal feeding problems and later drooling have been reported for TUBA1A-patients [5, 104, 121].
The prognosis of lissencephaly is extremely poor since most patients present with post-natal failure to thrive, feeding problems and repeated aspiration pneumoniae which are responsible for a shortened lifespan. Living patients present profound mental retardation, spastic quadriplegia and epilepsy. Status epilepticus may occur in the first post-natal days or later on, but may also be identified during pregnancy. Drug-resistant epilepsy comprises atypical absences, drop attacks, myoclonic, tonic and tonico-clonic seizures, partial epilepsy or West syndrome [44, 75].
Imaging studies
Apart from the absence of cortical circonvolutions, classical lissencephaly is characterized by increased cortical thickness (between 10 and 20 mm instead of 4 mm from birth). In order to help in determining the most likely genetic cause, Dobyns and Truwit have developed a classification considering both the severity of the anomalies and their anterior (a) or posterior (p) predominance: the spectrum varies from complete or near complete agyria (grades 1 and 2) to pachygyria (grade 4) and SCLH (grade 6). Intermediate grades consist of mixed agyria–pachygyria (grade 3) and mixed pachygyria–SCLH (grade 5) [40, 43]. As an example, LIS1 mutations usually give rise to agyria or pachygyria (grades 2–4 of severity), the parietal and occipital lobes being the most severely affected areas (p > a gradient) [21, 43, 131, 143]. On the contrary, lissencephaly due to DCX mutations is more severe in the frontal cortex (a > p gradient) [44, 109, 131]. However, rare males with DCX mutations have also been described with SCLH which is then more severe in anterior parts, and appears as an undulating band of gray matter in the frontal lobes beneath a nearly normal cortex [41]. The variant form of lissencephaly resulting from ARX loss-of-function mutations is characterized by a moderate increase in thickness of the cortex (up to 10 mm) with a p > a gradient associated with constant corpus callosum agenesis, severe congenital or post-natal microcephaly, olfactory hypoplasia and diffusely hypoplastic and dysplastic basal ganglia with cysts and abnormal white matter signals, contrasting with normal infratentorial structures. Ventriculomegaly is mild to moderate with enlarged trigone and posterior horns of the lateral ventricles [9, 42, 95, 101, 118, 126]. TUBA1A mutations are responsible for gyral malformations ranging from pachygyria (p > a gradient) to subtle SCLH. Other features include severe microcephaly, perisylvian pachygyria, dysgenesis of the corpus callosum, disorganization of the hippocampus, absence or hypoplasia of the anterior limb of the internal capsule, characteristic fullness of the striatum and colpocephaly, brainstem and cerebellar hypoplasia which mainly involves the inferior vermis. Very recently, nine patients with mutations in the WDR62 gene have been described. They all have extreme microcephaly, pachygyria, varying degrees of cortical thickening, a loss of gray–white matter junction and hypoplasia of the corpus callosum [12].
Ross and colleagues [142] have proposed a classification of LCH into four different subgroups depending on the severity of cortical, cerebellar and brainstem defects. The first group (LCHa) corresponds to rare mutations in LIS1 (3/4) or DCX (1/4) with a thick cortex and a mild hypoplastic but normally organized cerebellum. The second group (LCHb) is characterized by severe cerebellar hypoplasia and pachygyria with a thin cortex and anterior predominance of defects and results from mutations in RELN. The degree of cortical malformation varies from agyria to simplification of the gyral pattern and from near normal cortical thickness to marked thickening of the cortical gray matter. The cerebellum is small with no foliation and hypoplasia of the cerebellar hemispheres as well as of the inferior vermis. These abnormalities are usually associated with hippocampal dysplasia and hypoplastic brainstem [17, 18, 24, 85, 175]. LCHc includes microcephaly, agyria–pachygyria with severe cerebellar hypoplasia, cleft lip and palate, and a high mortality rate during the perinatal period. The last group, LCHd, corresponds to severe microcephaly, thick cortex, grades 1–2 agyria, diffuse cerebellar hypoplasia of both hemispheres and vermis as well as mild defects of the brainstem. Some patients with TUBA1A mutations belong to this group [5].
Nevertheless, irrespective of the type of lissencephaly (classical or variant) observed, some supratentorial and infratentorial abnormalities remain below the limits of resolution of MRI and are only detected by careful postmortem neuropathological examination (Fig. 1).
Fig. 1.
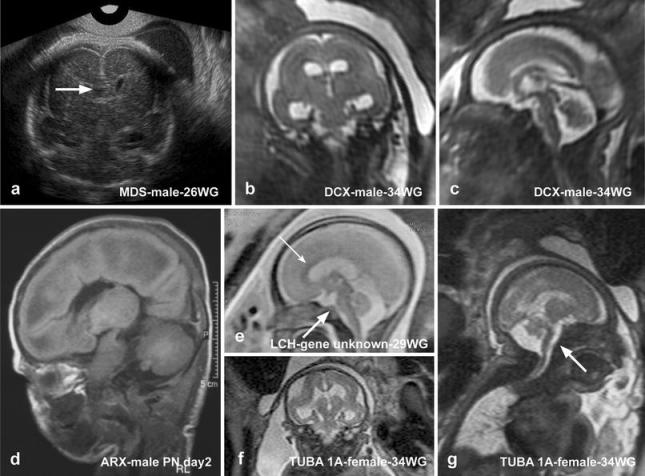
Ultrasonographic and MRI findings in the different type I lissencephaly subtypes: a Ultrasonographic pattern of MDS showing complete agyria with hypoplastic frontal lobes and dysmorphic corpus callosum and septum pellucidum (arrowhead). b, c MRI data in a DCX-mutated fetus displaying lissencephaly with moderate ventricular dilatation but no apparent infratentorial anomalies. d MRI performed at postnatal day 2 in a male newborn with XLAG syndrome, exhibiting pachygyria, more severe in the anterior regions and corpus callosum agenesis but with no infratentorial lesions. e MRI pattern on sagittal plane in a fetus with lissencephaly and cerebellar hypoplasia (genetic cause unknown). The corpus callosum is normal (thin arrow). The vermis is rudimentary and unfoliated, the primary fissure being absent. The brainstem is moderately hypoplastic (thick arrow). f MRI in a TUBA1A-mutated female fetus, showing severe microcephaly and ventriculomegaly. The vermis is hypoplastic especially in its inferior part, but remains foliated (g). The pons and medulla are severely flattened (arrow, g)
Neuropathology of type I lissencephaly
Lissencephaly associated with LIS1 mutations and Miller–Dieker syndrome
The first detailed neuropathological description was reported in 1956 by Crome who introduced the notion of a “four-layered” cortex. Since then, several cases have been reported [34, 39, 52, 55, 70, 78, 90, 107, 139, 168].
On gross examination, the brain weight is either normal or reduced. In the most severe forms, the cerebral hemispheres are smooth, with poorly defined central and Sylvian fissures (Fig. 2a). When present, the gyri are broad, small in number and coarse, with a failure or a delayed operculization of the Sylvian fissure. In less severe cases, primary temporal and occipital fissures may be observed and the hippocampal area, as well as the adjacent cortex, may have a normal gyration. The cerebellum is usually normal, rarely hypoplastic. On coronal sections, the cortical ribbon is thicker than in normal brain (up to 2 cm in width) and poorly delineated from the white matter which is markedly reduced, leading to an inverted proportion between the intermediate zone or white matter and the cortical ribbon (Fig. 2b). In MDS, the internal capsule, the claustrum, the corpus callosum and the cingular gyrus are not identified [39, 124, 168, 169].
Fig. 2.
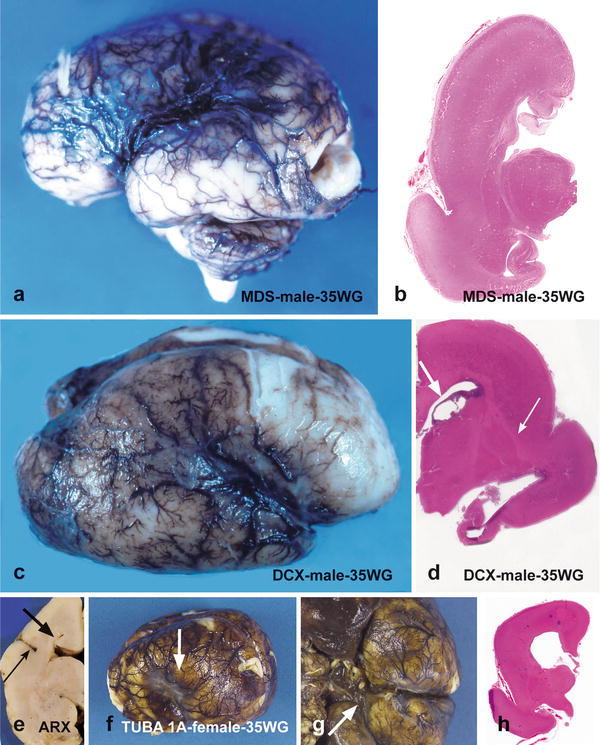
Macroscopic findings in the different type I lissencephaly subtypes: a left lateral view of the brain in MDS reveals complete agyria with rudimentary Sylvian fissure and absent central fissure, contrasting with the absence of macroscopical lesions at the infratentorial level (a). On coronal section passing through the hippocampi, there is no clear delineation between the cortex and the IZ (b). The deep gray nuclei are reduced in volume but normally shaped, the hippocampi are hypoplastic and the posterior arm of the internal capsule appears to be normal (b). A right lateral view of a DCX-mutated brain shows that the external surface is completely agyric, the Sylvian fissure only being observed (c). On coronal section passing through the hippocampi, the cortex is thick and several germinolysis cysts are present (d, thick arrow). The hippocampi are rudimentary, but the deep gray nuclei are not dysmorphic (d, thin arrow). A coronal section passing through the diencephalon in one case of XLAG syndrome reveals short, upwards displaced and dysmorphic Sylvian fissure (e, thin arrow). The corpus callosum is absent, with thick Probst bundles (e, thick arrow). In a left lateral view of TUBA1A-mutated brain, the Sylvian fissure is extremely short and vertically oriented (f, arrow). On the inferior view of the brain, olfactory bulbs are not visible (g, arrow). On coronal section passing through the diencephalon (h), no delimitation between the CP and the IZ is observed. The lateral ventricles are enlarged, with a bulging of the thalamic nuclei into the ventricular cavities (h)
On microscopic examination, the cortical plate exhibits the characteristic four-layered pattern (Fig. 3a). The molecular layer I contains Cajal–Retzius cells located close to the pia. Layer II is composed of densely packed pyramidal neurons which are mainly observed in its upper part, and underlined by an irregular sheet of granular cells (Fig. 3b). Layer III consists of scattered fusiform, rounded or multipolar neuronal elements. Layer IV is particularly poorly demarcated from the underlying white matter and composed of pleiomorphic neuronal cells, with misoriented pyramidal neurons. The reduced white matter contains multiple arrested post-mitotic neuroblasts. The remaining subependymal cell layer is also poorly demarcated from the deep white matter and sometimes contains periventricular heterotopias. In children of over 1 year of age, additional sublayers consisting of sheets of myelinated fibers with a radial arrangement and crossing the deeper layers may be recognized, in particular, in layers II and III [30, 52, 55, 70, 107, 122, 124]. The internal capsule is histologically observed, hypoplastic and irregular in shape. The septum which constantly contains aggregates of microcalcifications in MDS contains microcalcifications in 20% of cases of microdeletions but never in LIS1-mutated brains [39, 107]. The cytoarchitectony of the different hippocampal fields appears to be generally preserved but the dentate gyrus is occasionally distorted or thickened and composed of densely packed granular cells, conversely to the pyramidal cell layer where neuronal density is often decreased (Fig. 4a) [52, 55, 70, 107]. The deep gray nuclei are not dysmorphic in shape and location, even if decreased neuronal density is noted in the striatum [55] and the thalamus. Intense gliosis is dispersed throughout the white matter, and radial glia persists later in the posterior regions.
Fig. 3.
