

Abstract
Proteasomal processing is conducted by three individual catalytic subunits, namely β1, β2, and β5. Subunit-specific inhibitors are useful tools in dissecting the role of these individual subunits and are leads toward the development of antitumor agents. We here report that the presence of fluorinated phenylalanine derivatives in peptide based proteasome inhibitors has a profound effect on inhibitor potency and selectivity. Specifically, compound 4a emerges as one of the most β5 specific inhibitors known to date.
Introduction
The majority of all cytosolic and nuclear proteins in eukaryotic cells are degraded by the ubiquitin-proteasome pathway. In this system, proteins destined for degradation are modified with a polyubiquitin chain as a recognition tag for the 26S proteasome where proteolysis occurs. The 26S proteasome contains one or two 19S regulatory caps together with the proteolytically active, cylindrical 20S core. Within the mammalian constitutive 20S core, three pairs of proteolytically active sites are present displaying different substrate specificity. Of these, the β1 subunits (caspase-like) cleave after acidic residues, the β2 subunits (trypsin-like) cleave after basic residues, and the β5 subunits (chymotrypsin-like) cleave after bulky, hydrophobic residues.1,2 The peptidyl boronic acid proteasome inhibitor PS-341 (8)3 is used for the treatment of multiple myeloma and targets the β5 and β1 subunits. To study the role of the three individual active subunits, subunit-specific inhibitors are needed. Inhibitors with moderate to good selectivity for either one of the subunits have been developed,4 however there is still room for improvements.
The search for subunit selective inhibitors is predominantly conducted by either screening of natural products,5 rational design,6 or compound library building.4b,7 We observed that in these studies the effect of fluorine functionality in proteasome inhibitors is relatively uncharted.8 In contrast, fluorine has found wide interest in bioorganic and structural chemistry over the past decade and has become an important feature in drug design.9 This is predominantly due to the typical characteristics of fluorine (when bound to carbon) such as its comparable size to hydrogen, its electron withdrawing ability, superhydrophobicity of fluorocarbons, and self-association between fluorinated moieties. In protein structure design, introduction of fluorine can mimic functional groups, alter structural properties, and thereby (de)stabilize protein structures or function as recognitionmotifs.10 In addition, the beneficial 19F nuclear magnetic characteristics have found their use in structure analysis by (solid state) 19F NMR spectroscopy or 19F MRI to study, for example, protein aggregation.11
The set of fluorinated proteasome inhibitors prepared in the context of the here presented studies are depicted in Figure 1. Compounds 2a and 2b containing pentafluoroPhe (PheF5a) and 3,5-bis(trifluoromethyl)Phe (Phe(m-CF3)2), respectively, are based on compound 8 derivative 1 (having a comparable potency toward the β1 and β5 proteasome subunits with respect to 8)12 and differ in that the phenylalanine in 8 is replaced by the corresponding fluorinated analogue. In addition, incorporation of fluorinated phenylalanines at different positions in tripeptide epoxyketones2 led to compounds 3–6 in which systematically either one or both of the P2 and P3 positions were altered. We opted for the use of Phe(m-CF3)2) and PheF5 for the dual reason that these are readily available and that hydrophobic amino acids (that is the nonfluorinated analogues) are in principle accepted by all proteasome active sites. The epoxyketone electrophilic trap was selected based on the natural product epoxomicin. The epoxyketone war-head featured by epoxomicin displays a specific and selective reactivity toward the N-terminal threonine residue that makes up the proteasome catalytic active sites.2,5 For this reason, synthetic peptide epoxyketones are now much studied leads in medicinal chemistry studies in which the proteasome plays a role.13 The tripeptide epoxyketones 3–6 feature an azide moiety at the N-terminal end for future modifications (for instance, coupling to a fluorophore or biotin in either one- or two-step labeling experiments).14
Figure 1.

Synthesized fluorinated proteasome inhibitors. Indicated are the enzyme pockets (P1, P2, P3).
