

Abstract
We analyzed the whole genome sequences of a family of four, consisting of two siblings and their parents. Family-based sequencing allowed us to delineate recombination sites precisely, identify 70% of the sequencing errors, and identify very rare SNVs. We also directly estimated a human intergeneration mutation rate of ∼1.1×10-8 per position per haploid genome. Both offspring in this family have two recessive disorders--Miller syndrome, for which the gene was concurrently identified, and primary ciliary dyskinesia, for which causative genes have been previously identified. Family-based genome analysis enabled us to narrow the candidate genes for both of these Mendelian disorders to only four. Our results demonstrate the unique value of complete genome sequencing in families.
Keywords: whole genome sequencing, rare genetic disease, inheritance analysis, recessive models, de novo mutations, recombination hotspot, crossover, haploidentity, haploidentical block, inheritance state, inheritance vector, HMM, haplotype, Miller syndrome, POADS, DHODH, DNAH5, KIAA0556, CES1
Whole-genome sequences from four members of a family represent a qualitatively different type of genetic data than whole-genome sequences from individual or sets of unrelated genomes. They enable inheritance analyses that detect errors and permit the identification of precise locations of recombination events. This leads in turn to near-complete knowledge of inheritance states by precisely determining the parental chromosomal origins of sequence blocks in offspring. Confident predictions of inheritance states and haplotypes power analyses including identification of genomic features with non-classical inheritance patterns such as hemizygous deletions or copy number variants (CNVs). Identification of inheritance patterns in the pedigree permits the detection of ∼70% of sequencing errors and sharply reduces the search space for disease-causing variants. These analyses would be far less powerful in studies that had fewer markers (e.g., standard genotype or exome datasets) or that had sequence from fewer family members.
DNA from each family member was extracted from peripheral blood cells and sequenced by Complete Genomics Inc. with a nanoarray-based short-read sequencing-by-ligation technology (1) including an adaptation of the pairwise end-sequencing strategy (2). Reads were mapped to the NCBI reference genome (coverage averaged 40×; Fig. S1, Tables S1 & S2). Polymorphic markers employed for this analysis were SNVs with at least two variants among the four genotypes of the family, averaging 802 bp between markers. We observed 4,471,510 positions at which at least one family member had an allele that varied from the reference genome. This corresponds to a Watterson's theta (θW) of 9.5×10-4 per site for the two parents and the reference sequence (3), given the fraction of the genome successfully genotyped in each parent (Fig. S1). This is a close match to the estimate of θW = 9.3×10-4 we obtained by combining two previously published European genomes and the reference sequence (7). Of the 4.5 million variant positions, 3,665,772 were variable within the family – the rest were homozygous and identical in all four members. Comparisons to known SNVs show that 323,255 of these 3.7 million SNVs are novel.
For each meiosis in a pedigree, each base position in a resulting gamete will have inherited one of two parental alleles. The number of inheritance patterns of the segregation of alleles in gametes is therefore 2n, where n is the number of meioses in a pedigree. In a nuclear family of four, the Mendelian inheritance patterns can be grouped into four inheritance states for each variant position, with children receiving: 1) the same allele from both the mother and the father (identical), 2) the same allele from the mother but opposites from the father (haploidentical maternal), 3) the same allele from the father, but opposites from the mother (haploidentical paternal), and 4) opposites from both parents (nonidentical) (Fig. S2). Adjacent variant base pairs in alignments of the family genomes have the same inheritance state unless a recombination has occurred between these bases in one of the meioses. This delineates inheritance blocks.