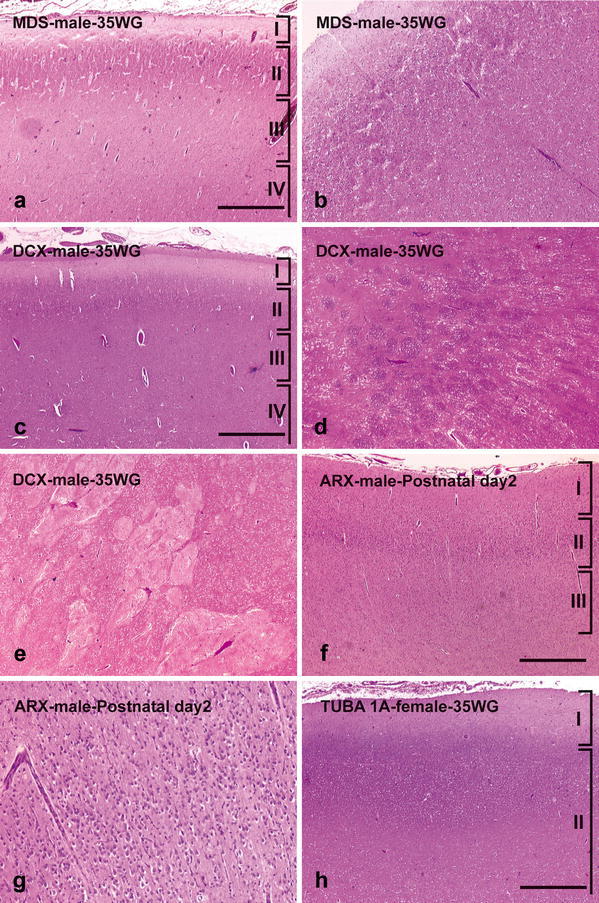
Main histological supratentorial alterations in the different subtypes of type I lissencephaly. In MDS, a characteristic four-layered cortex is observed (a) with the superficial part of the CP exhibiting no external granular cell layer, which is replaced by a paucicellular cell layer made up of pyramidal neurons (b). In a DCX-mutated brain, the superficial part of the CP contains normal layers I, II and III (c), whereas later born neurons and interneurons are arrested in the periventricular areas or the striatum, forming multiple nodules (d) which displace the dysmorphic and fragmented anterior limb of the internal capsule (e). In XLAG syndrome, the three-layered cortex (f) is exclusively composed of pyramidal neurons (g). A TUBA1A-mutated brain shows an unlayered cortex (h). Only layer I is well demarcated from the underlying structures, with a single band of neurons haphazardly scattered in the IZ and SVZ (h). Scale bars 920 μm
Fig. 4.
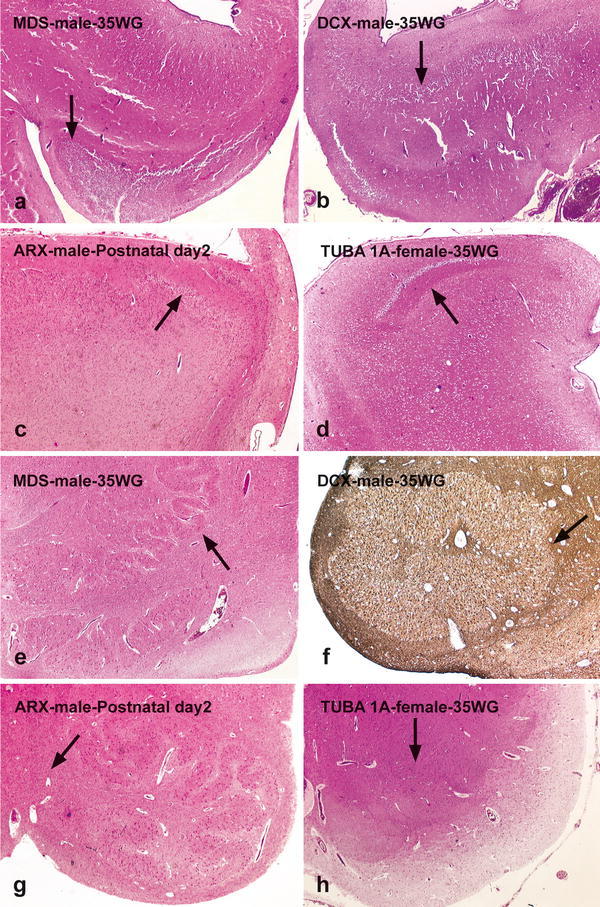
Histological abnormalities in the hippocampus and olivary nuclei of the different subtypes of type I lissencephaly. In MDS, the dentate gyrus is normally shaped but in a heterotopic position (arrow), with a decreased neuronal density in the pyramidal cell layer (a). In DCX-mutated brain, the dentate gyrus is hypoplastic and abnormally shaped (b, arrow). In XLAG syndrome, a rudimentary hippocampus is present with almost indiscernible dentate gyrus (c, arrow) and a pyramidal cell layer composed of small neuron clusters. In TUBA1A-mutated brain, the dentate gyrus is severely hypoplastic and non-convoluted (d, arrow), with extremely hypoplastic pyramidal cell layer. In MDS, the olivary nucleus is normally convoluted but markedly hypoplastic (e, arrow), due to arrested olivary neurons in the medullary dorsal nuclei and fascicles. A DCX-mutated brain shows an olivary pachygyria due to decreased neuronal density and extensive gliosis (f, arrow; GFAP immunostaining). In XLAG syndrome, there is a normal inferior olivary complex with absent cortico-spinal tract decussation (g). A TUBA1A-mutated brain shows hypoplastic and fragmented olivary nuclei (h, arrow)
Histological examination of infratentorial structures reveals diffuse decreased neuronal density of several brainstem nuclei at the mesencephalic and pontine levels [52]. Cortico-spinal tracts may be absent or fragmented and hypoplastic. In the medulla, the inferior olivary complex is particularly hypoplastic or dysplastic (Fig. 4e), with multiple heterotopic supernumerary foci of olivary neurons located in and between the dorsal nuclei and fascicles. The pyramids and their decussation are most often observed. In the cerebellum, multiple foci of Purkinje cells are disseminated in the deep white matter extending toward the middle cerebellar peduncles. Poorly convoluted and slightly fragmented dentate nuclei are often aberrantly located in the vicinity of the ventricular surface [55, 78, 107, 114].
Lissencephaly associated with DCX mutations in males
In contrary to LIS1, morphological descriptions of DCX syndrome are rare. The first neuropathological case to be reported was interpreted as a four-layered cortex similar to the LIS1-mutated cortex with infratentorial anomalies consisting of inferior olivary nuclei pachygyria [8]. In hemizygous DCX-mutated males, the brain surface is agyric or pachygyric, the anterior areas being more severely affected, with hypoplasia of the frontal lobes (Fig. 2c). On coronal sections, the corpus callosum displays various aspects described as thick, thin or absent, with a thickened septum pellucidum [8, 169]. The caudate nuclei are normal in shape and volume. Shortly before birth, several cysts of germinolysis occupy the walls of the lateral ventricules (Fig. 2d). Basal ganglia, internal capsule and claustrum are hardly discernible. The cortical ribbon is thick and poorly demarcated from the intermediate zone (IZ). The cerebellum and the brainstem are macroscopically normal.
On microscopic examination, the cortex displays various anomalies. It has first been described as a vague four-layered cortex coexisting with a band of ectopic neurons of diverse sizes and shapes organized in columns or clusters lying in a reduced rim of white matter, or as two-layered [8, 52, 141]. In fact, apart from layer I, all other layers show a different pattern from LIS1 cortex [52] with an ill-defined but six-layered cortex [114, 169]. Layer II consists of a thin band containing an admixture of pyramidal and granular neurons. Layer III is poorly delimited and paucicellular, made up of granule and immature neurons, and contains more pyramidal neurons than in LIS1-mutated cortex (Fig. 3c). The remaining layers IV, V, and VI are located within the white matter. In the superficial white matter, layer IV forms a thick ribbon of scattered, sometimes clustered pyramidal cells and immature neurons. Layer V is located in the intermediate zone and composed of large nests of arrested migrating neurons, arranged in a radially and columnar pattern [52, 114]. These cells seem to emerge from the layer VI which is formed by the juxtaposition of numerous nodules of varying size with clear-cut boundaries and widely distributed through the deep white matter and the striatum (Fig. 3d). Thalamic and hypothalamic nuclei are diffusely invaded by haphazardly dispersed heterotopic neurons, resulting in the formation of an indistinct cellular mass. The internal capsule is disrupted and displaced by the nodular heterotopias, resulting in the formation of coarse and irregular fascicles separated by nodules and columns (Fig. 3e). The hippocampal formation is hypoplastic (Fig. 4b). At last, as in LIS1-mutated brains, a prominent reactive astrocytic gliosis is observed in the cortex and white matter of the cerebral hemispheres, as well as in the basal ganglia [8, 114].
In the lower brainstem, the inferior olivary complex is pachygyric (Fig. 4f), and conversely to LIS1, ectopic olivary nuclei are infrequently observed. Bilateral and symmetric corticospinal tracts are identified. In the cerebellum, the dentate nuclei are hypoplastic. The main cerebellar anomaly consists in some cases in the presence of dispersed heterotopic Purkinje cells throughout the cerebellar white matter, or arranged in nodules of variable size [8, 52, 114, 169].
XLAG syndrome
The XLAG syndrome has been the first human disorder in which a failure in tangential migration of GABAergic interneurons has been demonstrated. Since the first neuropathological descriptions based on three families, only four additional cases have been reported so far in the literature [15, 52, 118, 128]. In all reported cases, the brain is smaller than in age-matched controls. The external surface is agyric in the occipital and parietal lobes, and pachygyric in the more anterior regions. Olfactory bulbs are absent most of the time. On coronal sections, the cortical ribbon is better delineated from the white matter than in LIS1- and DCX-mutated brains (Fig. 2e). The white matter, which is still not myelinated after 1 year of age, is atrophic. The internal capsule, fornices and corpus callosum are absent. Several diencephalic structures are not identified. The hippocampi are dysplastic and hypoplastic. The lateral ventricles are laterally bordered by thick Probst bundles. The third ventricle is enlarged without narrowing of the aqueduct of Sylvius. The boundaries of the striatum and basal ganglia are not apparent. Interhemispheric or occipital cysts may also be present. No infratentorial lesions are detected on macroscopic examination.
Histological analysis reveals in all cases the characteristic pattern of a three-layered cortex with a global decrease in neuron number affecting massively interneurons (Fig. 3f). The cortical plate is almost entirely composed of pyramidal cells (Fig. 3g). The molecular layer contains a few Cajal–Retzius cells lying under the pia, as well as foci of heterotopic small neurons. Leptomeningeal glioneuronal heterotopias may also be identified. Layer II is composed of numerous densely packed pyramidal neurons, sometimes forming nodules protruding into the molecular layer. Layer III, whose thickness increases in the posterior lobes, is irregular in width and less cellular, and contains small-to-medium-sized neurons, with among them randomly dispersed pyramidal cells of various sizes. The underlying atrophic white matter is diffusely gliotic and contains heterotopic neuronal clusters especially in the frontal lobes, as well as vessels circumscribed by enlarged Virchow–Robin spaces. In the diencephalon, striatal and thalamic structures are disorganized. The caudate nuclei are markedly reduced in size and cellularity, and so are the putamen, the pallidum and the thalami. Most hypothalamic nuclei are not identified and their absence has been correlated with some clinical hallmarks of the disease [118]. The hippocampus is rudimentary, especially the dentate gyrus (Fig. 4f).
Histological examination of the brainstem and cerebellum reveals several abnormalities. The cerebral peduncles and substantia nigra are dislocated, the corticospinal tracts are atrophic and fragmented from the cerebral peduncle level to the lower medulla where their decussation is generally not visualized (Fig. 4g). However, the cerebellum displays no lesion.