Results and Discussion
The C-terminally modified oligopeptides were produced following synthesis protocols we reported previously.7 The amino acids used were either commercially available or prepared following established procedures.15 See for full experimental data on the synthesis of the compounds the Supporting Information (SI). The inhibition potential of compounds 2a and 2b in comparison with their nonfluorinated analogue boronic ester 1 (the pinanediol analogue of the clinical drug 8) was assessed in a competition assay employing human embryonic kidney (HEK-293) cell lysates in combination with the fluorescent broad spectrum proteasome probe MV151 (9).12 Cell lysates were incubated with each of the three compounds at 0.05, 0.1, and 1 μM final concentrations prior to treatment with 0.5 μM final concentration of 9. The samples were denatured, resolved by SDS-PAGE, and the wet gel slabs were scanned on a fluorescence scanner. The gel image is shown in Figure 2. Lysates treated with the fluorescent probe display three bands that correspond to the three active subunits (β1, β2, and β5) as depicted in Figure 2 lane 1. The ability of a compound to inhibit the proteasome active sites is reflected by disappearance of the bands. As apparent from this image, the fluorinated compounds 2a and 2b are at least as potent as their nonfluorinated counterpart 1 showing complete inhibition of the β1 and β5 subunits between 0.1 and 1 μM. In addition, incorporation of fluorinated Phe leaves the selectivity of β1 and β5 over β2 subunits for this type of inhibitor unchanged. This result is also apparent from the activity (IC50) measurements of the inhibitors toward the different subunits in purified rabbit 26S proteasome as shown in Table 1. Experiments in murine EL4 cell lysates (mouse lymphoma cell line) which contain both the immuno- and constitutive proteasome subunits gave similar results (see SI).
Figure 2.

Characterization of the specificity of the fluorinated dipeptide boronates. Competition assay in HEK-293 lysate. Lysates were incubated for one hour with compounds 1, 2a, and 2b at the indicated final concentrations. Residual proteasome activity was labeled with 0.5 μM 9 for 1 h.
Table 1.
Activity (IC50 in μM) of 2–5 against the Three Active Constitutive 26S Proteasome Subunitsa
| compd | β1 nLPnLD | β2 RLR | β5 LLVY |
|---|---|---|---|
| 2a | 0.44 | >15 | 0.031 |
| 2b | 0.16 | 11 | 0.0030 |
| 3 | >15 | 1.8 | 0.0010 |
| 4a | >15 | >15 | 0.0020 |
| 4b | >15 | >15 | 0.10 |
| 5a | >15 | >15 | 0.20 |
| 5b | >15 | >15 | 0.13 |
Determined with the indicated subunit specific fluorogenic peptide substrates. All values are averages of two experiments.
Next, the inhibition properties of the seven epoxyketone containing compounds were determined in a similar competition assay against 9 at 1 and 100 μM final concentrations employing HEK-293 cell lysates (Figure 3) and by measuring inhibition of purified proteasomes for the most active compounds (Table 1). When comparing nonfluorinated compound 3 to the fluorinated ones (4–6), it becomes apparent that none of these compounds inhibit the β1 subunit at concentrations up to 100 μM and that introducing fluorines (in either position) leads to a decrease in the inhibition of the β2 subunit and hence to an increase in β5 specificity. When comparing Table 1 with Figure 3, it appears that the results from the two independent assays deviate in places. For example the β2-IC50 value for 4a is <15 μM (Table 1), whereas the majority of the corresponding band in Figure 3 is gone at 1 μM. Intrinsic differences in both assays, neither of which deliver ki values are at the basis of these small but distinct differences. The complementary assays, however, both show similar trends, which is why we prefer to report both.16 For instance, in Figure 3, there appears to be almost no difference in inhibition potential of β5 between the nonfluorinated compound 3 and either P2 or P3 fluorinated compounds 4a,b and 5a,b. Although there is a big difference in IC50 values, all compounds are at least submicromolar inhibitors.
Figure 3.

Characterization of specificity of fluorinated peptide epoxyketones. Competition assay in HEK-293 lysate. Lysates were incubated for 1 h with compounds 3, 4, 5, or 6 at the indicated final concentrations. Residual proteasome activity was labeled with 0.5 μM 9 for 1 h. N.B. compound 3a = 3b = 3.
