

Abstract
Anaplastic lymphoma kinase-positive, anaplastic large cell lymphoma (ALK+ ALCL) is an aggressive non-Hodgkin lymphoma of T/null immunophenotype that is most prevalent in children and young adults. The normal cellular counterpart of this malignancy is presumed to be the cytotoxic T lymphocyte (CTL), and this presumption is partly based on the observation that these tumour cells often express cytotoxic granules containing Granzyme B (GzB) and Perforin. Chromosomal translocations involving the gene encoding for the ALK tyrosine kinase are also characteristic of ALK+ ALCL, and the resulting fusion proteins (e.g. NPM-ALK) initiate signalling events important in ALK+ ALCL pathogenesis. These events include the elevated expression of JunB; an AP-1 family transcription factor that promotes ALK+ ALCL proliferation. In this report we demonstrate that JunB is a direct transcriptional activator of GzB and that GzB transcription is also promoted by NPM-ALK. We found that Perforin expression was not regulated by JunB, but was promoted by NPM-ALK in some cell lines and inhibited by it in others. In conclusion, our study makes the novel observation that signalling through NPM-ALK and JunB affect the expression of cytotoxic molecules in ALK+ ALCL. Moreover, these findings demonstrate the expression of GzB and Perforin in this lymphoma is not solely due its presumed CTL origin, but that oncogenic signalling is actively influencing the expression of these proteins.
Keywords: ALK+ ALCL, JunB, NPM-ALK, granzyme B, perforin
Introduction
Anaplastic lymphoma kinase-positive, anaplastic large cell lymphoma (ALK+ ALCL) is a T/null cell non-Hodgkin lymphoma (NHL) that accounts for 10-20% of NHL in children [1]. This lymphoma is thought to arise from an activated cytotoxic T lymphocyte (CTL) [1] given that greater than 90% of ALK+ ALCL tumours possess clonal T cell receptor gene rearrangements [2] and the fact these tumour cells often possess cytotoxic granules containing the serine protease, Granzyme B (GzB), and pore-forming protein, Perforin [2, 3]. A defining characteristic of ALK+ ALCL is the presence of chromosomal translocations involving the ALK tyrosine kinase gene [4], which result in the expression of ALK-containing fusion proteins (e.g. NPM-ALK) that are critical in the pathogenesis of this lymphoma. NPM-ALK activates the PI3K/Akt, Jak/STAT, JNK, MEK/ERK, Rac, and other signalling pathways that regulate ALK+ ALCL proliferation, survival, and migration [5, 6]. The MEK/ERK [7, 8] and PI3K/Akt [7] pathways contribute to the elevated expression of the JunB transcription factor in ALK+ ALCL.
JunB belongs to the AP-1 family of transcription factors which includes c-Jun and JunD, as well as members of the Fos/Fra, ATF, and Maf subfamilies of transcription factors [9, 10]. These proteins homo- and heterodimerize to form AP-1 transcriptional complexes which primarily regulate genes associated with proliferation and apoptosis [9, 10]. JunB is highly expressed in ALK+ ALCL [11-13], and transcriptional and translational mechanisms account for this elevated expression [7, 8]. The importance of JunB in this lymphoma is illustrated by the finding that reducingJunB expression in ALK+ ALCL cell lines impairs cellular proliferation [7].
While it is clear that JunB plays an important role in ALK+ ALCL, its transcriptional targets in this lymphoma are largely uncharacterized. JunB has been shown to promote CD30 expression in ALK+ ALCL [8, 14], but since sites recognized by AP-1 complexes are prevalent in the genome, we hypothesized that this transcription factor regulates the expression of many genes that contribute to the phenotype and/or pathogenesis of this lymphoma. In this report, we demonstrate that GzB is a JunB transcriptional target in ALK+ ALCL, and further show that NPM-ALK is required to promote GzB expression. We found that while Perforin expression is not regulated by JunB, its expression is either inhibited or promoted by NPM-ALK in a cell line-specific manner. These findings are the first to link oncogenic signalling directly to the expression of cytotoxic molecules in ALK+ ALCL.