Lissencephaly associated with TUBA1A mutations
Mutations in TUBA1A cause a wide spectrum of brain malformations ranging from laminar heterotopia or perisylvian pachygyria to agyria resembling the classical lissencephaly. At the present time, neuropathological descriptions are based on five fetal observations in which prenatal ultrasound and MRI examination revealed severe cortical dysgenesis from the second trimester of the pregnancy [47, 108, 133]. Neuropathological examination showed lesions which, similar to children, involve five brain structures including the neocortex, the hippocampus, the corpus callosum, the brainstem and the cerebellum.
On macroscopic examination, the brain and infratentorial weights are below the 5th percentile. The brain surface is either agyric or pachygyric, with a postero-anterior gradient of severity. Sylvian fissures are missing or are extremely short and vertically oriented (Fig. 2f). The olfactory tracts are usually absent (Fig. 2g). The cerebellum is also hypoplastic and complete or partial agenesis of the vermis may be found. In one of the cases, the inferior part of the cerebellar hemispheres exhibited a micronodular pattern, whereas the cerebellar folia were normal in its superior part [108]. On coronal sections, ventricular dilatation is diffuse, and there is an absence of demarcation between gray and white matter (Fig. 2h). The corpus callosum is described as completely or partially absent, thick, or thin. Probst bundles are infrequently observed. Basal ganglia are abnormally oriented.
Histological examination of the cerebral hemispheres reveals a quasi absence of lamination forming a two-layered cortex (Fig. 3h). Layer I contains scarce and misplaced Cajal–Retzius cells, and is underlined by a single band of neurons extending from the inferior limit of the marginal zone to the periventricular zone. Nevertheless, histological examination makes it possible in almost all cases to distinguish a roughly ordered six-layered cortical plate in the less affected areas, including a layer II with a poor density of immature, often misoriented or inverted pyramidal neurons, and a paucicellular layer III consisting of scattered granular cells. The deepest layers consist of numerous and haphazardly dispersed immature neuronal cells filling the IZ and SVZ, occasionally with a nodular or columnar arrangement. In the hippocampus, the dentate gyrus is absent, hypoplastic or discontinuous and not properly folded. The pyramidal cell layer of the Ammons’ horn is thick or hypoplastic (Fig. 4d). In the parietal and occipital cortices, cortical lamination is more apparent. Basal ganglia are hypo- and dysplastic, sometimes asymmetric and most often fragmented, in particular, the pallidum. Thalamic nuclei appear prominent and displaced, and contain an indistinct mass of neurons. Caudate and hypothalamic nuclei are dramatically reduced in size and shape. The internal capsule is hypoplastic and fragmented with aberrantly directed fascicles, or not identified. Anterior commissure, fornices and septum pellucidum may be missing. In the mesencephalon, the pons and the medulla, neurons are most often scattered in the neuropil, with hardly any organization. Neuronal density of the pontine nuclei is globally decreased. Most of the brainstem nuclei are indiscernible. Corticospinal tracts are hypoplastic and aberrantly placed. In the medulla, olivary nuclei are agyric and sometimes reduced to small foci of immature neuronal cells (Fig. 4h), the vast majority of them being arrested in the dorsal part of the medulla and of the pons, as well as in the inferior cerebellar peduncles. In the cerebellum, dentate nuclei are also pachygyric and fragmented, forming scattered and small nests of neurons in the white matter. In the cerebellar cortex, Purkinje cells are reduced in number, some of them being arrested in the cerebellar white matter, arranged in small clusters or in streaks intermingled with hypoplastic deep cerebellar nuclei [108].
To summarize, type I lissencephaly which constitutes a primary defect in neuronal migration during development and which has traditionally been considered as a disorder of radial migration also results from non-radial migration impairment (Fig. 5). Nevertheless, genes implicated in type I lissencephaly are not only crucial for appropriate migration toward the cortex but also for other developmental events including cell proliferation, neuronal differentiation, axonal pathfinding and transport, as well as connectivity and myelination. Irrespective of the gene involved, neuropathological features argue for more complex underlying pathogenic mechanisms and are summarized in Table 1.
Fig. 5.
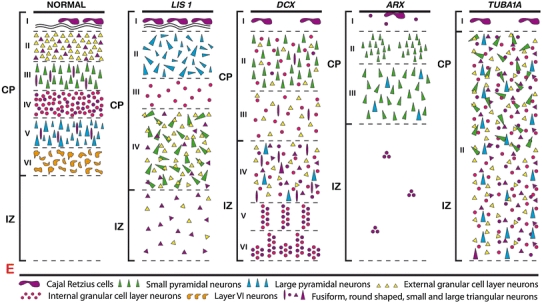
Schematic representation of the localization of different neuronal populations in the cerebral mantle of normal and lissencephalic brains, highlighting the differences of cortical lamination defects. CP cortical plate, IZ intermediate zone, E ependyma
Table 1.
Morphological lesions in supra- and infratentorial structures in the different types of classical lissencephaly resulting from tangential versus radial migration and from abnormalities of differentiation, including axonal pathfinding, connectivity and myelination
| Brain structure | Gene | Radial (R)/tangential (T) migration abnormalities | Differentiation deficits |
|---|---|---|---|
| Cortex | LIS1 | Transient external subpial layer persistence (T) | Immature cortical neurons |
| Absent lamination, four-layered cortex, absence of external granular cell layer (R) | |||
| CR+ interneuron decrease in the upper layers (R/T) | |||
| Heterotopic CR+ interneurons in layer IV, IZ and VZ (T) | |||
| DCX | Scattered Cajal–Retzius cells in layer I (T) | ||
| Two- or four-layered or ill-defined six-layered cortex (R/T) | |||
| ARX | Three-layered cortex, pyramidal type exclusively (T) | ||
| Absence of CR+ interneurons (T) | |||
| Absence of layers II and IV (R/T) | |||
| Rare dispersed Cajal–Retzius cells in layer I (T) | |||
| TUBA1A | Scattered Cajal–Retzius cells in layer I (T) | Immature cortical neurons | |
| Two-layered cortex or unlayered (R/T) | |||
| Absence of layer II (R/T) | |||
| CR+ interneuron decrease in the cortex (R/T) | |||
| Misoriented neurons (R) | |||
| Hippocampus | LIS1 | Hypoplastic (R/T) | |
| DCX | Disorganized, dysplastic (R/T) | ||
| ARX | Rudimentary, dysplastic (R/T) | ||
| TUBA1A | Hypoplastic and dysplastic (R/T) | ||
| IZ/WM | LIS1 | Persistent radial glia (R) | Internal capsule hypoplasia |
| Multiple arrested neuronal cells (R) | Corpus callosum and septum pellucidum abnormalities | ||
| Periventricular heterotopias (R/T) | Immature neurons | ||
| Arrested CR+ and CB+ interneurons (T) | |||
| Premature disappearance of EGL (T) | |||
| DCX | Layers IV, V and VI in the deep WM (R) | Dysmorphic corpus callosum | |
| Columnar and nodular periventricular heterotopias (R/T) | Immature neurons- disorganized dendritic network | ||
| CR+ and CB+ interneurons in the IZ/WM and SVZ (T) | |||
| ARX | Dispersed neurons or in clusters in the IZ/WM (R) | Absent olfactory bulbs | |
| Rare CR+ cells in the IZ/SVZ (T) | Agenesis of the corpus callosum | ||
| Heterotopic immature neurons in the IZ | |||
| TUBA1A | Heterotopic pyramidal neurons and interneurons (dispersed or in clusters) (R/T) | Absent olfactory tracts and bulbs | |
| Persistent radial glia (R) | Agenesis of the anterior commissure | ||
| Thin optic nerves and chiasm | |||
| Dysplastic corpus callosum | |||
| Absence, hypoplasia or fragmentation of the internal capsule. Immature neurons in the cortex | |||
| Basal ganglia | LIS1 | Decreased neuronal density in the striatum, pallidum and thalami (R/T) | |
| CR+ and CB+ interneuron decrease in the striatum (T) | |||
| DCX | Fragmentation of the striatum by nodular and columnar heterotopias (R/T) | ||
| ARX | Absent diencephalic nuclei, quasi absence of CR+ and CB+ interneurons in the striatum (R/T) | ||
| TUBA1A | Hypoplastic and dysplastic hypothalamic nuclei and basal ganglia (R/T) | ||
| Cerebellum | LIS1 | Heterotopic Purkinje cells in the WM (R) | Axonal swellings in the WM |
| Fragmented dentate nuclei (T) | |||
| DCX | Heterotopic Purkinje cells (R) | Axonal swellings in the WM | |
| Dentate nuclei hypoplasia (T) | |||
| ARX | Normal | ||
| TUBA1A | Purkinje cell migration abnormalities (R) | Axonal swellings in the WM | |
| Dentate nuclei pachygyria (T) | Dystrophic Purkinje cell dendritic trees | ||
| Brainstem | LIS1 | Neuronal density decrease in the brainstem nuclei (R) | Hypoplastic corticospinal tracts |
| Heterotopic olivary neurons (T) | Decussation abnormalities | ||
| DCX | Neuronal density decrease in the brainstem nuclei (R) | ||
| Dysplastic brainstem nuclei (R) | |||
| Olivary nuclei pachygyria (T) | |||
| ARX | Substantia nigra abnormalities (R) | Corticospinal tract hypoplasia | |
| Decussation abnormalities | |||
| TUBA1A | Quasi absence of cranial nerve nuclei (R) | Corticospinal tract hypoplasia, absence or dysplasia | |
| Neuronal density decrease in the pontine nuclei (T) | Brainstem hypoplasia | ||
| Hypoplastic and heterotopic olivary nuclei (T) |
Molecular basis of type I lissencephalies
LIS1
LIS1, located at 17p13.3, consists of 11 exons and encodes an atypical microtubule-associated protein (MAP) characterized by the presence of 7 WD40 repeat domains [144]. LIS1 haploinsufficiency resulting either from intragenic mutations or deletions encompassing LIS1 alone generally causes isolated lissencephaly and more rarely SCLH or LCH [22, 111, 113, 132, 143, 153, 166]. Larger deletions encompassing LIS1 and the neighboring genes are responsible for MDS [19, 42]. Interestingly, large duplications encompassing the region of the MDS are responsible for mild to moderate mental retardation, hypotonia and dysmorphism but without lissencephaly [140].
To date, 70 intragenic heterozygous mutations of the LIS1 gene have been described [22, 143, 166]. They usually occur de novo and familial recurrences are rare with an autosomal dominant mode of inheritance [22]. The majority of mutations are truncating mutations (84%) whereas missense mutations distributed along the whole coding sequence account for 16% of the mutations only (Fig. 6). No evident genotype–phenotype correlations have been found: neither the nature (missense/truncating) nor the position of the mutation is correlated with the clinical severity, the degree of agyria or cortical thickness [143, 166]. Clinical presentation only correlates with the degree of agyria and cortical thickening. Intragenic LIS1 mutations or genomic rearrangements are identified in 85–87% of the patients affected with classical lissencephaly with a p > a gradient and therefore constitute the most frequent cause of this form of lissencephaly [80, 117]. However, somatic mosaicism which has been estimated as high as 14% might hamper the detection of LIS1 mutations [166].
Fig. 6.