Interestingly, the presence of fluorine substituents at both P2 and P3 positions (6a,b, Figure 3) has a dramatic effect on the inhibition. This effect is most pronounced for the Phe-(m-CF3)2 analogues (b series). In general, the PheF5 compounds (a series) are more active against the β5 subunit than their hexafluoro-Phe analogues (b series). Experiments in the lysates of murine EL4 cells gave similar results, but the difference in inhibition potential between the compounds was even more pronounced (see SI). Thus, it appears that introduction of Phe(F5) in the P2 position generates a highly specific inhibitor of the β5 site. This most potent and β5 selective inhibitor (compound 4a) was further investigated. The inhibitory potential at much lower concentrations (1 nM to 1 μM) is shown in Figure 4 (competition assay against 9). Already, at 5 nM, inhibition of β5 is apparent, and between 100 and 250 nM, all β5 subunits are saturated while β1 and β2 are unaffected or even upregulated (a phenomenon which is not fully understood but has been observed by others as well).17 Having an IC50 value of 2 nM for the β5 subunit against >15 μM for β1 and β2, this compound is one of the most β5 selective inhibitors known to date. For instance, this compound compares well with NC005, a β5 selective inhibitor we recently found.6 Comparison of nonfluorinated compound 3 with 4a reveals that both compounds are equally active toward the β5 subunit. Enhanced selectivity for β5 arises by the dramatic drop in activity for the β2 subunit when fluorine is introduced, as in 4a.
Figure 4.
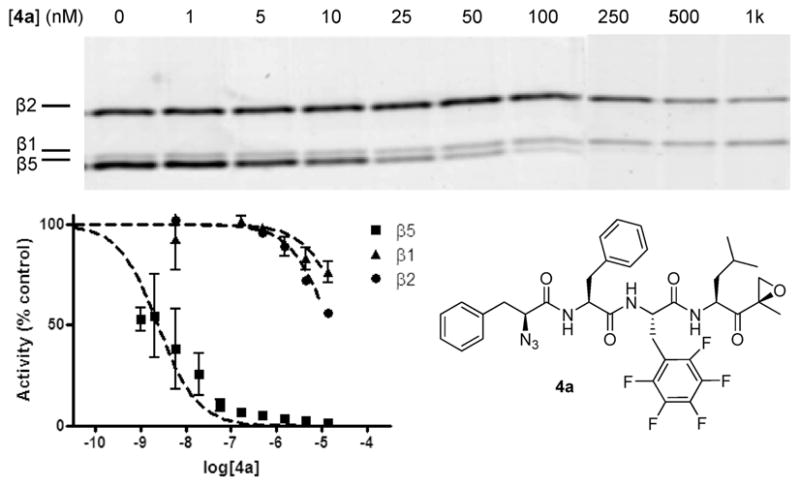
Characterization of specificity of compound 4a. Upper panel: Competition assay in HEK-293 lysate. Lysates were incubated for 1 h with compound 4a at the indicated final concentrations. Residual proteasome activity was labeled with 0.5 μM 9 for 1 h (This image was created from two separate gel images which, together, show a larger concentration series. The original images are shown in Figure S-4 in the SI). Lower panel: Remaining subunit activity after inhibition with compound 4a determined with fluorogenic peptides.
For the direct labeling of β5, a new fluorescent probe was made by reacting compound 4a with Bodipy-FL-alkyne18 in a Cu(I) mediated Huisgen 1,3-dipolar cycloaddition, giving green fluorescent probe 7 (Figure 5). The potential of this probe to label the β5 subunit was explored in a competition assay against 9 as explained before. The wet gel slab was scanned on a fluorescence scanner at two different wavelengths, allowing visualization of one of the two fluorescent dyes at a time. Figure 5A shows the read-out at 520 nm visualizing the appearance of one band: labeling of the β5 subunit by compound 7. The labeling is already visible at a concentration of 1 nM, and the subunit appears to be saturated (no more increase in the bands intensity) between 50 and 100 nM. At this point, only a faint β2 band is visible.19 The fluorescence read-out at 560 nm in Figure 5B (displaying labeling with 9 of remaining activities) reveals that the β5 band disappears while leaving the remaining subunit-bands intact.
Figure 5.

Novel probe for the β5 site. Competition assay in HEK-293 lysate. Lysates were incubated for 1 h with compound 7 at the indicated final concentrations. Residual proteasome activity was labeled with 0.5 μM 9 for 1 h. Fluorescence readout at (A) λex 488 nm, λem 520 nm (compound 7) and (B) λex 532 nm, λem 560 nm (compound 9). Reagents and conditions: (a) Bodipy-FL-alkyne,14 10 mol % CuSO4, 15 mol % sodiumascorbate, toluene/H2O/t-BuOH 1/1/1, 80 °C, yield 87%.