Materials and methods
Antibodies and other reagents
The anti-tubulin monoclonal antibody (mAb) was from Calbiochem (San Diego, CA). The mAbs against JunB, GzB, and Perforin were from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-β-actin and anti-FLAG M2 mAbs were from Sigma-Aldrich (Mississauga, ON, Canada), while the anti-NPM mAb was from Thermo Scientific (Waltham, MA). Short interfering RNA (siRNA) oligonucleotides (pooled and individual) were purchased from Dharmacon RNAi Technologies (Lafayette, CO). The human GzB proximal promoter (-728 to +59 relative to the transcriptional start site) was PCR amplified from Karpas 299 cells and cloned into the pGL2 basic luciferase vector (Promega; Madison, WI). The AP-1 mutant construct was generated by mutating the AP-1 consensus sequence from TGAG-TCA to TTAGTTA. The JunB cDNAwas PCR amplified and cloned into the pFLAG-CMV2 vector to generate the Flag-tagged JunB construct.
Cell lines and electroporations
Cell lines were cultured in RPMI 1640 media supplemented with 10% heat-inactivated fetal calf serum, 1 mM sodium pyruvate, 2 mM sodium glutamate, and 50 μM 2-mercaptoethanol. 4×106 cells were electroporated with 100 nM of siRNA as previously described [15], and incubated for 48 h at 37°C prior to lysis. For luciferase assays, 1×107 cells were electroporated with the indicated pGL2 luciferase constructs (10 μg), a constitutively expressed Renilla luciferase construct (1 μg) (controls for transfection efficiency), and 100 nM siRNA, and then incubated for 24 h at 37°C prior to analysis. For the JunB over-expression experiment, cells were transfected with luciferase constructs along with a Flag-tagged JunB construct or empty vector (5 μg).
Cell lysis and western blotting
Cells were lysed in Nonidet P-40 lysis buffer [15] containing protease inhibitor cocktail (Sigma-Aldrich), 1 mM phenylmethylsulfon-ylfluoride, and 1 mM sodium orthovanadate. The protein concentration of cleared lysates was determined using the BCA Protein Assay kit (Thermo Scientific). Equivalent protein amounts were resolved on SDS-PAGE gels then transferred to nitrocellulose membranes for western blotting. Western blots were visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Reprobed blots were stripped in 0.1%TBST, pH 2. Quantitative western blotting was performed on a LI-COR Odyssey Infrared Imager (LI-COR Biosciences; Lincoln, NE). For quantification, protein levels were normalized to actin or tubulin levels in each sample and expressed relative to non-targeting control siRNA-treated cells (which were set at 100%).
Quantitative RT-PCR (qRT-PCR)
Total RNA was collected using the RNeasy mini kit (Qiagen; Mississauga, ON), subjected to DNase I digestion, and reverse transcribed to cDNA using the Superscript II Reverse Transcriptase System (Invitrogen; Burlington, ON, Canada). qRT-PCR was performed using PerfeCTa SYBR Green FastMix (Quanta Biosciences; Gaithersburg, MD) with an Eppendorf Mastercycler realplex4 thermal cycler. GzB and Perforin expression were normalized to β-actin and expression levels determined using the ΔΔ-CT method [16]. Results are displayed relative to non-targeting control siRNA-transfected cells which were set at 100%.
Luciferase assays
1×106 cells were assayed in triplicate for GzB promoter-driven firefly luciferase and constitutive Renilla luciferase activity on a BMG Labtech Plate Reader using the Dual-Glo Luciferase Assay System (Promega). The ratio of firefly to Renilla luciferase activity was determined for each sample and triplicate measurements were averaged.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were collected using the ProteoJET cytoplasmic and nuclear protein extraction kit (Fermentas; Burlington, ON, CA). EMSAs were performed using the LightShift chemiluminescent EMSA kit (Thermo Scientific) essentially as outlined in the manufacturer's protocol.
Results
GzB protein levels are reduced in JunB siRNA–treated ALK+ ALCL cell lines
Using quantitative mass spectrometry we identified GzB as a protein that was down-regulated in ALK+ ALCL cell lines treated with JunB siRNA (results not shown). Since GzB expression is a well-known phenotypic characteristic of ALK+ ALCL [2, 3], we were intrigued by this finding. Western blotting lysates from the Karpas 299 and SUP-M2 ALK+ ALCL cell lines treated with either pooled JunB or non-targeting (control) siRNA confirmed that reducing JunB expression resulted in decreased GzB protein levels (Figure 1A and B). To rule out potential off-targeting effects of the pooled JunB siRNAs, we performed similar experiments using individual siRNAs directed against JunB. We found that GzB protein levels were reduced in cells treated with each of the four individual JunB siRNAs (Figure 1C). Moreover, the decrease in GzB levels directly correlated with the degree of JunB silencing observed with the individual siRNAs. Taken together, these results demonstrate that JunB promotes the expression of GzB in ALK+ ALCL.