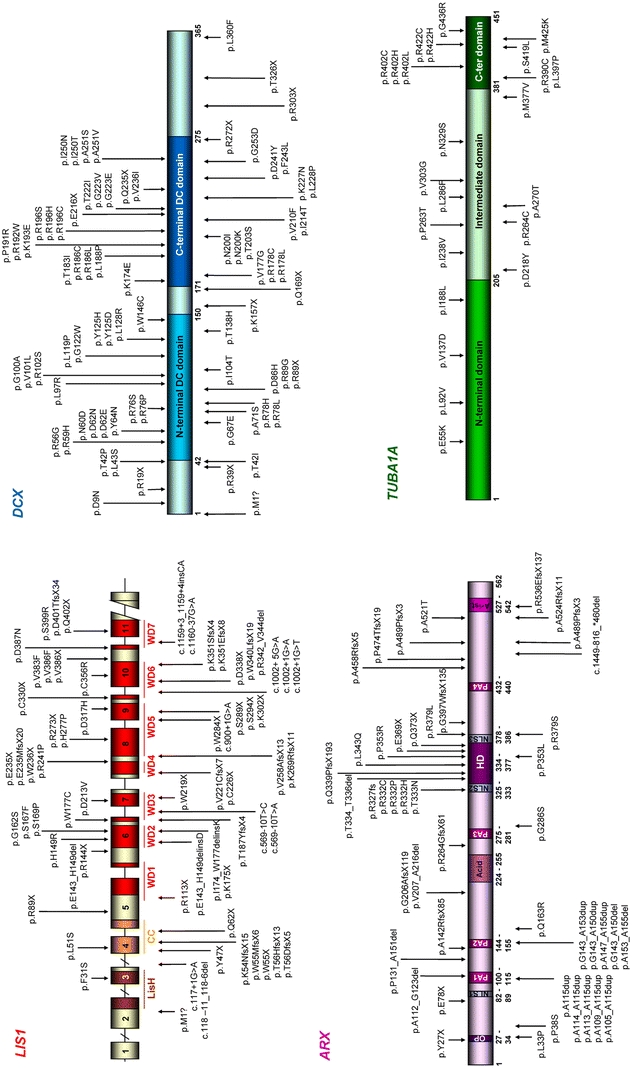
Summary of the mutations identified in DCX, LIS1, TUBA1A and ARX: for each gene, mutations are shown on a schematic representation of the protein, except for LIS1 which represents the gene since a number of mutations are localized outside the exons. Exonic mutations are indicated at protein level in order to observe the predicted consequences of the mutations on the protein and intronic mutations are indicated at nucleotidic level as no protein effect can be predicted. For ARX, mutations figured above the protein are responsible for malformation phenotypes (group 1, see text) whereas mutations shown below are responsible for mental retardation with or without epilepsy (group 2). The accession numbers used for the nomenclature are: for DCX cDNA, NM_178152.1; for LIS1 cDNA, NM_000430.3; for TUBA1 cDNA, NM_006009.2; for ARX cDNA, NM_139058.2
LIS1 was initially identified as the non-catalytic subunit of platelet-activating factor acetylhydrolase (PAF-AH), a heterotrimeric enzyme that inactivates the lipid messenger PAF [79]. In addition, Lis1 was shown to be associated with microtubules [144] and is present in regions of high microtubule density in cells, especially at the centrosome and microtubule-organizing centre (MTOC) [48, 155]. Several studies in different organisms have revealed that Lis1 is part of a conserved evolutionary pathway that regulates cytoplasmic dynein, the main cytoplasmic microtubule-minus end-directed motor protein, involved in several cellular processes such as vesicle transport, nuclear migration and organization of the mitotic spindle (see for reviews [76, 167, 172]) (Fig. 7).
Fig. 7.
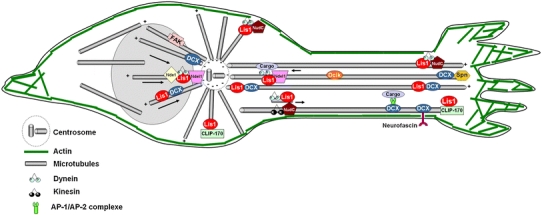
Schematic representation of the localization and role of Dcx, Lis1 and their protein partners in neuronal migration. During migration, the centrosome, which is normally positioned in front of the nucleus, moves into the leading process, rapidly followed by the translocation of the nucleus toward the centrosome. Lis1 associates physically with Nde1, Ndel1 and mNudC, a tyrosine kinase, which all, in turn, interact with cytoplasmic dynein. Lis1 also binds to other proteins involved in the microtubule network, including CLIP-170, which is usually localized at the plus end of growing microtubules. Lis1, Dcx and their protein partners play a major role in nucleokinesis by coupling the centrosome with the nucleus. Lis1 and Dcx are also found at the extremities of neuronal processes where they potentially regulate their stabilization as well as axonal elongation and cell adhesion
DCX
The DCX gene, which encodes Doublecortin, was identified in Xq22.3 [37, 65, 156]. Hemizygous DCX mutations in males lead to classical lissencephaly whereas heterozygous mutations in females lead to SCLH [36], which is probably due to random inactivation of one of the X chromosomes resulting in two genetically distinct populations of cells that segregate in a cell-autonomous fashion. Hence, affected females display a mosaic phenotype that is intermediate in severity characterized by epilepsy and mild to moderate intellectual disability, usually correlating with the thickness of the band and the degree of pachygyria [67, 74, 111]. The DCX gene contains seven exons and mutations have been described in approximately 38% of males with classical lissencephaly and 85% in females with SCLH [3, 37, 65, 68, 109, 115, 132, 145]. In contrast to LIS1, missense mutations of DCX are more common than truncating mutations, since they represent nearly 80% of the mutations [109].
The Doublecortin protein is characterized by the presence of two evolutionary conserved tandem repeats which define a new family of MAP and constitute microtubule-binding modules [53, 66]. Each of these two domains binds to tubulin but both repeats are necessary for microtubule polymerization and stabilization [25, 86, 119, 120]. Mutations are clustered in the two microtubule-binding domains and most of them have been shown to impair microtubule polymerization both in vitro and in vivo [145, 159] (Fig. 6). Missense mutations in the N-terminal microtubule-binding domain have been suggested to cause a more severe lissencephaly phenotype compared with those located in the C-terminal domain [109], probably due to their different properties: the N-terminal domain of DCX binds only to assembled microtubules, whereas the C-terminal domain binds to both microtubules and unpolymerized tubulin [100].
Intragenic deletions encompassing one or more DCX exons have also been identified [80, 117]. Therefore, the screening for partial deletions and duplications is necessary to complete sequencing analysis to ensure an accurate molecular diagnosis of this condition. In addition, a high rate of mosaicism has been reported [3, 68], which complicates the genetic counseling in affected families, and a significant recurrence risk exists even if the mother is not a carrier.
ARX
The ARX gene, located at Xp22.13, was identified in 2002 as a novel homeobox transcription factor with striking homology to the Drosophila gene aristaless. In addition to a prd-type homeodomain, ARX contains four polyalanine tracts, an octapeptide and a C-terminal aristaless domain. Mutations in ARX were first identified in non-syndromic X-linked mental retardation (XLMR) [11] and in X-linked West syndrome [95, 157]. Since then, more than 60 distinct mutations have been described and broadened the spectrum of phenotypes, ranging from severe developmental abnormalities of the brain to syndromic and non-syndromic forms of XLMR with intrafamilial and interfamilial variability (reviewed in [61, 64, 151]) (Fig. 6). The resulting phenotypes can be divided into two groups. The first group includes XLAG [42, 101, 126], hydranencephaly with abnormal genitalia (MIM#300215) and Proud syndrome (MIM#300004), characterized by XLMR, agenesis of the corpus callosum and abnormal genitalia [95]. The second group gathers together phenotypes with mental retardation, epilepsy and dystonia without cerebral or visceral malformations (reviewed in [61, 64, 151]). Phenotype/genotype correlation studies have suggested that truncating mutations or missense mutations within the homeobox DNA-binding domain lead to the more severe phenotypes whereas missense mutations outside the homeobox or expansions/deletions of polyalanine tracts lead to phenotypes without any malformations (Fig. 6) [95, 150].
Arx knock-out mice display a phenotype very similar to the human XLAG [101]. Studies of the role of Arx in these mice as well as in different other models have indicated that it is essential for important developmental processes such as regionalization, proliferation, cell differentiation and neuronal migration (see for review [58]).
TUBA1A
In 2007, Keays et al. [97] reported the identification of a mouse mutant with abnormal laminar architecture of the hippocampus and cerebral cortex resulting from impaired radial neuronal migration. The causative mutation was identified in the guanosine triphosphate (GTP) binding pocket of one of the α-tubulin gene (tuba1a). The similarity of anatomical and behavioral phenotypes between these mice and the Lis1 [51] and Dcx knockout mice [29, 94] led these authors to screen a cohort of patients with brain anomalies and identified several heterozygous de novo mutations in the TUBA1A gene (previously called TUBA3), located at 12q13.12 [5, 97, 133].
All the 38 TUBA1A mutations detected so far are de novo heterozygous missense mutations (Fig. 6). Four recurrent mutations were described: c.790C>T (p.R264C), c.1204C>T (p.R402H), c.1205G>A (p.R402C) and c.1265G>A (p.R422H). Interestingly, three mutations affect the same amino acid, the arginine in position 402: c.1204C>T (p.R402H), c.1205G>A (p.R402C) and c.1205G>T (p.R402L) [5, 97, 104, 121, 133]. Genotype–phenotype correlation studies reveal that p.R402C causes a phenotype similar to LIS1 deletions or intragenic mutations, whereas p.R402H resembles MDS. In contrast, the effects of the other mutations differ dramatically from the LIS1-associated phenotype, with more variable severity, a > p lissencephaly and hypoplasia of the corpus callosum and cerebellum, and in some cases LCHc or d [104]. TUBA1A mutations account for 1–4% of lissencephaly and are therefore considered as a rare cause of lissencephaly [104, 121].
Functional studies of some of these mutations have suggested that several of them lead to defective interactions in the tubulin heterodimer assembly pathway whereas others may alter TUBA1A three-dimensional conformation and/or compromise its interaction with MAPs or microtubule motors such as kinesin [104, 160, 161]. Other tubulin genes that are highly expressed during central nervous system development represent candidate genes for agyria/pachygyria. More recently, two studies have implicated three other tubulin genes (TUBB2B, TUBA8 and TUBB3) in bilateral asymmetrical polymicrogyria, polymicrogyria with optic nerve hypoplasia and a spectrum of disorders including polymicrogyria with pontocerebellar hypoplasia, respectively [1, 89, 134, 162]. These findings further emphasize that disruption of microtubule dynamics seems to underlie a large spectrum of neuronal migration disorders including lissencephaly, pachygyria and polymicrogyria [88].
WDR62
Very recently, five recessive missense and truncating mutations have been described in the WDR62 gene in nine patients from different consanguineous marriages, using a whole-exome sequencing approach. These mutations result in microcephaly, pachygyria with cortical thickening and hypoplasia of the corpus callosum [12].
WDR62 has been shown to be enriched in neural progenitors of the VZ and SVZ of mouse and human fetal brain [12]. Although the function of this gene is not yet known, it has been shown to localize in the nucleus, but not at the centrosome like other known microcephaly genes, and interacts with c-Jun N-terminal kinase (JNK), thus potentially playing a role in the mitogen-activated protein kinase (MAPK) signaling cascade [12, 171]. More studies are required to assess the frequency of WDR62 mutations in cortical malformations and fully understand its role in corticogenesis.
RELN and VLDLR
To date, only two splice-site mutations in the RELN gene have been identified in two consanguineous families with an autosomal recessive form of lissencephaly with cerebellar hypoplasia and defects of the hippocampus and brainstem [24, 85]. The human phenotype is reminiscent of what is observed in homozygous reeler mice, which display ataxia, cerebellar hypoplasia with a decreased number of Purkinje cells, inverted layering of the cortex and abnormal axonal connectivity due to abnormal neuronal migration (see for review [33]). An additional consanguineous pedigree has been reported with an homozygous apparently balanced reciprocal translocation, t(7;12)(q22;p13) disrupting the RELN gene at 7q22.1 [175]. As only three consanguineous families have been described, the clinical phenotype might be wider, and to our knowledge, no neuropathological description is presently available. Patients display hypotonia, nystagmus, severe mental retardation and generalized epilepsy [85].
Reelin encodes a large glycoprotein that controls cell–cell interactions critical for cell positioning in the brain [31, 83, 127]. Secreted by Cajal–Retzius cells, Reelin acts on migrating cortical neurons by binding to Vldlr and Apoer2 (apolipoprotein E receptor 2) receptors [31, 32, 82, 163], and triggers off the recruitment of a cytoplasmic adaptor molecule, mDab1, to the receptors and its phosphorylation by non-receptor tyrosine kinases of the Src-family: Src and Fyn [4, 14, 92, 105] (Fig. 8). Mutations in mDab1 exhibit a phenotype very similar to the reeler one, further supporting the notion that these proteins participate in the same pathway [87, 149, 170]. More recently, a homozygous deletion encompassing VLDLR has been reported in affected individuals with non-progressive ataxia, mental retardation, inferior cerebellar hypoplasia and mild gyral simplification in the Hutterite population [17, 18]. As VLDLR is part of the Reelin signaling pathway, it is not surprising that these two genes result in similar phenotypes.