Many algorithms can identify the boundaries of blocks, and theory-driven implementations are in wide use (4-6). For our complete genome sequence data, we developed an algorithm to identify all states, including non-Mendelian states. One non-Mendelian state will occur in regions where highly similar sequences are inadvertently compressed computationally (e.g., during sequence assembly of CNVs). In such a “compression block,” many positions will appear to be heterozygous in all individuals, regardless of the inheritance patterns of the positions contributing to the compression. Other non-Mendelian patterns are seen in regions prone to errors in sequence calling or assembly, or that have inherited hemizygous deletions. For both of these patterns, many positions will be observed as Mendelian inheritance errors (MIEs). Our algorithm identified six states: one for each of the four Mendelian inheritance states, one for a compression state, and one for an MIE-prone state (7). We identified 1.5% of the genome in this pedigree as 409 compression blocks and 1.7% as 126 error-prone blocks. Since these blocks are a source of false positives for recombination predictions, SNVs, and disease candidate alleles, it is important to identify them (Fig. 1). The power to precisely determine inheritance state boundaries is striking in families of at least four, and would be reduced had we sequenced fewer individuals (Fig. 2). Meiotic gene conversions could in principle be recognized in the same way as inheritance blocks: they would be indistinguishable from a short region flanked by meiotic recombinations in the same meiosis. We found that the great majority of candidate gene conversions regions were caused by reads mismapped to repetitive DNA, like CNVs or satellites, and did not conclusively identify gene conversion regions.
Figure 1.
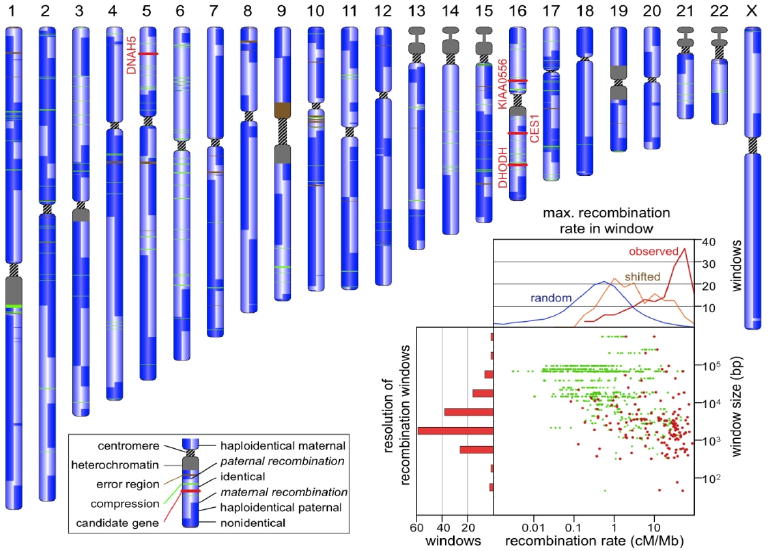
The landscape of recombination. Each chromosome in this schematic karyotype is used to represent information abstracted from the four corresponding chromosomes of the two children in the pedigree. It is vertically split to indicate the inheritance state from the father (left half) and mother (right half) as shown in the key. The three compound heterozygous (DHODH, DNAH5, KIAA0556) and one recessive (CES1) candidate gene, depicted by red bands, lie in “identical” blocks. Inset: Scatterplot of HapMap recombination rates (in centimorgans per megabase, cM/Mb) within the predicted crossover regions. The maximum value of cM/Mb found in each window is shown in red. The left hand histogram shows the size distribution of recombination windows (log10 value: -0.58 ± 0.92). The upper graph shows the cM/Mb distribution for the observed maximal values (red), for similarly sized windows shifted by 6 kb (orange), and for similarly sized windows randomly chosen from the entire genome (blue). Note that a shift of 6 kb from the observed locations eliminates the correlation with hotspots. Of 155 recombination windows, 92 contained a HapMap site with >10 cM/Mb. Only five randomly picked windows are expected to contain such high recombination rates.
Figure 2.