Figure 1.
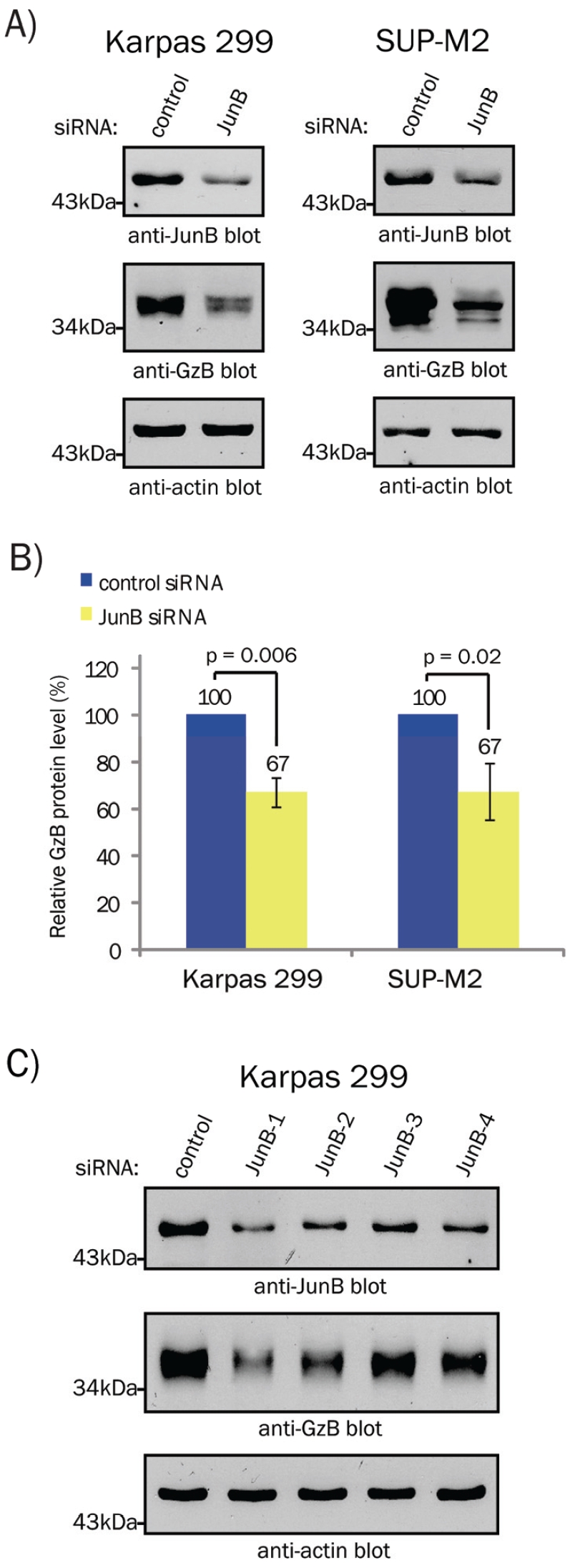
JunB promotes GzB protein expression in ALK+ ALCL. A. Western blot analysis of JunB (upper panel) and GzB (middle panel) expression in cell lysates of cells transfected with 100 nM of pooled non-targeting (control) or JunB siRNAs. The anti-actin western blot (lower panel) demonstrates equivalent protein loading. B. Quantification of western blots in A using the LI-COR Odyssey Infrared imager. The results shown are the mean and standard deviation of three independent experiments. p values were obtained using paired, one-tailed t-tests. C. Western blot analysis of JunB (upper panel) and GzB (middle panel) expression in lysates from Karpas 299 cells transfected with 100 nM of a non-targeting (control) or individual JunB siRNAs. The anti-actin western blot (lower panel) demonstrates equivalent protein loading. Molecular mass markers are indicated to the left of the blots.