Fig. 8.
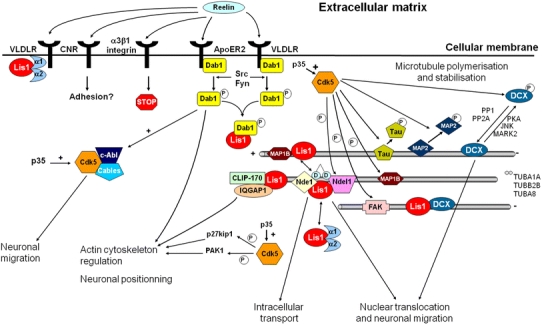
Schematic representation of the possible relationships existing between the different genes involved in lissencephaly type I. A potential common link between all the pathways involved in neuronal migration defects may be the Cdk5 kinase. Targeted disruption of cdk5 or of its neuronal-specific activator p35 in mice induces a cortical phenotype very similar to the one observed in reeler, with an inversion of the normal inside-out lamination. Several molecular targets of Cdk5 which are involved in cytoskeletal dynamics in migrating neurons have been identified, including Dcx, Ndel1, Tau and MAP1B. Dcx phosphorylation by Cdk5 weakens its interaction with microtubules and appears to be critical for migration. Similarly, phosphorylation of Ndel1 by Cdk5 regulates its subcellular localization and association with the dynein complex
Alterations of cytoskeletal dynamics in type I lissencephaly
Defects of neuron generation and/or cell proliferation
In most cases of type I lissencephaly, the brain is of small size, suggesting some defects in neuron generation and/or cell proliferation. Accordingly, Lis1 has been found to be required for interkinetic nuclear migration and cell division of cortical VZ neuroblasts [164], through the regulation of the mitotic spindle orientation which determines the cleavage plane [154, 174]. In addition, mutant mice for Lis1 or Nde1 show a thinner cortex [50, 84] and human neural precursors from a fetus with MDS demonstrated diminished rates of cell proliferation and increased cell death [148]. As the timing of the last division affects the final destination of a cortical neuron, it is thus possible that the altered distribution of neurons observed in human lissencephaly might also result from a defect in cell division [130].
Similarly, in utero electroporation in mice to knock-down or overexpress Arx in the developing cortex show that Arx is important to maintain progenitor proliferation [60]. These results are in agreement with the microcephaly observed in the cortex of Arx knockout mice [101] and human patients with XLAG [42, 95, 101, 126].
Unlike Lis1, Arx or Wdr62, Dcx and Tuba1a proteins are described as specifically expressed in post-mitotic migrating and differentiating neurons during development [26, 53, 66, 69]. Regarding Tuba1a, it is conceivable that the microcephaly phenotype may be the result of cell death either due to abnormal neuronal migration or due to a failure of differentiation, and, in particular, of synaptogenesis and synapse remodeling, responsible for a defect of connectivity between cells. Otherwise, a study has recently described spindle orientation abnormalities similar to Lis1 mutants in radial glial cells of Dcx knock-out mice, which, in turn, lead to moderate proliferation defects [135], suggesting that Dcx may play a critical role in neurogenesis through unknown mechanisms.
Alteration of cytoskeletal dynamics results in defects of neuronal migration
Consistent with an important role for Lis1 in neuronal migration, mice with decreased levels of Lis1 exhibit dose-dependent disorganization of cortical layers, hippocampus, cerebellum and olfactory bulbs [20, 62, 84]. In vitro analyses of heterozygous cerebellar granule neurons show slower migration and display a lengthening of the distance between the centrosome and the nucleus, suggesting that Lis1 is required for nuclear movement by coupling the nucleus with the centrosome [13, 84, 158]. Similarly, cell-autonomous reductions of Lis1, Ndel1 or dynein by RNAi also affect neuronal migration in vivo by affecting soma translocation [152, 164, 165]. A few studies have also reported that Lis1 plays a role in cortical interneuron migration in both mouse and human [114, 116, 123, 129].
The role of Dcx in radial neuronal migration has also been extensively studied in both mouse and rat models [6, 29, 38, 94, 103, 136]. Tangential migration defects have also been reported more recently for olfactory bulb interneurons migrating from the SVZ to the olfactory bulb through the rostral migratory stream, and cortical interneurons which migrate from the medial ganglionic eminences [59, 93, 102]. The role of Dcx in neuronal migration is not very well understood yet. Dcx is supposed to function with Lis1 and dynein to mediate nucleus–centrosome coupling [158]. Overexpression of Dcx or Lis1 increases migration rates and Dcx overexpression can rescue the coupling defect caused by reduced levels of Lis1 or by dynein inhibition. Accordingly, both physical and genetic interactions between Lis1 and Dcx have been reported [23, 135].
In the developing cortex, Arx is expressed in tangentially migrating interneurons emanating from the GE, but not in radially migrating neurons. The strong degree of colocalization between Arx and GABA as well as the absence of interneurons documented in the cortex of XLAG patients [15, 52, 114, 128] and in Arx mutant mice [27, 101] have led to the idea that XLAG syndrome was the result of defective tangential migration [96]. In addition, Arx expression in cortical progenitors has been shown to be critical for radial migration [60]. RNAi-mediated inactivation of Arx in cortical progenitors results in decreased motility of radially migrating neurons, with an accumulation of round cells in the SVZ/IZ, with very few or no processes [60]. Similar cell morphology defects have been reported following inactivation or gain-of-function of proteins which interact with the cytoskeleton or are involved in neuronal polarity [81], suggesting that Arx may play a role in cell morphology through the cytoskeleton regulation.
Descriptions of the phenotype of the mutant mice [97] or human adult and fetus cases with TUBA1A mutations strongly suggest that, similar to LIS1 and DCX, this gene is critical for neuronal radial and tangential migrations [5, 47, 108, 121, 133]. Similarly, the role of TUBB2B in neuronal migration has been demonstrated by both neuropathological observations of neuronal heterotopias in the hemispheric and cerebellar white matter and the arrest of migrating neurons in the IZ following RNAi-mediated inactivation of tubb2b [89].
The commonly accepted explanation for the inverted cortex in reeler mice is that in the absence of Reelin, the newly arrived neurons fail to penetrate the preplate. As a result, the preplate is not split into the marginal zone and the subplate and newly born neurons cannot bypass previously deposited neurons and thus accumulate in an outside-in manner (see for review [18, 138]). Several studies have shown that, in particular, mDab1 is necessary for neurons to cross previously deposited neuronal layers and that, in turn, mDab1 should be down-regulated in neurons to properly terminate their migration and allow their siblings to bypass them [49, 77]. In addition to ApoER2 and VLDLR, Reelin has also been found to bind to α3β1-integrin [45] and protocadherins [147]. The relative importance and interactions of these Reelin receptors are currently unknown. α3β1-Integrin has been shown to be involved in the interaction between the migrating neuron and radial glia, and binding of Reelin induces detachment of neurons which stop migrating [45]. All available data have recently led to the “detach and go” model, in which Reelin may stimulate both the detachment from radial glia and the translocation of the cell soma to the top of the developing cortical plate [28].
Alteration of the cytoskeleton results in defects of neuronal differentiation
Several studies have demonstrated a role for dynein in the regulation of intracellular transport, suggesting that Lis1 may participate in the retrograde transport of vesicles and organelles from neuronal processes toward the nucleus. Atypical swellings have been reported in axons of Drosophila Lis1-deficient neurons [112] and in Purkinje cell dendritic trees of a fetus with MDS [114] suggestive of axonal transport abnormalities. More recently, it has been proposed that the Lis1/Ndel1/dynein complex also influences the transport of plus-end-directed microtubules (anterograde transport) to the periphery of the migrating neuron, hence aiding the extension of the leading process [173] (Fig. 7). Similarly, Dcx has been reported to promote neuronal intracellular transport [38], probably by providing stable tracks for the plus-end-directed kinesins [120] and may be involved in clathrin-vesicular trafficking [56]. It is not yet clear whether all these functions are related to neuronal migration and axonal growth but it is coherent to think that defective vesicle transport during development may result in impaired membrane expansion at the leading process.
Most cases of type I lissencephaly display corpus callosum defects, which may result from impaired axonal elongation due to abnormal cytoskeletal dynamics. For example, Lis1 is necessary to stabilize polymerized microtubules in new branches [71] and in Lis1 knock-out mice, tangentially migrating cells exhibit a longer leading process and a reduced number of branches [123]. In addition, Dcx is enriched at the extremities of neuronal processes [54, 57, 63, 146], where microtubules play important roles in regulating process elongation and movement direction (Fig. 7). Similar to Lis1, a role in leading process stabilization or organization through the formation of stable microtubules has been described for Dcx [10, 38, 59, 94, 103, 158]. Descriptions of the phenotype of the mutant mice [97, 101] or human cases with ARX or TUBA1A mutations also strongly suggest that, similar to LIS1 and DCX, these two genes are involved in multiple important developmental processes including neuronal differentiation, axonal growth, protein transport and synaptogenesis.
Finally, several recent studies have reported abnormal network activity and synaptogenesis in the hippocampus and cortex of Dcx and Lis1 mutant mice, thus providing useful models to study seizures and the altered cognitive function in type I lissencephaly [2, 51, 91, 98, 106, 125].
Concluding remarks
All the genes which are implicated in classical lissencephalies have been shown to regulate both radial and tangential migration as well as cell proliferation and differentiation. Their products mediate a wide range of cellular functions including signal transduction, cell adhesion and motility but somehow or other they seem to regulate actin cytoskeleton or microtubule dynamics (Fig. 7). The observation that genes encoding two MAPs (LIS1 and DCX) and at least three tubulin isoforms (TUBA1A, TUBB2B, TUBB3) are responsible for lissencephaly/pachygyria disorders emphasizes the role of microtubule dynamics in the pathophysiology of type I lissencephaly.