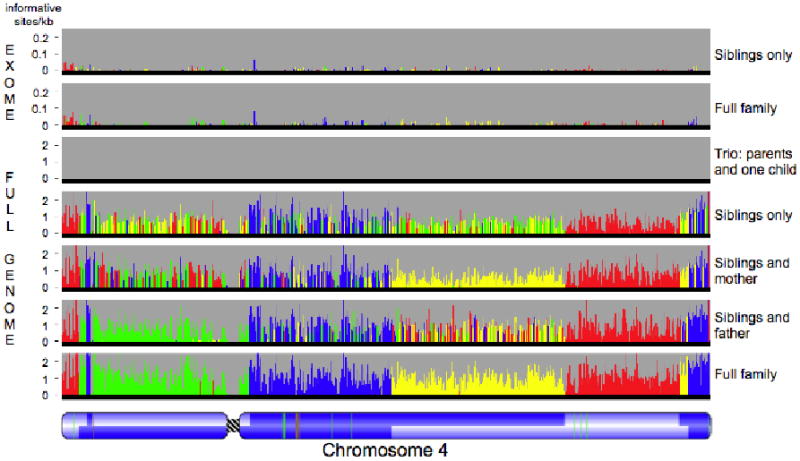
Power of four. Inheritance states illustrated for a single chromosome in six scenarios representing restrictions of the dataset to the exome (for two siblings only or for the full family) or to subsets of the family (parents and one child, two siblings, siblings and one parent), compared to inheritance state consistency analysis (ISCA) with full data from all four family members. The most supported state for each bin is shown as a color; the height of each histogram bar is proportional to the number of informative markers supporting that state. The father has two regions of homozygosity (thin red lines, bottom panel) on the short arm of the chromosome, where it is not possible to distinguish the haploidentical maternal from identical states (Fig. S2A, panel b). These regions are undetected when the mother's genotypes are missing, because all markers positions in the region are uninformative (second to bottom panel). A pedigree of two parents and one child has only one inheritance state, and so provides no information on recombination. Red, identical; blue, nonidentical; green, haploidentical maternal; yellow, haploidentical paternal. Chromosome structure is annotated as in Fig. 1.
Recombination in maternal meioses is thought to occur 1.7 times more frequently than in paternal meioses (8). We inferred 98 crossovers in maternal and 57 in paternal meioses (count includes both offspring), consistent with this estimate. The median resolution of the 155 crossover sites was 2.6 kb, with a few sites localized within a 30-bp window (Fig. 1). Crossover sites were significantly correlated with hotspots of recombination as inferred from HapMap data, where a hotspot is defined as a region with ≥10 cM/Mb; 92 of the 155 recombinations took place in a hotspot.
By identifying inconsistencies across the 22% of the genomes of the two children in “identical” blocks, for which they are effectively twins, we computed an error rate of 1.0×10-5. We also determined error rate by other methods, including resequencing, which gave similar estimates, ranging from 8.1×10-6 to 1.1×10-5 (7). Furthermore, ∼70% of the errors in a four person pedigree can be detected as apparent MIEs and inconsistencies in inheritance state blocks, so the effective base-pair error rate in the context of a pedigree is ∼3×10-6.
Analysis of the mutation rate, including germline and early embryonic somatic mutations, requires highly accurate sequence data. Even with such data, however, most apparent aberrations in allele inheritance will be due to errors in the data and not mutation. Our data had thousands of such false-positive candidates for each true de novo mutation. Our initial data encompassed 2.3 billion bases and contained 49,720 candidate MIEs consistent with the presence of a single-nucleotide mutation. After excluding sites in MIE-prone and compression states as well as sites that were unsuitable for probe design, 33,937 potential mutations among 1.83 billion bases remained. We resequenced each of these candidates, applying a stringent base-calling algorithm to confirm 28 candidates as de novo mutations. In a final confirmation step, we verified all 28 mutations with mass spectrometry (7; Table S3), corresponding to a mutation rate of 3.8×10-9 per position per generation per haploid genome.