JunB promotes GzB transcription in ALK+ ALCL
Since JunB is a transcription factor, we next examined whether it promotes GzB transcription. qRT-PCR demonstrated that GzB mRNA levels were reduced in ALK+ ALCL cell lines treated with JunB siRNA (Figure 2A). We also investigated whether JunB could promote the expression of a luciferase reporter construct under control of the human GzB proximal promoter. The human GzB promoter contains an AP -1 binding site as well as CRE, Ikaros, CBF, Ets, and NF-AT recognition sites [17]. This AP-1 site is conserved in mice and is required for maximal GzB promoter activity in activated murine and human T cells [18, 19]. Treatment of Karpas 299 cells with JunB siRNA (Figure 2B) or mutation of the AP-1 site in the promoter (Figure 2C) significantly reduced GzB promoter luciferase activity. We also found that over-expression of JunB enhanced GzB promoter luciferase activity in Karpas 299 cells (Figure 2D). We next performed EMSAs to test whether JunB can bind the AP-1 site in the GzB promoter. One or more proteins present in Karpas 299 nuclear extract associated with a biotin-labeled probe based on the AP-1 site in the GzB promoter (Figure 2E). Moreover, this binding was out-competed by a molar excess of an unlabeled GzB AP-1 probe, but not by an unlabeled probe with a mutation in the AP-1 site (Figure 2E). We also observed an almost complete super-shift of the protein/probe complex by the addition of an anti-JunB antibody, but not by the addition of an irrelevant control antibody (Figure 2E). This latter result demonstrates that not only is JunB present in these probe/protein complexes, but that almost all probe/protein complexes contain JunB. In sum, these findings suggest that JunB binds the AP-1site in the GzB promoter and functions as a direct activator of GzB transcription in ALK+ ALCL.
Figure 2.

JunB promotes GzB transcription in ALK+ ALCL. A. qRT-PCR analysis of GzB mRNA levels in Karpas 299 (left) and SUP-M2 (right) cells transfected with 100 nM of the indicated pooled siRNAs. Note: these experiments were performed on the mRNA extracted from cells transfected in Figure 1. B. Luciferase activity was measured in Karpas 299 cells transfected with a promoter-less luciferase construct (pGL2 basic) or a luciferase construct under control of the GzB proximal promoter (pGL2-GzB promoter) and 100 nM of the indicated pooled siRNAs. Luciferase activity is expressed relative to the activity present in cells co-transfected with the pGL2-GzB promoter and control siRNA (which was set at 100%). A western blot demonstrating the efficiency of JunB knock-down is shown to the right. C. Karpas 299 cells were transfected with the pGL2 basic, pGL2-GzB promoter, or AP-1 mutant pGL2-GzB promoter luciferase constructs and luciferase activity was measured 24 h post-transfection. Luciferase activity is expressed relative to the activity present in the pGL2-GzB promoter transfected cells (which was set at 100%). D. Luciferase activity was measured in Karpas 299 cells transfected with the pGL2-GzB promoter luciferase construct and Flag-tagged JunB (+) or empty vector (-) (top). Luciferase activity is expressed relative to the activity present in the pGL2-GzB promoter and empty vector–transfected cells (which was set at 100%). Anti-Flag and anti-JunB western blots demonstrate JunB over-expression (bottom). Molecular mass markers are indicated to the left of blots. E. EMSAs were performed with Karpas 299 nuclear extracts using a biotinylated probe based on the sequence of the AP-1 site in the GzB proximal promoter (GzB AP-1 probe). For competitor experiments, a 200-fold molar excess of unlabeled GzB AP-1 probe (wt competitor) or an unlabeled GzB AP-1 probe with a mutation in the AP-1 binding site (AP-1 mutant competitor) were included in the reaction. For super-shift experiments, 1 µg of the indicated antibody (Ab) was included in the reaction. In all experiments, the quantification represents the mean and standard deviation of three independent experiments. p values were determined using paired, one-tailed t-tests.
Perforin levels are not regulated by JunB in ALK+ ALCL
Perforin is another component of the cytotoxic granules found in ALK+ ALCL [2, 3], and since the Perforin promoter contains an AP-1 binding site [17], we postulated it may also be regulated by JunB in this lymphoma. However, western blotting experiments revealed that Perforin protein levels were not significantly altered in cells treated with JunB siRNA (Figure 3). Therefore, while JunB is an important transcriptional regulator of GzB in ALK+ ALCL, it does not regulate Perforin in this lymphoma.
Figure 3.

JunB does not regulate Perforin expression in ALK+ ALCL. Western blot (left) and quantification (right) of Perforin expression in cell lysates from Karpas 299 (A) and SUP-M2 (B) cells transfected with the 100 nM of the indicated pooled siRNAs. Results are the mean and standard deviation of three independent experiments. There was no statistical difference in Perforin levels between control and JunB siRNA-treated samples (paired, two-tailed t-tests; p > 0.15).