Acknowledgments
The authors wish to thank Esther Le Roy for the iconography, Robert Faure for his help in editing the manuscript and apologize to colleagues whose work is not cited in this review for a reason of limited space.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Abdollahi MR, Morrison E, Sirey T, et al. Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am J Hum Genet. 2009;85:737–744. doi: 10.1016/j.ajhg.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackman JB, Aniksztejn L, Crepel V, et al. Abnormal network activity in a targeted genetic model of human double cortex. J Neurosci. 2009;29:313–327. doi: 10.1523/JNEUROSCI.4093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aigner L, Uyanik G, Couillard-Despres S, et al. Somatic mosaicism and variable penetrance in doublecortin-associated migration disorders. Neurology. 2003;60:329–332. doi: 10.1001/archneur.60.3.329. [DOI] [PubMed] [Google Scholar]
- 4.Arnaud L, Ballif BA, Forster E, Cooper JA. Fyn tyrosine kinase is a critical regulator of disabled-1 during brain development. Curr Biol. 2003;13:9–17. doi: 10.1016/S0960-9822(02)01397-0. [DOI] [PubMed] [Google Scholar]
- 5.Bahi-Buisson N, Poirier K, Boddaert N, et al. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J Med Genet. 2008;45:647–653. doi: 10.1136/jmg.2008.058073. [DOI] [PubMed] [Google Scholar]
- 6.Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- 7.Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65:1873–1887. doi: 10.1212/01.wnl.0000183747.05269.2d. [DOI] [PubMed] [Google Scholar]
- 8.Berg MJ, Schifitto G, Powers JM, et al. X-linked female band heterotopia-male lissencephaly syndrome. Neurology. 1998;50:1143–1146. doi: 10.1212/wnl.50.4.1143. [DOI] [PubMed] [Google Scholar]
- 9.Berry-Kravis E, Israel J. X-linked pachygyria and agenesis of the corpus callosum: evidence for an X chromosome lissencephaly locus. Ann Neurol. 1994;36:229–233. doi: 10.1002/ana.410360216. [DOI] [PubMed] [Google Scholar]
- 10.Bielas SL, Serneo FF, Chechlacz M, et al. Spinophilin facilitates dephosphorylation of doublecortin by PP1 to mediate microtubule bundling at the axonal wrist. Cell. 2007;129:579–591. doi: 10.1016/j.cell.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bienvenu T, Poirier K, Friocourt G, et al. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum Mol Genet. 2002;11:981–991. doi: 10.1093/hmg/11.8.981. [DOI] [PubMed] [Google Scholar]
- 12.Bilguvar K, Ozturk AK, Louvi A, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010;467:207–210. doi: 10.1038/nature09327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bix GJ, Clark GD. Platelet-activating factor receptor stimulation disrupts neuronal migration In vitro. J Neurosci. 1998;18:307–318. doi: 10.1523/JNEUROSCI.18-01-00307.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bock HH, Herz J. Reelin activates SRC family tyrosine kinases in neurons. Curr Biol. 2003;13:18–26. doi: 10.1016/S0960-9822(02)01403-3. [DOI] [PubMed] [Google Scholar]
- 15.Bonneau D, Toutain A, Laquerriere A, et al. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann Neurol. 2002;51:340–349. doi: 10.1002/ana.10119. [DOI] [PubMed] [Google Scholar]
- 16.Bouchet C, Gonzales M, Vuillaumier-Barrot S, et al. Molecular heterogeneity in fetal forms of type II lissencephaly. Hum Mutat. 2007;28:1020–1027. doi: 10.1002/humu.20561. [DOI] [PubMed] [Google Scholar]
- 17.Boycott KM, Flavelle S, Bureau A, et al. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet. 2005;77:477–483. doi: 10.1086/444400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boycott KM, Bonnemann C, Herz J, et al. Mutations in VLDLR as a cause for autosomal recessive cerebellar ataxia with mental retardation (dysequilibrium syndrome) J Child Neurol. 2009;24:1310–1315. doi: 10.1177/0883073809332696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruno DL, Anderlid BM, Lindstrand A, et al. Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J Med Genet. 2010;47:299–311. doi: 10.1136/jmg.2009.069906. [DOI] [PubMed] [Google Scholar]
- 20.Cahana A, Escamez T, Nowakowski RS, et al. Targeted mutagenesis of Lis1 disrupts cortical development and LIS1 homodimerization. Proc Natl Acad Sci USA. 2001;98:6429–6434. doi: 10.1073/pnas.101122598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardoso C, Leventer RJ, Matsumoto N, et al. The location and type of mutation predict malformation severity in isolated lissencephaly caused by abnormalities within the LIS1 gene. Hum Mol Genet. 2000;9:3019–3028. doi: 10.1093/hmg/9.20.3019. [DOI] [PubMed] [Google Scholar]
- 22.Cardoso C, Leventer RJ, Dowling JJ, et al. Clinical and molecular basis of classical lissencephaly: mutations in the LIS1 gene (PAFAH1B1) Hum Mutat. 2002;19:4–15. doi: 10.1002/humu.10028. [DOI] [PubMed] [Google Scholar]
- 23.Caspi M, Atlas R, Kantor A, Sapir T, Reiner O. Interaction between LIS1 and doublecortin, two lissencephaly gene products. Hum Mol Genet. 2000;9:2205–2213. doi: 10.1093/oxfordjournals.hmg.a018911. [DOI] [PubMed] [Google Scholar]
- 24.Chang BS, Duzcan F, Kim S, et al. The role of RELN in lissencephaly and neuropsychiatric disease. Am J Med Genet B Neuropsychiatr Genet B. 2007;144:58–63. doi: 10.1002/ajmg.b.30392. [DOI] [PubMed] [Google Scholar]
- 25.Cierpicki T, Kim MH, Cooper DR, Derewenda U, Bushweller JH, Derewenda ZS. The DC-module of doublecortin: dynamics, domain boundaries, and functional implications. Proteins. 2006;64:874–882. doi: 10.1002/prot.21068. [DOI] [PubMed] [Google Scholar]
- 26.Coksaygan T, Magnus T, Cai J, et al. Neurogenesis in Talpha-1 tubulin transgenic mice during development and after injury. Exp Neurol. 2006;197:475–485. doi: 10.1016/j.expneurol.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 27.Colombo E, Collombat P, Colasante G, et al. Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J Neurosci. 2007;27:4786–4798. doi: 10.1523/JNEUROSCI.0417-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper JA. A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 2008;31:113–119. doi: 10.1016/j.tins.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Corbo JC, Deuel TA, Long JM, et al. Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J Neurosci. 2002;22:7548–7557. doi: 10.1523/JNEUROSCI.22-17-07548.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crome L. Pachygyria. J Pathol Bacteriol. 1956;71:335–352. doi: 10.1002/path.1700710208. [DOI] [PubMed] [Google Scholar]
- 31.D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 32.D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/S0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 33.D’Arcangelo G. Reelin mouse mutants as models of cortical development disorders. Epilepsy Behav. 2006;8:81–90. doi: 10.1016/j.yebeh.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Daube JR, Chou SM. Lissencephaly: two cases. Neurology. 1966;16:179–191. doi: 10.1212/wnl.16.2_part_1.179. [DOI] [PubMed] [Google Scholar]
- 35.de Wit MC, Lequin MH, de Coo IF, et al. Cortical brain malformations: effect of clinical, neuroradiological, and modern genetic classification. Arch Neurol. 2008;65:358–366. doi: 10.1001/archneur.65.3.358. [DOI] [PubMed] [Google Scholar]
- 36.des Portes V, Pinard JM, Smadja D, et al. Dominant X linked subcortical laminar heterotopia and lissencephaly syndrome (XSCLH/LIS): evidence for the occurrence of mutation in males and mapping of a potential locus in Xq22. J Med Genet. 1997;34:177–183. doi: 10.1136/jmg.34.3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.des Portes V, Pinard JM, Billuart P, et al. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/S0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 38.Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49:41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 39.Dobyns WB, Curry CJ, Hoyme HE, Turlington L, Ledbetter DH. Clinical and molecular diagnosis of Miller–Dieker syndrome. Am J Hum Genet. 1991;48:584–594. [PMC free article] [PubMed] [Google Scholar]
- 40.Dobyns WB, Truwit CL. Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics. 1995;26:132–147. doi: 10.1055/s-2007-979744. [DOI] [PubMed] [Google Scholar]
- 41.Dobyns WB, Andermann E, Andermann F, et al. X-linked malformations of neuronal migration. Neurology. 1996;47:331–339. doi: 10.1212/wnl.47.2.331. [DOI] [PubMed] [Google Scholar]
- 42.Dobyns WB, Berry-Kravis E, Havernick NJ, Holden KR, Viskochil D. X-linked lissencephaly with absent corpus callosum and ambiguous genitalia. Am J Med Genet. 1999;86:331–337. doi: 10.1002/(SICI)1096-8628(19991008)86:4<331::AID-AJMG7>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 43.Dobyns WB, Truwit CL, Ross ME, et al. Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology. 1999;53:270–277. doi: 10.1212/wnl.53.2.270. [DOI] [PubMed] [Google Scholar]
- 44.Dobyns WB. The clinical patterns and molecular genetics of lissencephaly and subcortical band heterotopia. Epilepsia. 2010;51(Suppl 1):5–9. doi: 10.1111/j.1528-1167.2009.02433.x. [DOI] [PubMed] [Google Scholar]
- 45.Dulabon L, Olson EC, Taglienti MG, et al. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/S0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- 46.Encha-Razavi F, Larroche JC, Roume J, Gonzales M, Kondo HC, Mulliez N. Lethal familial fetal akinesia sequence (FAS) with distinct neuropathological pattern: type III lissencephaly syndrome. Am J Med Genet. 1996;62:16–22. doi: 10.1002/(SICI)1096-8628(19960301)62:1<16::AID-AJMG4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 47.Fallet-Bianco C, Loeuillet L, Poirier K, et al. Neuropathological phenotype of a distinct form of lissencephaly associated with mutations in TUBA1A. Brain. 2008;131:2304–2320. doi: 10.1093/brain/awn155. [DOI] [PubMed] [Google Scholar]
- 48.Faulkner NE, Dujardin DL, Tai CY, et al. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- 49.Feng L, Allen NS, Simo S, Cooper JA. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 2007;21:2717–2730. doi: 10.1101/gad.1604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44:279–293. doi: 10.1016/j.neuron.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 51.Fleck MW, Hirotsune S, Gambello MJ, et al. Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J Neurosci. 2000;20:2439–2450. doi: 10.1523/JNEUROSCI.20-07-02439.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Forman MS, Squier W, Dobyns WB, Golden JA. Genotypically defined lissencephalies show distinct pathologies. J Neuropathol Exp Neurol. 2005;64:847–857. doi: 10.1097/01.jnen.0000182978.56612.41. [DOI] [PubMed] [Google Scholar]
- 53.Francis F, Koulakoff A, Boucher D, et al. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–256. doi: 10.1016/S0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- 54.Francis F, Meyer G, Fallet-Bianco C, et al. Human disorders of cortical development: from past to present. Eur J Neurosci. 2006;23:877–893. doi: 10.1111/j.1460-9568.2006.04649.x. [DOI] [PubMed] [Google Scholar]
- 55.Friede RL. Developmental neuropathology. 2. Berlin: Springer; 1989. pp. 330–334. [Google Scholar]
- 56.Friocourt G, Chafey P, Billuart P, et al. Doublecortin interacts with mu subunits of clathrin adaptor complexes in the developing nervous system. Mol Cell Neurosci. 2001;18:307–319. doi: 10.1006/mcne.2001.1022. [DOI] [PubMed] [Google Scholar]
- 57.Friocourt G, Koulakoff A, Chafey P, et al. Doublecortin functions at the extremities of growing neuronal processes. Cereb Cortex. 2003;13:620–626. doi: 10.1093/cercor/13.6.620. [DOI] [PubMed] [Google Scholar]
- 58.Friocourt G, Poirier K, Rakic S, Parnavelas JG, Chelly J. The role of ARX in cortical development. Eur J Neurosci. 2006;23:869–876. doi: 10.1111/j.1460-9568.2006.04629.x. [DOI] [PubMed] [Google Scholar]