Conclusions
In summary, the effect of incorporation of fluorinated Phe in proteasome inhibitors was studied. We found that substitution of nonfluorinated Phe in compound 8 analogue 1 with fluorinated versions does not affect its selectivity for the different active subunits, whereas the potency is slightly increased for the Phe(m-CF3)2 version. In addition, we found that the effect of incorporation of fluorinated Phe in peptide epoxyketone proteasome inhibitors depends on the site of substitution. Fluorination of both the P2 and P3 sites decreases potency dramatically, however, fluorinated Phe at the P2 position hardly affects the potency but instead yields much more β5 selective inhibitors. Comparison of the results obtained with the boronic esters with those from the epoxyketone studies invites the tentative conclusion that β2 is most sensitive toward fluorine substituents. Compound 1 is ineffective toward β2 and introduction of fluorines into this sequence has no apparent effect. When comparing epoxyketone 3 with 4–6, however, the major difference is that the latter, fluorinated analogues leave β2 largely intact. Further studies, for instance, making use of different fluorine amino acids, are needed to substantiate this finding. Finally, compound 4a was identified as one of the most β5 selective inhibitors known to date and was converted to a β5 selective fluorescent probe (7), which can be used to label and visualize the β5 subunit selectively.
Experimental Section
All compounds tested are >95% pure on the basis of LC-MS. LC-MS analysis was performed on a Jasco HPLC system with a Phenomenex Gemini 3 μm C18 50 mm × 4.60 mm column (detection simultaneously at 214 and 254 nm), coupled to a PE Sciex API 165 mass spectrometer with ESI.
Compounds 2a and 2b were synthesized via the same method described in literature for compound 1.12
General Procedure for the Synthesis of Compounds 3–6
Hydrazine hydrate (20 equiv) was added to N3Phe-Phe-PheOMe (1 equiv) in MeOH (20 mL/mmol) and refluxed until TLC analysis revealed complete consumption of the starting material (usually after 3 h). Toluene was added, and the mixture was concentrated under reduced pressure followed by coevaporation with toluene (2×). The acylhydrazide (1 equiv) was dissolved in a 10/1 (v/v) mixture of dichloromethane/N,N-dimethylformamide (DCM/DMF) (10 mL/mmol) and cooled to −35 °C under argon. To this were added tert-butylnitrite (1.1 equiv) and HCl (2.8 equiv as a 4Msolution in 1,4-dioxane) and the mixture was stirred for 3 h at −35 °C. Next, a mixture of the deprotected amine-epoxyketone war-head (1.1 equiv, as a trifluoroacetic acid (TFA) salt) and N,N-diisopropylethylamine (DiPEA) (5 equiv) in DMF (1 mL) were added. The reaction was slowly warmed to room temperature and stirred for another 12 h before being diluted with DCM and extracted with 1 M HCl (2×), saturated NaHCO3 (2×) and brine. After drying (MgSO4) and concentrating, the obtained crude product was purified by column chromatography, applying a 1%→15% MeOH/DCM eluent system.
Competition Experiments
Whole cell lysates of HEK-293T were made by sonication in 3 volumes of lysis buffer containing 50 mM Tris pH 7.5, 1 mM dithiothreitol (DTT), 5 mM MgCl2, 250 mM sucrose, and 2 mM ATP. Protein concentration was determined by the Bradford assay. Cell lysates (13.5 μg total protein) were exposed to the inhibitors for 1 h prior to incubation with 9 (0.5 μM) for 1 h at 37 °C. Reaction mixtures were boiled with Laemmli’s buffer containing β-mercaptoethanol for 3 min before being resolved on 12.5% SDS-PAGE. In-gel detection of residual proteasome activity was performed in the wet gel slabs directly on a Typhoon variable mode imager (Amersham Biosciences) using the Cy3/Tamra settings (λex 532, λem 560 nm).
IC50 Determinations
Purified 26S proteasome (~10 ng/mL) was incubated with various concentrations of inhibitors at 37 °C for 30 min in the assay buffer (50 mM Tris-HCl, pH 7.5, 40 mM KCl, 2 mM EDTA, 1 mM DTT, 100 μM ATP, 50 μg/mL BSA). In the meantime, 100 μM solution of the fluorogenic peptide substrates (Suc-LLVY-7-amido-4-methyl-coumarin (amc) for the β5 site, Ac-nLPnLD-amc for the β1 site, and Ac-RLR-amc or Ac-RQR-amc for the β2 site) in the assay buffer were pre-incubated at 37 °C. Immediately after the end of this incubation, an aliquot of the inhibitor-treated proteasome was mixed with the substrate, and fluorescence of released amc was measured continuously for 30 min at 37 °C. The rate of reaction was determined from the slope of the reaction progress curves. Mock-treated proteasomes served as control. Residual activity in inhibitor treated samples were plotted against concentration of inhibitors, and IC50 values were determined from these plots.