NPM-ALK regulates GzB and perforin expression in ALK+ ALCL
Since NPM-ALK promotes the expression of JunB [7, 14], we examined whether NPM-ALK also promotes GzB expression. Knock-down of NPM-ALK in Karpas 299 and SUP-M2 cells resulted in reduced GzB protein and mRNA levels (Figures 4A and B, respectively). Reducing ALK expression also reduced GzB promoter luciferase activity (Figure 4C). Consistent with a previous study [14], we found that JunB levels were decreased in cells treated with ALK siRNA (Figure 4A).
Figure 4.

NPM-ALK promotes GzB expression in ALK+ ALCL. A. Western blots (left) and quantification (right) of GzB and JunB expression in lysates from Karpas 299 or SUP-M2 cells transfected with 100 nM of the indicated pooled siRNAs. Note: the anti-NPM blots demonstrate NPM-ALK silencing. Molecular mass markers are indicated to the left of the blots. B. qRT-PCR analysis of GzB mRNA expression in Karpas 299 and SUP-M2 cells transfected with the indicated siRNAs. Note: these experiments were performed on the mRNA extracted from cells transfected in A. C. GzB promoter luciferase activity was measured in Karpas 299 cells transfected with 100nM of non-targeting (control) or ALK siRNA. Luciferase activity is expressed relative to the activity present in cells transfected with the pGL2-GzB promoter and control siRNA (which was set at 100%). In all experiments, the quantification represents the mean and standard deviation of three independent experiments. p values were obtained using paired, one-tailed t-tests.
We also investigated whether NPM-ALK influences Perforin expression. In Karpas 299 cells, Perforin protein (Figure 5A) and mRNA (Figure 5B) levels were decreased in ALK siRNA-treated cells. In contrast, NPM-ALK knock-down in SUP-M2 cells led to a significant increase in Perforin protein and mRNA levels (Figures 5A and B, respectively). In light of these results, we examined how NPM-ALK knock-down affected Perforin levels in the SU-DHL-1 ALK+ ALCL cell line. Similar to SUP-M2 cells, we found that SU-DHL-1 cells treated with ALK siRNA had increased Perforin protein and mRNA levels (Figure 5C and D, respectively). NPM-ALK promoted GzB and JunB expression in SU-DHL-1 cells (Figure 5C and D), as was observed in Karpas 299 and SUP-M2 cells. These results demonstrate that while NPM-ALK is an important activator of GzB transcription in ALK+ ALCL, this oncogene can either promote or repress the expression of Perforin.
Figure 5.

NPM-ALK regulates Perforin expression in ALK+ ALCL. A, Western blots (left) and quantification (right) of Perforin expression in lysates from Karpas 299 or SUP-M2 cells transfected with 100 nM of non-targeting (control) or ALK siRNA. B, qRT-PCR analysis of Perforin mRNA expression in Karpas 299 and SUP-M2 cells transfected with 100 nM of non-targeting (control) or ALK siRNA. Note: these experiments were performed on the mRNA extracted from cells transfected in A. C, Western blots (left) and quantification (right) of the indicated proteins in lysates from SU-DHL -1 cells transfected with 100 nM of non-targeting (control) or ALK siRNA. D, qRT-PCR analysis of Perforin and GzB mRNA expression in SU-DHL-1 cells transfected with 100 nM of non-targeting (control) or ALK siRNA. Note: these experiments were performed on the mRNA extracted from cells transfected in C. Molecular mass markers are indicated to the left of the western blots. In all experiments, the quantification represents the mean and standard deviation of three independent experiments. p values were obtained using paired, one-tailed t-tests. Note: the anti-NPM blots demonstrate NPM-ALK silencing.
Discussion
The expression of GzB and Perforin is a common feature of ALK+ ALCL and one piece of evidence suggesting that these tumour cells are derived from a CTL. However, our results reveal that the expression of these proteins is not just a remnant of the suggested CTL origin of this lymphoma. We demonstrate that the NPM-ALK oncogene and JunB transcription factor promote the transcription of GzB in ALK+ ALCL cell lines, which we believe helps explain why the majority of ALK+ ALCL tumours are positive for GzB expression [2, 3, 20]. We found that Perforin expression was not regulated by JunB, but was either promoted or repressed by NPM-ALK in a cell line-specific manner. Taken together, these novel findings demonstrate that oncogenic signalling influences the expression of cytotoxic molecules in this lymphoma.