- 59.Friocourt G, Liu JS, Antypa M, Rakic S, Walsh CA, Parnavelas JG. Both doublecortin and doublecortin-like kinase play a role in cortical interneuron migration. J Neurosci. 2007;27:3875–3883. doi: 10.1523/JNEUROSCI.4530-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friocourt G, Kanatani S, Tabata H, et al. Cell-autonomous roles of ARX in cell proliferation and neuronal migration during corticogenesis. J Neurosci. 2008;28:5794–5805. doi: 10.1523/JNEUROSCI.1067-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Friocourt G, Parnavelas JG. Mutations in ARX result in several defects involving GABAergic neurons. Front Cell Neurosci. 2010;4:4. doi: 10.3389/fncel.2010.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gambello MJ, Darling DL, Yingling J, Tanaka T, Gleeson JG, Wynshaw-Boris A. Multiple dose-dependent effects of Lis1 on cerebral cortical development. J Neurosci. 2003;23:1719–1729. doi: 10.1523/JNEUROSCI.23-05-01719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gdalyahu A, Ghosh I, Levy T, et al. DCX, a new mediator of the JNK pathway. EMBO J. 2004;23:823–832. doi: 10.1038/sj.emboj.7600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gecz J, Cloosterman D, Partington M. ARX: a gene for all seasons. Curr Opin Genet Dev. 2006;16:308–316. doi: 10.1016/j.gde.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 65.Gleeson JG, Allen KM, Fox JW, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/S0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 66.Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/S0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 67.Gleeson JG, Luo RF, Grant PE, et al. Genetic and neuroradiological heterogeneity of double cortex syndrome. Ann Neurol. 2000;47:265–269. doi: 10.1002/1531-8249(200002)47:2<265::AID-ANA22>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 68.Gleeson JG, Minnerath S, Kuzniecky RI, et al. Somatic and germline mosaic mutations in the doublecortin gene are associated with variable phenotypes. Am J Hum Genet. 2000;67:574–581. doi: 10.1086/303043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gloster A, El Bizri H, Bamji SX, Rogers D, Miller FD. Early induction of Talpha1 alpha-tubulin transcription in neurons of the developing nervous system. J Comp Neurol. 1999;405:45–60. doi: 10.1002/(SICI)1096-9861(19990301)405:1<45::AID-CNE4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 70.Golden JA. Lissencephaly, Chap 4. In: Golden JA, Harding BN, editors. Neurodevelopmental pathology. Basel: ICN Neuropathol Press; 2004. [Google Scholar]
- 71.Gopal PP, Simonet JC, Shapiro W, Golden JA. Leading process branch instability in Lis1+/− nonradially migrating interneurons. Cereb Cortex. 2010;20:1497–1505. doi: 10.1093/cercor/bhp211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gressens P, Kosofsky BE, Evrard P. Cocaine-induced disturbances of corticogenesis in the developing murine brain. Neurosci Lett. 1992;140:113–116. doi: 10.1016/0304-3940(92)90694-3. [DOI] [PubMed] [Google Scholar]
- 73.Gressens P. Mechanisms and disturbances of neuronal migration. Pediatr Res. 2000;48:725–730. doi: 10.1203/00006450-200012000-00004. [DOI] [PubMed] [Google Scholar]
- 74.Guerrini R, Moro F, Andermann E, et al. Nonsyndromic mental retardation and cryptogenic epilepsy in women with doublecortin gene mutations. Ann Neurol. 2003;54:30–37. doi: 10.1002/ana.10588. [DOI] [PubMed] [Google Scholar]
- 75.Guerrini R, Filippi T. Neuronal migration disorders, genetics, and epileptogenesis. J Child Neurol. 2005;20:287–299. doi: 10.1177/08830738050200040401. [DOI] [PubMed] [Google Scholar]
- 76.Gupta A, Tsai LH, Wynshaw-Boris A. Life is a journey: a genetic look at neocortical development. Nat Rev Genet. 2002;3:342–355. doi: 10.1038/nrg799. [DOI] [PubMed] [Google Scholar]
- 77.Hammond V, Howell B, Godinho L, Tan SS. Disabled-1 functions cell autonomously during radial migration and cortical layering of pyramidal neurons. J Neurosci. 2001;21:8798–8808. doi: 10.1523/JNEUROSCI.21-22-08798.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Harding BN. Malformations of the nervous system. In: Adams JH, Duchen LW, editors. Greenfield’s neuropathology, 6th edn. New York: Oxford University Press; 1997. [Google Scholar]
- 79.Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase [corrected] Nature. 1994;370:216–218. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- 80.Haverfield EV, Whited AJ, Petras KS, Dobyns WB, Das S. Intragenic deletions and duplications of the LIS1 and DCX genes: a major disease-causing mechanism in lissencephaly and subcortical band heterotopia. Eur J Hum Genet. 2009;17:911–918. doi: 10.1038/ejhg.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heng JI, Chariot A, Nguyen L. Molecular layers underlying cytoskeletal remodelling during cortical development. Trends Neurosci. 2010;33:38–47. doi: 10.1016/j.tins.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 82.Hiesberger T, Trommsdorff M, Howell BW, et al. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/S0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 83.Hirotsune S, Takahara T, Sasaki N, et al. The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat Genet. 1995;10:77–83. doi: 10.1038/ng0595-77. [DOI] [PubMed] [Google Scholar]
- 84.Hirotsune S, Fleck MW, Gambello MJ, et al. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- 85.Hong SE, Shugart YY, Huang DT, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93–96. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- 86.Horesh D, Sapir T, Francis F, et al. Doublecortin, a stabilizer of microtubules. Hum Mol Genet. 1999;8:1599–1610. doi: 10.1093/hmg/8.9.1599. [DOI] [PubMed] [Google Scholar]
- 87.Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 88.Jaglin XH, Chelly J. Tubulin-related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet. 2009;25:555–566. doi: 10.1016/j.tig.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 89.Jaglin XH, Poirier K, Saillour Y, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet. 2009;41:746–752. doi: 10.1038/ng.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jellinger K, Rett A. Agyria–pachygyria (lissencephaly syndrome) Neuropediatrie. 1976;7:66–91. doi: 10.1055/s-0028-1091611. [DOI] [PubMed] [Google Scholar]
- 91.Jones DL, Baraban SC. Inhibitory inputs to hippocampal interneurons are reorganized in Lis1 mutant mice. J Neurophysiol. 2009;102:648–658. doi: 10.1152/jn.00392.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jossin Y, Ogawa M, Metin C, Tissir F, Goffinet AM. Inhibition of SRC family kinases and non-classical protein kinases C induce a reeler-like malformation of cortical plate development. J Neurosci. 2003;23:9953–9959. doi: 10.1523/JNEUROSCI.23-30-09953.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kappeler C, Saillour Y, Baudoin JP, et al. Branching and nucleokinesis defects in migrating interneurons derived from doublecortin knockout mice. Hum Mol Genet. 2006;15:1387–1400. doi: 10.1093/hmg/ddl062. [DOI] [PubMed] [Google Scholar]
- 94.Kappeler C, Dhenain M, Phan Dinh TF, et al. Magnetic resonance imaging and histological studies of corpus callosal and hippocampal abnormalities linked to doublecortin deficiency. J Comp Neurol. 2007;500:239–254. doi: 10.1002/cne.21170. [DOI] [PubMed] [Google Scholar]
- 95.Kato M, Das S, Petras K, et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum Mutat. 2004;23:147–159. doi: 10.1002/humu.10310. [DOI] [PubMed] [Google Scholar]
- 96.Kato M, Dobyns WB. X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”. J Child Neurol. 2005;20:392–397. doi: 10.1177/08830738050200042001. [DOI] [PubMed] [Google Scholar]
- 97.Keays DA, Tian G, Poirier K, et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128:45–57. doi: 10.1016/j.cell.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kerjan G, Koizumi H, Han EB, et al. Mice lacking doublecortin and doublecortin-like kinase 2 display altered hippocampal neuronal maturation and spontaneous seizures. Proc Natl Acad Sci USA. 2009;106:6766–6771. doi: 10.1073/pnas.0812687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kerner B, Graham JM, Jr, Golden JA, Pepkowitz SH, Dobyns WB. Familial lissencephaly with cleft palate and severe cerebellar hypoplasia. Am J Med Genet. 1999;87:440–445. doi: 10.1002/(SICI)1096-8628(19991222)87:5<440::AID-AJMG14>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 100.Kim MH, Cierpicki T, Derewenda U, et al. The DCX-domain tandems of doublecortin and doublecortin-like kinase. Nat Struct Biol. 2003;10:324–333. doi: 10.1038/nsb918. [DOI] [PubMed] [Google Scholar]
- 101.Kitamura K, Yanazawa M, Sugiyama N, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- 102.Koizumi H, Higginbotham H, Poon T, Tanaka T, Brinkman BC, Gleeson JG. Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat Neurosci. 2006;9:779–786. doi: 10.1038/nn1704. [DOI] [PubMed] [Google Scholar]
- 103.Koizumi H, Tanaka T, Gleeson JG. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 104.Kumar RA, Pilz DT, Babatz TD, et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum Mol Genet. 2010;19:2817–2827. doi: 10.1093/hmg/ddq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kuo G, Arnaud L, Kronstad-O’Brien P, Cooper JA. Absence of Fyn and Src causes a reeler-like phenotype. J Neurosci. 2005;25:8578–8586. doi: 10.1523/JNEUROSCI.1656-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lapray D, Popova IY, Kindler J et al (2010) Spontaneous epileptic manifestations in a DCX knockdown model of human double cortex. Cereb Cortex [Epub ahead of print] [DOI] [PubMed]
- 107.Larroche JC. Cytoarchitectonic abnormalities (abnormalities of cell migration) In: Vinken PJ, Bruyn GW, editors. Handbook of clinical neurology. Amsterdam: North-Holland Publishing; 1977. pp. 479–506. [Google Scholar]
- 108.Lecourtois M, Poirier K, Friocourt G, et al. Human lissencephaly with cerebellar hypoplasia due to mutations in TUBA1A: expansion of the foetal neuropathological phenotype. Acta Neuropathol. 2010;119:779–789. doi: 10.1007/s00401-010-0684-z. [DOI] [PubMed] [Google Scholar]
- 109.Leger PL, Souville I, Boddaert N, et al. The location of DCX mutations predicts malformation severity in X-linked lissencephaly. Neurogenetics. 2008;9:277–285. doi: 10.1007/s10048-008-0141-5. [DOI] [PubMed] [Google Scholar]
- 110.Leventer RJ, Phelan EM, Coleman LT, Kean MJ, Jackson GD, Harvey AS. Clinical and imaging features of cortical malformations in childhood. Neurology. 1999;53:715–722. doi: 10.1212/wnl.53.4.715. [DOI] [PubMed] [Google Scholar]
- 111.Leventer RJ, Cardoso C, Ledbetter DH, Dobyns WB. LIS1 missense mutations cause milder lissencephaly phenotypes including a child with normal IQ. Neurology. 2001;57:416–422. doi: 10.1212/wnl.57.3.416. [DOI] [PubMed] [Google Scholar]
- 112.Liu Z, Steward R, Luo L. Drosophila Lis1 is required for neuroblast proliferation, dendritic elaboration and axonal transport. Nat Cell Biol. 2000;2:776–783. doi: 10.1038/35041011. [DOI] [PubMed] [Google Scholar]
- 113.Lo NC, Chong CS, Smith AC, Dobyns WB, Carrozzo R, Ledbetter DH. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997;6:157–164. doi: 10.1093/hmg/6.2.157. [DOI] [PubMed] [Google Scholar]
- 114.Marcorelles P, Laquerriere A, Adde-Michel C, et al. Evidence for tangential migration disturbances in human lissencephaly resulting from a defect in LIS1, DCX and ARX genes. Acta Neuropathol. 2010;120:503–515. doi: 10.1007/s00401-010-0692-z. [DOI] [PubMed] [Google Scholar]
- 115.Matsumoto N, Leventer RJ, Kuc JA, et al. Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur J Hum Genet. 2001;9:5–12. doi: 10.1038/sj.ejhg.5200548. [DOI] [PubMed] [Google Scholar]
- 116.McManus MF, Nasrallah IM, Pancoast MM, Wynshaw-Boris A, Golden JA. Lis1 is necessary for normal non-radial migration of inhibitory interneurons. Am J Pathol. 2004;165:775–784. doi: 10.1016/S0002-9440(10)63340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mei D, Lewis R, Parrini E, et al. High frequency of genomic deletions—and a duplication—in the LIS1 gene in lissencephaly: implications for molecular diagnosis. J Med Genet. 2008;45:355–361. doi: 10.1136/jmg.2007.056507. [DOI] [PubMed] [Google Scholar]