Supplementary Material
Acknowledgments
This work was supported by The Netherlands Organization for Scientific Research (NWO), The Netherlands Genomics Centre Initiative (NGI) and The NCI (grant RO1CA124634). We thank Hans van den Elst and Nico Meeuwenoord for HPLC and LC-MS assistance.
Footnotes
Abbreviations: Phe(F5), pentafluorophenylalanine; Phe(m-CF3)2, 3,5-bis(trifluoromethyl)phenylalanine, HEK-293, human embryonic kidney cell lysates; EL-4, mouse lymphoma cell line; DiPEA, N,N-diisopropylethylamine; DTT, dithiothreitol; Tris, tris(hydroxymethyl)-aminomethane; amc, 7-amido-4-methylcoumarin; TFA, trifluoroacetic acid; DMF, N,N-dimethylformamide; DCM, dichloromethane.
Supporting Information Available: Complete synthetic details and characterization of all compounds and additional biological experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Borissenko L, Groll M. 20S Proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 2.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 3.Adams J, Behnke M, Chen SW, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, Stein RL. Potent and selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorg Med Chem Lett. 1998;8:333–338. doi: 10.1016/s0960-894x(98)00029-8.For the structure of compound 8, see SI.
- 4.Examples of selective inhibitors for (a) β1: Van Swieten PF, Samuel E, Hernáandez RO, van den Nieuwendijk AMCH, Leeuwenburgh MA, van der Marel GA, Kessler BM, Overkleeft HS, Kisselev AF. A cell-permeable inhibitor and activity-based probe for the caspase-like activity of the proteasome. Bioorg Med Chem Lett. 2007;17:3402–3405. doi: 10.1016/j.bmcl.2007.03.092.β2: Nazif T, Bogyo M. Global analysis of proteasomal substrate specificity using positional-scanning libraries of covalent inhibitors. Proc Natl Acad Sci USA. 2001;98:2967–2972. doi: 10.1073/pnas.061028898.β5: Elofsson M, Splittgerber U, Myung J, Mohan R, Crews CM. Towards subunit-specific proteasome inhibitors: synthesis and evaluation of peptide α′,β′-epoxyketones. Chem Biol. 1999;6:811–822. doi: 10.1016/s1074-5521(99)80128-8.
- 5.Kim KB, Myung J, Sin N, Crews CM. Proteasome inhibition by the natural products epoxomicin and dihydroeponemycin: insights into specificity and potency. Bioorg Med Chem Lett. 1999;9:3335–3340. doi: 10.1016/s0960-894x(99)00612-5. [DOI] [PubMed] [Google Scholar]
- 6.Britton M, Lucas MM, Downey SL, Screen M, Pletnev AA, Verdoes M, Tokhunts RA, Amir O, Goddard AL, Pelphrey PM, Wright DL, Overkleeft HS, Kisselev AF. Selective inhibitor of proteasome’s caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem Biol. 2009;16:1278–1289. doi: 10.1016/j.chembiol.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verdoes M, Florea BI, van der Linden WA, Renou D, van den Nieuwendijk AMCH, van der Marel GA, Overkleeft HS. Mixing of peptides and electrophilic traps gives rise to potent, broad-spectrum proteasome inhibitors. Org Biomol Chem. 2007;5:1416–1426. doi: 10.1039/b702268a. [DOI] [PubMed] [Google Scholar]
- 8.Formicola L, Maréechal X, Basse N, Bouvier-Durand M, Bonnet-Delpon D, Milcent T, Reboud-Ravaux M, Ongeri S. Novel fluorinated pseudopeptides as proteasome inhibitors. Bioorg Med Chem Lett. 2009;19:83–86. doi: 10.1016/j.bmcl.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 9.(a) Berkowitz DB, Karukurichi KR, de la Salud-Bea R, Nelson DL, McCune CD. Use of fluorinated functionality in enzyme inhibitor development: mechanistic and analytical advantages. J Fluor Chem. 2008;129:731–742. doi: 10.1016/j.jfluchem.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Molteni M, Pesenti C, Sani M, Volonterio A, Zanda M. Fluorinated peptidomimetics: synthesis, conformational and biological features. J Fluor Chem. 2004;125:1735–1743. [Google Scholar]; (c) Zanda M. Trifluoromethyl group: an effective xenobiotic function for peptide backbone modification. New J Chem. 2004;28:1401–1411. [Google Scholar]
- 10.For examples see: Zheng H, Comeforo K, Gao J. Expanding the fluorous arsenal: tetrafluorinated phenylalanines for protein design. J Am Chem Soc. 2009;131:18–19. doi: 10.1021/ja8062309.Holmgren SK, Taylor KM, Bretscher LE, Raines RT. Code for collagen's stability deciphered. Nature. 1998;392:666–667. doi: 10.1038/33573.Jäackel C, Salwiczek M, Koksch B. Fluorine in a native protein environment; how the spatial demand and polarity of fluoroalkyl groups affect protein folding. Angew Chem, Int Ed. 2006;45:4198–4203. doi: 10.1002/anie.200504387.