Since the raw estimate of 3.8×10-9 does not account for the true mutations that were not conclusively identified by resequencing, we estimated a false-negative rate by applying the base-calling algorithm to five Mb of independent resequencing data, divided into 25 randomly selected regions of the genome. A comparison of the resequencing data with the complete genome sequence for the same regions provided a de novo mutation false negative rate of 0.662 (95% C.I. 0.644 – 0.680). Adjusting for the false-negative rate produced an unbiased mutation rate estimate of 1.1×10-8 per position per haploid genome, corresponding to approximately 70 new mutations in each diploid human genome (95% confidence interval of 6.8×10-9 to 1.7×10-8) (7). In great apes, CpG sites are reported to mutate at a rate eleven times higher than other sites (9). We observed 5 CpG mutations, closely matching this estimate. Of the remaining 23 mutations, seven were transversions and sixteen were transitions. This yields a transition-to-transversion ratio of 2.3 (Table S3), once again similar to a previous estimate of 2.2 for non-CpG sites (10).
Although both the observed transition-to-transversion ratio and the proportion of CpG mutations in our data match predictions, our estimated human mutation rate is lower than previous estimates, the most widely cited of which is 2.5×10-8 per generation (10) based on three parameters: a human-chimpanzee nucleotide divergence per site (Kt) of 0.013, a species divergence time of five million years ago, and an ancestral effective population size of 10,000. More recent estimates indicate a nucleotide divergence of 0.012 (9), species divergence time between six and seven million years ago (11-15), and ancestral effective population size between 40,000 and 148,000 (16-19). With these parameter ranges and a generation length of 15 to 25 years, the mutation rate estimate is between 7.6×10-9 and 2.2×10-8 per generation, which is consistent with our intergenerational estimate of 1.1×10-8. Our estimate is within one standard deviation (SD) of an earlier estimate of 1.7×10-8 (SD: 9×10-9) based on 20 disease-causing loci (20). The rate we report is for autosomes, and should be lower than that of the Y chromosome, as in the male germline more cell divisions occur per generation. Though our rate differs approximately as expected from the recently reported estimate of 3.0×10-8 (95% CI: 8.9×10-9 – 7.0×10-8) for the Y chromosome, the error rates make this difference not significant (21).
Genomic inheritance analysis facilitates the identification of alleles that cause genetic disorders. Because genome sequences from a family of four provide near-exact determination of inheritance state boundaries, the number of false-positive disease gene candidates is greatly reduced compared to analyses lacking the context of a pedigree or complete genome sequence (Tables S3 & S4, Fig. 3). Two disorders in this family, Miller syndrome and primary ciliary dyskinesia, which affect both offspring but neither parent, provided an opportunity to test this application. A parsimonious explanation is that each phenotype arises from defects in a single gene, or a site regulating a single gene. The inheritance mode is undetermined, but a recessive mode is more consistent with observed data. We therefore examined each candidate variant by testing each of three inheritance modes: dominant, simple recessive, or compound heterozygote (a subcategory of recessive).
Figure 3.
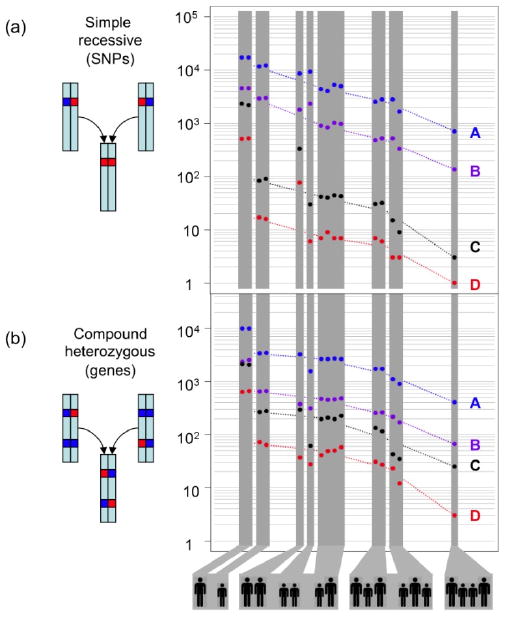
The power of family genome inheritance analysis. The number of false-positive candidates drops exponentially as the number of family members increases. (A) Number of candidate SNVs consistent with a simple recessive inheritance mode. (B) Number of candidate genes consistent with a compound heterozygous model. The different groupings of parents (large silhouettes) and children (small silhouettes) are depicted below. Dashed lines join the average values of each grouping. For this figure, probably detrimental includes missense, nonsense, splice defect, and non-initiation; possibly detrimental also includes UTR, non-coding, and splice-region. A block of SNVs such that all SNPs in the block are within 5 kb of another SNV in the block is counted only once, as together these are likely to encode at most one phenotype. A: all possibly detrimental SNVs; B: all probably detrimental SNVs; C: rare possibly detrimental SNVs; D: rare probably detrimental SNVs.