In addition to JunB, c-Jun is also highly expressed in ALK+ ALCL [11, 21]. c-Jun has also been reported to promote ALK+ ALCL proliferation [22], although there is not complete agreement on this point [7]. We were only able to significantly knock-down c-Jun levels using high levels of siRNA (600 nM) which could cause off-targeting events [23]. However, even using these high levels of siRNA, we found that c-Jun knock-down had no effect on GzB protein or mRNA levels (results not shown). We also could not detect c-Jun binding to the AP-1 site in the GzB proximal promoter by EMSA (results not shown). These results argue that c-Jun is not an important transcriptional activator of GzB in ALK+ ALCL. We also found that c-Jun did not regulate Perforin expression in this lymphoma (results not shown). This result and the findings of Figure 3 argue that if the AP-1 site in the Perforin promoter is important in ALK+ ALCL, it is responsive to AP-1 transcriptional complexes that do not contain JunB or c-Jun.
Since NPM-ALK promotes JunB expression ([7, 14] and Figures 4A and 5C), we postulate that NPM-ALK likely promotes GzB transcription in large part through JunB. However, NPM-ALK knock-down had a more dramatic affect on GzB expression than JunB knock-down (compare Figures 1 and 2 with Figure 4), so it is likely that NPM-ALK activates other signalling pathways that promote GzB transcription. One possibility is the Jak/STAT pathway. NPM-ALK activates Jak/STAT signalling [5, 6] and since this pathway promotes GzB expression in other cell types [24-27], it may play a role in promoting GzB expression in ALK+ ALCL.
NPM-ALK was found to promote Perforin expression in Karpas 299 cells, but interestingly repressed Perforin expression in SUP-M2 and SU-DHL-1 cells (Figure 5). We postulate that the repression of Perforin expression by NPM-ALK in SUP-M2 and SU-DHL-1 cells may be due to the ability of NPM-ALK to silence gene expression by promoting DNA methylation [28, 29]. NPM-ALK up-regulates the expression of DNA methyltrans-ferases which methylate the promoters of several T cell-associated genes (e.g. CD3ε, SLP-76, ZAP-70, LAT) thereby inhibiting the expression of these genes [28]. We found that Perforin levels were elevated after treatment of SUP-M2 and SU-DHL-1 cells (but not Karpas 299 cells) with the DNA methyltransferase inhibitor, 5-aza-2'-deoxycytidine, consistent with the possibility that NPM-ALK may limit Perforin expression by DNA methylation in these cell lines (results not shown). Why NPM-ALK differentially regulates Perforin gene expression in ALK+ ALCL cell lines is unclear. However, these results are consistent with the observation that other genes known to be silenced by NPM-ALK through DNA methylation are not uniformly silenced in ALK+ ALCL patient samples and cell lines [28].
The fact that GzB expression is actively promoted by oncogenic signalling in ALK+ ALCL raises the question as to whether GzB could be important in the pathogenesis of this lymphoma. A recent study found that ∼75% of urothelial carcinomas and about half of the bladder cancer cell lines tested express GzB [30]. This study further demonstrated that GzB knock-down in these bladder cancer lines significantly inhibited their invasion through ma-trigel [30]. Whether ALK+ ALCL cell lines use GzB to similarly promote invasion through matrix is something that we are currently investigating. This could be one reason for why this lymphoma is widely disseminated in patients at clinical presentation [31-33].
Acknowledgments
We would like to thank Dr. Troy Baldwin and Zubair Mohammed for critically reading the manuscript and Drs. Catherine Ewen and Chris Bleackley for valuable reagents. Quantitative mass spectrometry experiments were performed at the University of Victoria/Genome BC Proteomics Centre.
This work was funded by operating grants from the British Columbia Proteomics Network (BCPN) and the Natural Sciences and Engineering Research Council (NSERC), and start-up funds from the University of Alberta (to R.J.I). J.D.P. is a recipient of a NSERC studentship.