- 118.Miyata R, Hayashi M, Miyai K, Akashi T, Kato M, Kohyama J. Analysis of the hypothalamus in a case of X-linked lissencephaly with abnormal genitalia (XLAG) Brain Dev. 2009;31:456–460. doi: 10.1016/j.braindev.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 119.Moores CA, Perderiset M, Francis F, Chelly J, Houdusse A, Milligan RA. Mechanism of microtubule stabilization by doublecortin. Mol Cell. 2004;14:833–839. doi: 10.1016/j.molcel.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 120.Moores CA, Perderiset M, Kappeler C, et al. Distinct roles of doublecortin modulating the microtubule cytoskeleton. EMBO J. 2006;25:4448–4457. doi: 10.1038/sj.emboj.7601335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Morris-Rosendahl DJ, Najm J, Lachmeijer AM, et al. Refining the phenotype of alpha-1a Tubulin (TUBA1A) mutation in patients with classical lissencephaly. Clin Genet. 2008;74:425–433. doi: 10.1111/j.1399-0004.2008.01093.x. [DOI] [PubMed] [Google Scholar]
- 122.Munchoff C, Noetzel H. Uber eine nahezu totale agyrie bei einem 6 jahre alt gewordenen knaben. Acta Neuropathol. 1965;4:469–475. doi: 10.1007/BF00688508. [DOI] [Google Scholar]
- 123.Nasrallah IM, McManus MF, Pancoast MM, Wynshaw-Boris A, Golden JA. Analysis of non-radial interneuron migration dynamics and its disruption in Lis1+/− mice. J Comp Neurol. 2006;496:847–858. doi: 10.1002/cne.20966. [DOI] [PubMed] [Google Scholar]
- 124.Norman MG, McGillivray BC, Kalousek DK et al (1995) Neuronal migration disorders and cortical dysplasia. In: Congenital malformations of the brain. Oxford University Press, Oxford, pp 223–277
- 125.Nosten-Bertrand M, Kappeler C, Dinocourt C, et al. Epilepsy in Dcx knockout mice associated with discrete lamination defects and enhanced excitability in the hippocampus. PLoS One. 2008;3:e2473. doi: 10.1371/journal.pone.0002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ogata T, Matsuo N, Hiraoka N, Hata JI. X-linked lissencephaly with ambiguous genitalia: delineation of further case. Am J Med Genet. 2000;94:174–176. doi: 10.1002/1096-8628(20000911)94:2<174::AID-AJMG11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 127.Ogawa M, Miyata T, Nakajima K, et al. The reeler gene-associated antigen on Cajal–Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 128.Okazaki S, Ohsawa M, Kuki I, et al. Aristaless-related homeobox gene disruption leads to abnormal distribution of GABAergic interneurons in human neocortex: evidence based on a case of X-linked lissencephaly with abnormal genitalia (XLAG) Acta Neuropathol. 2008;116:453–462. doi: 10.1007/s00401-008-0382-2. [DOI] [PubMed] [Google Scholar]
- 129.Pancoast M, Dobyns W, Golden JA. Interneuron deficits in patients with the Miller–Dieker syndrome. Acta Neuropathol. 2005;109:400–404. doi: 10.1007/s00401-004-0979-z. [DOI] [PubMed] [Google Scholar]
- 130.Pawlisz AS, Mutch C, Wynshaw-Boris A, Chenn A, Walsh CA, Feng Y. Lis1-Nde1-dependent neuronal fate control determines cerebral cortical size and lamination. Hum Mol Genet. 2008;17:2441–2455. doi: 10.1093/hmg/ddn144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pilz DT, Matsumoto N, Minnerath S, et al. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum Mol Genet. 1998;7:2029–2037. doi: 10.1093/hmg/7.13.2029. [DOI] [PubMed] [Google Scholar]
- 132.Pilz DT, Kuc J, Matsumoto N, et al. Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1. Hum Mol Genet. 1999;8:1757–1760. doi: 10.1093/hmg/8.9.1757. [DOI] [PubMed] [Google Scholar]
- 133.Poirier K, Keays DA, Francis F, et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A) Hum Mutat. 2007;28:1055–1064. doi: 10.1002/humu.20572. [DOI] [PubMed] [Google Scholar]
- 134.Poirier K, Saillour YY, Bahi-Buisson N et al (2010) Mutations in the neuronal beta tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 135.Pramparo T, Youn YH, Yingling J, Hirotsune S, Wynshaw-Boris A. Novel embryonic neuronal migration and proliferation defects in Dcx mutant mice are exacerbated by Lis1 reduction. J Neurosci. 2010;30:3002–3012. doi: 10.1523/JNEUROSCI.4851-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ramos RL, Bai J, LoTurco JJ. Heterotopia formation in rat but not mouse neocortex after RNA interference knockdown of DCX. Cereb Cortex. 2006;16:1323–1331. doi: 10.1093/cercor/bhj074. [DOI] [PubMed] [Google Scholar]
- 137.Reiner O, Carrozzo R, Shen Y, et al. Isolation of a Miller–Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 138.Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- 139.Robain O, Deonna T. Pachygyria and congenital nephrosis disorder of migration and neuronal orientation. Acta Neuropathol. 1983;60:137–141. doi: 10.1007/BF00685358. [DOI] [PubMed] [Google Scholar]
- 140.Roos L, Jonch AE, Kjaergaard S, et al. A new microduplication syndrome encompassing the region of the Miller–Dieker (17p13 deletion) syndrome. J Med Genet. 2009;46:703–710. doi: 10.1136/jmg.2008.065094. [DOI] [PubMed] [Google Scholar]
- 141.Ross ME, Allen KM, Srivastava AK, et al. Linkage and physical mapping of X-linked lissencephaly/SBH (XLIS): a gene causing neuronal migration defects in human brain. Hum Mol Genet. 1997;6:555–562. doi: 10.1093/hmg/6.4.555. [DOI] [PubMed] [Google Scholar]
- 142.Ross ME, Swanson K, Dobyns WB. Lissencephaly with cerebellar hypoplasia (LCH): a heterogeneous group of cortical malformations. Neuropediatrics. 2001;32:256–263. doi: 10.1055/s-2001-19120. [DOI] [PubMed] [Google Scholar]
- 143.Saillour Y, Carion N, Quelin C, et al. LIS1-related isolated lissencephaly: spectrum of mutations and relationships with malformation severity. Arch Neurol. 2009;66:1007–1015. doi: 10.1001/archneurol.2009.149. [DOI] [PubMed] [Google Scholar]
- 144.Sapir T, Elbaum M, Reiner O. Reduction of microtubule catastrophe events by LIS1, platelet-activating factor acetylhydrolase subunit. EMBO J. 1997;16:6977–6984. doi: 10.1093/emboj/16.23.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sapir T, Horesh D, Caspi M, et al. Doublecortin mutations cluster in evolutionarily conserved functional domains. Hum Mol Genet. 2000;9:703–712. doi: 10.1093/hmg/9.5.703. [DOI] [PubMed] [Google Scholar]
- 146.Schaar BT, McConnell SK. Cytoskeletal coordination during neuronal migration. Proc Natl Acad Sci USA. 2005;102:13652–13657. doi: 10.1073/pnas.0506008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Senzaki K, Ogawa M, Yagi T. Proteins of the CNR family are multiple receptors for Reelin. Cell. 1999;99:635–647. doi: 10.1016/S0092-8674(00)81552-4. [DOI] [PubMed] [Google Scholar]
- 148.Sheen VL, Ferland RJ, Harney M, et al. Impaired proliferation and migration in human Miller–Dieker neural precursors. Ann Neurol. 2006;60:137–144. doi: 10.1002/ana.20843. [DOI] [PubMed] [Google Scholar]
- 149.Sheldon M, Rice DS, D’Arcangelo G, et al. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- 150.Sherr EH. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): one gene leads to many phenotypes. Curr Opin Pediatr. 2003;15:567–571. doi: 10.1097/00008480-200312000-00004. [DOI] [PubMed] [Google Scholar]
- 151.Shoubridge C, Fullston T, Gecz J. ARX spectrum disorders: making inroads into the molecular pathology. Hum Mutat. 2010;31:889–900. doi: 10.1002/humu.21288. [DOI] [PubMed] [Google Scholar]
- 152.Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, Tsai LH. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron. 2004;44:263–277. doi: 10.1016/j.neuron.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 153.Sicca F, Kelemen A, Genton P, et al. Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology. 2003;61:1042–1046. doi: 10.1212/wnl.61.8.1042. [DOI] [PubMed] [Google Scholar]
- 154.Siller KH, Serr M, Steward R, Hays TS, Doe CQ. Live imaging of Drosophila brain neuroblasts reveals a role for Lis1/dynactin in spindle assembly and mitotic checkpoint control. Mol Biol Cell. 2005;16:5127–5140. doi: 10.1091/mbc.E05-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Smith DS, Niethammer M, Ayala R, et al. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- 156.Sossey-Alaoui K, Hartung AJ, Guerrini R, et al. Human doublecortin (DCX) and the homologous gene in mouse encode a putative Ca2+-dependent signaling protein which is mutated in human X-linked neuronal migration defects. Hum Mol Genet. 1998;7:1327–1332. doi: 10.1093/hmg/7.8.1327. [DOI] [PubMed] [Google Scholar]
- 157.Stromme P, Mangelsdorf ME, Shaw MA, et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet. 2002;30:441–445. doi: 10.1038/ng862. [DOI] [PubMed] [Google Scholar]
- 158.Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol. 2004;165:709–721. doi: 10.1083/jcb.200309025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG. Patient mutations in doublecortin define a repeated tubulin-binding domain. J Biol Chem. 2000;275:34442–34450. doi: 10.1074/jbc.M007078200. [DOI] [PubMed] [Google Scholar]
- 160.Tian G, Kong XP, Jaglin XH, Chelly J, Keays D, Cowan NJ. A pachygyria-causing alpha-tubulin mutation results in inefficient cycling with CCT and a deficient interaction with TBCB. Mol Biol Cell. 2008;19:1152–1161. doi: 10.1091/mbc.E07-09-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tian G, Jaglin XH, Keays DA, Francis F, Chelly J, Cowan NJ. Disease-associated mutations in TUBA1A result in a spectrum of defects in the tubulin folding and heterodimer assembly pathway. Hum Mol Genet. 2010;19:3599–3613. doi: 10.1093/hmg/ddq276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tischfield MA, Baris HN, Wu C, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140:74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Trommsdorff M, Gotthardt M, Hiesberger T, et al. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/S0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 164.Tsai JW, Chen Y, Kriegstein AR, Vallee RB. LIS1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages. J Cell Biol. 2005;170:935–945. doi: 10.1083/jcb.200505166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tsai JW, Bremner KH, Vallee RB. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat Neurosci. 2007;10:970–979. doi: 10.1038/nn1934. [DOI] [PubMed] [Google Scholar]
- 166.Uyanik G, Morris-Rosendahl DJ, Stiegler J, et al. Location and type of mutation in the LIS1 gene do not predict phenotypic severity. Neurology. 2007;69:442–447. doi: 10.1212/01.wnl.0000266629.98503.d0. [DOI] [PubMed] [Google Scholar]
- 167.Vallee RB, Tsai JW. The cellular roles of the lissencephaly gene LIS1, and what they tell us about brain development. Genes Dev. 2006;20:1384–1393. doi: 10.1101/gad.1417206. [DOI] [PubMed] [Google Scholar]
- 168.Van Allen M, Clarren SK. A spectrum of gyral anomalies in Miller-Dieker (lissencephaly) syndrome. J Pediatr. 1983;102:559–564. doi: 10.1016/S0022-3476(83)80184-X. [DOI] [PubMed] [Google Scholar]
- 169.Viot G, Sonigo P, Simon I, et al. Neocortical neuronal arrangement in LIS1 and DCX lissencephaly may be different. Am J Med Genet A. 2004;126:123–128. doi: 10.1002/ajmg.a.20569. [DOI] [PubMed] [Google Scholar]
- 170.Ware ML, Fox JW, Gonzalez JL, et al. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron. 1997;19:239–249. doi: 10.1016/S0896-6273(00)80936-8. [DOI] [PubMed] [Google Scholar]
- 171.Wasserman T, Katsenelson K, Daniliuc S, Hasin T, Choder M, Aronheim A. A novel c-Jun N-terminal kinase (JNK)-binding protein WDR62 is recruited to stress granules and mediates a nonclassical JNK activation. Mol Biol Cell. 2010;21:117–130. doi: 10.1091/mbc.E09-06-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wynshaw-Boris A, Pramparo T, Youn YH, Hirotsune S (2010) Lissencephaly: mechanistic insights from animal models and potential therapeutic strategies. Semin Cell Dev Biol [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 173.Yamada M, Toba S, Yoshida Y, et al. LIS1 and NDEL1 coordinate the plus-end-directed transport of cytoplasmic dynein. EMBO J. 2008;27:2471–2483. doi: 10.1038/emboj.2008.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yingling J, Youn YH, Darling D, et al. Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell. 2008;132:474–486. doi: 10.1016/j.cell.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zaki M, Shehab M, El Aleem AA, et al. Identification of a novel recessive RELN mutation using a homozygous balanced reciprocal translocation. Am J Med Genet A. 2007;143:939–944. doi: 10.1002/ajmg.a.31667. [DOI] [PubMed] [Google Scholar]