The two recessive modes require that both offspring have identical, dysfunctional variants for which the parents are heterozygous, and which may come either from the same position (simple recessive), or occur at distinct positions within the same gene (compound heterozygote). Genes consistent with these two recessive modes must lie in “identical” inheritance blocks, since both offspring are affected, limiting the search space to the 22% of the genome in these blocks. Since the phenotypes are rare, they are likely to be encoded by rare variants, which further limits the possibilities. Only two missense SNVs in the CES1 gene matched the simple recessive mode (Table S4), while three genes fit the compound heterozygote mode: DHODH, DNAH5, and KIAA0556 (Fig. 1). A small number of possibly detrimental variants outside exons also matched the simple recessive mode: two in highly conserved regions, one in an intronic sequence near a splice site, five in non-protein coding transcripts, and one in a UTR. Concurrent with this study, the core exomes of the two affected offspring were sequenced along with those of two unrelated individuals with Miller syndrome (22). Compared to that study of only affected individuals, our analysis of just two affected offspring and their unaffected parents reduced the number of gene candidates in the core exome from nine to four; had we not sequenced the parents, we would have had 34 rather than four candidates (Fig. 3 & Table S5). The exome study supported DHODH as the primary gene for Miller syndrome. DNAH5 had been previously identified as a cause of primary ciliary dyskinesia, and so was likely the cause in these offspring as well (23).
Family genome analysis can clearly be effective for finding candidate genes encoding Mendelian traits because sequence accuracy is enhanced. In addition, delineation of recombination sites identifies inherited chromosome segments precisely, and reduces the chromosomal search space for candidate genes (in this case to 22% of the genome). The ability to identify large effects of very rare alleles in small pedigrees can complement the power of genome-wide association studies in identifying weak effects of common alleles in large populations. An unknown fraction of important phenotypes in humans are encoded by non-exonic variants identified only by whole-genome sequencing. When the cost of recruiting additional families is expensive relative to sequencing costs, sequencing genomes of families will be an economical strategy for the identification of many disease-causing genes. Constraining searches to very rare variants can provide considerable power, as recently demonstrated for Freeman-Sheldon syndrome and congenital chloride diarrhea (24, 25). De novo mutations can be assayed, either as we have reported here, or through family sequencing of more than two generations. As our knowledge of gene function increases, we will be able to use the power of family genome analysis rapidly to identify disease-gene candidates. These data, along with relevant environmental and medical information, will characterize the integrated medical records of the future.