References
- 1.Delsol G FB, Muller-Hermelink H, Campo E, Jaffe E, Gascoyne R, Stein H, Kinney M. Anaplastic Large Cell Lymphoma (ALCL), ALK-positive. In: Swerdlow S CE, Harris N, Jaffe E, Pileri S, Stein H, Thiele J, Vardiman J, editors. Lyon: International Agency for Research on Cancer (IARC); 2008. pp. 312–316. [Google Scholar]
- 2.Foss HD, Anagnostopoulos I, Araujo I, Assaf C, Demel G, Kummer JA, Hummel M, Stein H. Anaplastic large-cell lymphomas of T-cell and null-cell phenotype express cytotoxic molecules. Blood. 1996;88:4005–4011. [PubMed] [Google Scholar]
- 3.d'Amore ES, Menin A, Bonoldi E, Bevilacqua P, Cazzavillan S, Donofrio V, Gambini C, Forni M, Gentile A, Magro G, Boldrini R, Pillon M, Rosolen A, Alaggio R. Anaplastic large cell lymphomas: a study of 75 pediatric patients. Pediatr Dev Pathol. 2007;10:181–191. doi: 10.2350/06-04-0082.1. [DOI] [PubMed] [Google Scholar]
- 4.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 5.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 6.Amin HM, Lai R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood. 2007;110:2259–2267. doi: 10.1182/blood-2007-04-060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Staber PB, Vesely P, Haq N, Ott RG, Funato K, Bambach I, Fuchs C, Schauer S, Linkesch W, Hrzenjak A, Dirks WG, Sexl V, Bergler H, Kadin ME, Sternberg DW, Kenner L, Hoefler G. The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood. 2007;110:3374–3383. doi: 10.1182/blood-2007-02-071258. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe M, Sasaki M, Itoh K, Higashihara M, Umezawa K, Kadin ME, Abraham LJ, Watanabe T, Horie R. JunB induced by constitutive CD30-extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase signaling activates the CD30 promoter in anaplastic large cell lymphoma and reed-sternberg cells of Hodgkin lymphoma. Cancer Res. 2005;65:7628–7634. doi: 10.1158/0008-5472.CAN-05-0925. [DOI] [PubMed] [Google Scholar]
- 9.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 10.Shaulian E. AP-1–The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell Signal. 2010;22:894–899. doi: 10.1016/j.cellsig.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, Bommert K, Mechta-Grigoriou F, Stein H, Dorken B, Scheidereit C. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002;21:4104–4113. doi: 10.1093/emboj/cdf389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rassidakis GZ, Thomaides A, Atwell C, Ford R, Jones D, Claret FX, Medeiros LJ. JunB expression is a common feature of CD30+ lymphomas and lymphomatoid papulosis. Mod Pathol. 2005;18:1365–1370. doi: 10.1038/modpathol.3800419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szremska AP, Kenner L, Weisz E, Ott RG, Passegue E, Artwohl M, Freissmuth M, Stoxreiter R, Theussl HC, Parzer SB, Moriggl R, Wagner EF, Sexl V. JunB inhibits proliferation and transformation in B-lymphoid cells. Blood. 2003;102:4159–4165. doi: 10.1182/blood-2003-03-0915. [DOI] [PubMed] [Google Scholar]
- 14.Hsu FY, Johnston PB, Burke KA, Zhao Y. The expression of CD30 in anaplastic large cell lymphoma is regulated by nucleophosmin-anaplastic lymphoma kinase-mediated JunB level in a cell type-specific manner. Cancer Res. 2006;66:9002–9008. doi: 10.1158/0008-5472.CAN-05-4101. [DOI] [PubMed] [Google Scholar]
- 15.Ingham RJ, Raaijmakers J, Lim CS, Mbamalu G, Gish G, Chen F, Matskova L, Ernberg I, Winberg G, Pawson T. The Epstein-Barr virus protein, latent membrane protein 2A, co-opts tyrosine kinases used by the T cell receptor. J Biol Chem. 2005;280:34133–34142. doi: 10.1074/jbc.M507831200. [DOI] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent developments in the transcriptional regulation of cytolytic effector cells. Nat Rev Immunol. 2004;4:900–911. doi: 10.1038/nri1490. [DOI] [PubMed] [Google Scholar]
- 18.Hanson RD, Grisolano JL, Ley TJ. Consensus AP-1 and CRE motifs upstream from the human cytotoxic serine protease B (CSP-B/CGL-1) gene synergize to activate transcription. Blood. 1993;82:2749–2757. [PubMed] [Google Scholar]
- 19.Babichuk CK, Bleackley RC. Mutational analysis of the murine granzyme B gene promoter in primary T cells and a T cell clone. J Biol Chem. 1997;272:18564–18571. doi: 10.1074/jbc.272.30.18564. [DOI] [PubMed] [Google Scholar]