- 11.(a) Flaherty DP, Walsh SM, Kiyota T, Dong Y, Ikezu T, Vennerstrom JL. Polyfluorinated bis-styrylbenzene β-amyloid plaque binding ligands. J Med Chem. 2007;50:4986–4992. doi: 10.1021/jm070085f. [DOI] [PubMed] [Google Scholar]; (b) Wadhwani P, Büurck J, Strandberg E, Mink C, Afonin S, Ulrich AS. Using a strerically restrictive amino acid as a 19F NMR label to monitor and to control peptide aggregation in membranes. J Am Chem Soc. 2008;130:16515–16517. doi: 10.1021/ja804928q. [DOI] [PubMed] [Google Scholar]
- 12.Verdoes M, Florea BI, Menendez-Benito V, Maynard CJ, Witte MD, van der Linden WA, van den Nieuwendijk AMCH, Hofmann T, Berkers CR, van Leeuwen FWB, Groothuis TA, Leeuwenburgh MA, Ovaa H, Neefjes JJ, Filippov DV, van der Marel GA, Dantuma NP, Overkleeft HS. A fluorescent broad-spectrum proteasome inhibitor for labeling proteasomes in vitro and in vivo. Chem Biol. 2006;13:1217–1226. doi: 10.1016/j.chembiol.2006.09.013.For the structure of compound 9, see SI.
- 13.(a) Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J, Shwonek P, Parlati F, Demo SD, Bennett MK, Kirk CJ, Groettrup M. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med. 2009;15:781–788. doi: 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]; (b) Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, Bennett MK. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007;67:6383–6391. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 14.Van Swieten PF, Leeuwenburgh MA, Kessler BM, Overkleeft HS. Bioorthogonal organic chemistry in living cells: novel strategies for labeling biomolecules. Org Biomol Chem. 2005;3:20–27. doi: 10.1039/b412558d. [DOI] [PubMed] [Google Scholar]
- 15.Park HG, Jeong BS, Yoo MS, Lee JH, Park MK, Lee YJ, Kim MJ, Jew SS. Highly enantioselective and practical cinchona-derived phase-transfer catalysis for the synthesis of α-amino acids. Angew Chem, Int Ed. 2002;41:3036–3038. doi: 10.1002/1521-3773(20020816)41:16<3036::AID-ANIE3036>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 16.The fluorescence intensity of the bands in Figure 3 was quantified to get an impression of the residual proteasome subunit activity. These values are shown in Table 2 in the SI.
- 17.Kisselev AF, Garcia-Calvo M, Overkleeft HS, Peterson E, Pennington MW, Ploegh HL, Thornberry NA, Goldberg AL. The caspase-like sites of proteasomes, their substrate specificity, new inhibitors and substrates, and allosteric interactions with the trypsin-like sites. J Biol Chem. 2003;278:35869–35877. doi: 10.1074/jbc.M303725200. [DOI] [PubMed] [Google Scholar]
- 18.Verdoes M, Hillaert U, Florea BI, Sae-Heng M, Risseeuw MDP, Filippov DV, van der Marel GA, Overkleeft HS. Acetylene functionalized BODIPY dyes and their application in the synthesis of activity based proteasome probes. Bioorg Med Chem Lett. 2007;17:6169–6171. doi: 10.1016/j.bmcl.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 19.The IC50 values of compound 7 for each active subunit were determined in the same manner as for the other compounds: β1>10 μM, β2>10 μM, β5 0.30 μM.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.