Supplementary Material
Acknowledgments
This study was supported by the University of Luxembourg Institute for Systems Biology Program and by these NIH grants: Center for Systems Biology GM076547 (L.H. and L.R.), RO1GM081083 (A.F.S. and G.G.), R01HL094976 and R21HG004749 (J.S.), RC2HG005608 (M.B. and J.S.), and R01HD048895 (M.B.). H. Tabor assisted with ethical review. J. Xing performed the PCA analysis. H. Mefford performed CNV analysis. A. Bigham and K. Buckingham evaluated candidate genes in unrelated individuals. D. Ballinger, A. Sparks, A. Halpern, and G. Nilsen assisted with sequencing and analysis. R. Bressler, S. Dee, and D. Mauldin assisted with bioinformatics. S. Ng and R. Qiu performed the capture array. S. Bloom obtained the resequencing data on the Illumina Genome Analyzer. M. Janer and S. Li performed Sequenom analysis. D. Cox commented on an early version of the manuscript. R. Durbin and D. Altshuler granted permission for our use of 1000genomes SNP data. The dbGAP accessions can be found at www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs000244.v1.p1 (accession phs000244.v1.p1).
References and Notes
- 1.Drmanac R, et al. Science. 2009 November 5;:1181498. [Google Scholar]
- 2.Roach JC, Boysen C, Wang K, Hood L. Genomics. 1995 Mar 20;26:345. doi: 10.1016/0888-7543(95)80219-c. [DOI] [PubMed] [Google Scholar]
- 3.Watterson GA. Theor Popul Biol. 1975 Apr;7:256. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 4.Donnelly KP. Theor Popul Biol. 1983 Feb;23:34. doi: 10.1016/0040-5809(83)90004-7. [DOI] [PubMed] [Google Scholar]
- 5.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Am J Hum Genet. 1996 Jun;58:1347. [PMC free article] [PubMed] [Google Scholar]
- 6.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Nat Genet. 2002 Jan;30:97. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 7.Supplemental Online Material.
- 8.Petkov PM, Broman KW, Szatkiewicz JP, Paigen K. Trends Genet. 2007 Nov;23:539. doi: 10.1016/j.tig.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 9.Chimpanzee Sequencing and Analysis Consortium. Nature. 2005 Sep 1;437:69. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 10.Nachman MW, Crowell SL. Genetics. 2000 Sep;156:297. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haile-Selassie Y. Nature. 2001 Jul 12;412:178. doi: 10.1038/35084063. [DOI] [PubMed] [Google Scholar]
- 12.Haile-Selassie Y, Asfaw B, White TD. Am J Phys Anthropol. 2004 Jan;123:1. doi: 10.1002/ajpa.10330. [DOI] [PubMed] [Google Scholar]
- 13.Haile-Selassie Y, Suwa G, White TD. Science. 2004 Mar 5;303:1503. doi: 10.1126/science.1092978. [DOI] [PubMed] [Google Scholar]
- 14.Deino AL, Tauxe L, Monaghan M, Hill A. J Hum Evol. 2002 Jan-Feb;42:117. doi: 10.1006/jhev.2001.0521. [DOI] [PubMed] [Google Scholar]
- 15.Brunet M, et al. Nature. 2002 Jul 11;418:145. [Google Scholar]
- 16.Chen FC, Li WH. Am J Hum Genet. 2001 Feb;68:444. doi: 10.1086/318206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burgess R, Yang Z. Mol Biol Evol. 2008 Sep;25:1979. doi: 10.1093/molbev/msn148. [DOI] [PubMed] [Google Scholar]
- 18.Takahata N. Jpn J Genet. 1993 Dec;68:539. doi: 10.1266/jjg.68.539. [DOI] [PubMed] [Google Scholar]
- 19.Wall JD. Genetics. 2003 Jan;163:395. doi: 10.1093/genetics/163.1.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondrashov AS. Hum Mutat. 2003 Jan;21:12. doi: 10.1002/humu.10147. [DOI] [PubMed] [Google Scholar]
- 21.Xue Y, et al. Curr Biol. 2009 Sep 15;19:1453. doi: 10.1016/j.cub.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng SB, et al. Nat Genet. 2010 Jan;42:30. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olbrich H, et al. Nat Genet. 2002 Feb;30:143. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 24.Ng SB, et al. Nature. 2009 Sep 10;461:272. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi M, et al. Proc Natl Acad Sci U S A. 2009 Oct 27; [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.