- 20.Dukers DF, ten Berge RL, Oudejans JJ, Pulford K, Hayes D, Misere JF, Ossenkoppele GJ, Jaspars LH, Willemze R, Meijer CJ. A cytotoxic phenotype does not predict clinical outcome in anaplastic large cell lymphomas. J Clin Pathol. 1999;52:129–136. doi: 10.1136/jcp.52.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drakos E, Leventaki V, Schlette EJ, Jones D, Lin P, Medeiros U, Rassidakis GZ. c-Jun expression and activation are restricted to CD30+ lymphoproliferative disorders. Am J Surg Pathol. 2007;31:447–453. doi: 10.1097/01.pas.0000213412.25935.e4. [DOI] [PubMed] [Google Scholar]
- 22.Leventaki V, Drakos E, Medeiros LJ, Lim MS, Elenitoba-Johnson KS, Claret FX, Rassidakis GZ. NPM-ALK oncogenic kinase promotes cell-cycle progression through activation of JNK/cJun signaling in anaplastic large-cell lymphoma. Blood. 2007;110:1621–1630. doi: 10.1182/blood-2006-11-059451. [DOI] [PubMed] [Google Scholar]
- 23.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 24.Hagn M, Schwesinger E, Ebel V, Sontheimer K, Maier J, Beyer T, Syrovets T, Laumonnier Y, Fabricius D, Simmet T, Jahrsdorfer B. Human B cells secrete granzyme B when recognizing viral antigens in the context of the acute phase cytokine IL-21. J Immunol. 2009;183:1838–1845. doi: 10.4049/jimmunol.0901066. [DOI] [PubMed] [Google Scholar]
- 25.Jahrsdorfer B, Vollmer A, Blackwell SE, Maier J, Sontheimer K, Beyer T, Mandel B, Lunov O, Tron K, Nienhaus GU, Simmet T, Debatin KM, Weiner GJ, Fabricius D. Granzyme B produced by human plasmacytoid dendritic cells suppresses T-cell expansion. Blood. 2010;115:1156–1165. doi: 10.1182/blood-2009-07-235382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manyak CL, Norton GP, Lobe CG, Bleackley RC, Gershenfeld HK, Weissman IL, Kumar V, Sigal NH, Koo GC. IL-2 induces expression of serine protease enzymes and genes in natural killer and nonspecific T killer cells. J Immunol. 1989;142:3707–3713. [PubMed] [Google Scholar]
- 27.DeBlaker-Hohe DF, Yamauchi A, Yu CR, Horvath-Arcidiacono JA, Bloom ET. IL-12 synergizes with IL-2 to induce lymphokine-activated cytotoxicity and perforin and granzyme gene expression in fresh human NK cells. Cell Immunol. 1995;165:33–43. doi: 10.1006/cimm.1995.1184. [DOI] [PubMed] [Google Scholar]
- 28.Ambrogio C, Martinengo C, Voena C, Tondat F, Riera L, di Celle PF, Inghirami G, Chiarle R. NPM-ALK oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res. 2009;69:8611–8619. doi: 10.1158/0008-5472.CAN-09-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han Y, Amin HM, Frantz C, Franko B, Lee J, Lin Q, Lai R. Restoration of shp1 expression by 5-AZA-2'-deoxycytidine is associated with down-regulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell lymphoma. Leukemia. 2006;20:1602–1609. doi: 10.1038/sj.leu.2404323. [DOI] [PubMed] [Google Scholar]
- 30.D'Eliseo D, Pisu P, Romano C, Tubaro A, De Nunzio C, Morrone S, Santoni A, Stoppacciaro A, Velotti F. Granzyme B is expressed in urothelial carcinoma and promotes cancer cell invasion. Int J Cancer. 2009;21:21. doi: 10.1002/ijc.25135. [DOI] [PubMed] [Google Scholar]
- 31.Brugieres L, Deley MC, Pacquement H, Meguerian-Bedoyan Z, Terrier-Lacombe MJ, Robert A, Pondarre C, Leverger G, Devalck C, Rodary C, Delsol G, Hartmann O. CD30(+) anaplastic large-cell lymphoma in children: analysis of 82 patients enrolled in two consecutive studies of the French Society of Pediatric Oncology. Blood. 1998;92:3591–3598. [PubMed] [Google Scholar]
- 32.Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf-Peeters C, Verhoef G, Menestrina F, Todeschini G, Paulli M, Lazzarino M, Giardini R, Aiello A, Foss HD, Araujo I, Fizzotti M, Pelicci PG, Flenghi L, Martelli MF, Santucci A. ALK+ lymphoma: clinico-pathological findings and outcome. Blood. 1999;93:2697–2706. [PubMed] [Google Scholar]
- 33.Gascoyne RD, Aoun P, Wu D, Chhanabhai M, Skinnider BF, Greiner TC, Morris SW, Connors JM, Vose JM, Viswanatha DS, Coldman A, Weisenburger DD. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood. 1999;93:3913–3921. [PubMed] [Google Scholar]