

Abstract
Aims
Nitric oxide triggers cGMP-dependent kinase-mediated phosphorylation of the actin regulator vasodilator stimulated phosphoprotein (VASP) at residue serine239. The function of this phosphorylation for smooth muscle cell (SMC) adhesion, spreading, matrix contraction, and invasion is not well understood.
Methods and Results
We reconstituted VASP deficient SMC with wild-type VASP (wt-VASP) or VASP-mutants that mimic “locked” serine239 phosphorylation (S239D-VASP) or “blocked” serine239 phosphorylation (S239A-VASP). Collagen gel contraction was reduced in S239D-VASP compared to S239A-VASP and wt-VASP expressing cells and nitric oxide stimulation decreased gel-contraction of wt-VASP reconstituted SMC. Invasion of collagen was enhanced in S239D-VASP and nitric oxide-stimulated wild-type SMCs compared to S239A-VASP expressing cells. Expression of S239D-VASP impaired SMC attachment to collagen, reduced the number of membrane protrusions, and caused cell rounding compared to expression of S239A-VASP. Treatment of wt-VASP reconstituted SMCs with nitric oxide exerted similar effects as expression of S239D-VASP. As unstimulated cells were spreading on collagen S239A-VASP and wt-VASP localized to actin fibers whereas S239D-VASP was enriched in the cytosol.
Conclusions
Nitric oxide interferes with SMC invasion and contraction of collagen matrices. This requires phosphorylation of VASP on serine239, which reduces VASP binding to actin fibers. These findings support the conclusion that VASP phosphorylation at serine239 regulates cytoskeleton remodeling.
Keywords: VASP, phosphorylation, SMC contraction, SMC matrix invasion
Introduction
Nitric oxide (NO), first characterized as an important arterial vasodilator (Cohen, 1995; Loscalzo and Welch, 1995; Umans and Levi, 1995), also regulates other smooth muscle cell (SMC) functions that depend on actin organization. These include cell migration (Dubey and et al., 1995; Sarkar and et al., 2002; Chang and et al., 2002), adhesion (Hassid and et al., 1999; Pelletier and et al., 2005), and proliferation (Garg and Hassid, 1989; Ignarro and et al., 2001; Chen and et al., 2004).
NO effects are mediated in part by the activation of soluble guanylyl cyclase (sGC), which increases tissue levels of the second messenger cGMP that in turn activates cGMP-dependent protein kinase (PKG). Among the substrates of PKG is the actin binding protein, vasodilator-stimulated phosphoprotein (VASP) (Reinhard and et al., 2001). VASP belongs to the Ena/VASP family that is known to be involved in regulating cytoskeletal dynamics such as actin filament nucleation (Huttelmaier and et al., 1999), profilin and G-actin recruitment (Reinhard and et al., 1995; Walders-Harbeck and et al., 2002), anti-capping activity (Bear and et al., 2002; Barzik and et al., 2005), filament detachment (Samarin and et al., 2003), and filament bundling (Schirenbeck and et al., 2006). VASP consists of three functional domains. The Ena/VASP homology 1 (EVH-1) domain binds specifically to poly-proline sequences in target proteins such as zyxin, vinculin and Fyb/SLAP and helps to recruit Ena/VASP to the leading edge of cells (Reinhard and et al., 2001; Krause and et al., 2003; Kwiatkowski and Gertler, 2003). The central proline-rich region binds SH3 and WW domains of various signaling molecules as well as profilin, an abundant actin-monomer carrier (Reinhard and et al., 2001; Krause and et al., 2003; Kwiatkowski and Gertler, 2003). Finally, the Ena/VASP homology 2 (EVH-2) domain comprises globular and filamentous actin-binding sites (GAB and FAB) that are involved in actin bundling (Schirenbeck and et al., 2006), and a C-terminal coiled-coil (CC) region that promotes VASP tetramerization (Bachmann and et al., 1999; Reinhard and et al., 2001; Kwiatkowski and Gertler, 2003; Kühnel and et al., 2004). Three phosphorylation sites have been identified in human VASP: serine157, serine239 and threonine278. In mouse VASP, the conserved phosphorylation sites are serine153, serine235, and threonine274. Serine239 is the preferred target for the nitric oxide (NO)/cGMP/PKG pathway, while serine157 is preferentially phosphorylated by cAMP-dependent protein kinase and protein kinase C (Butt and et al., 1994; Smolenski and et al., 2000; and Chitaley and et al., 2004). Threonine278 is not phosphorylated by PKG in intact cells and is a site for AMP-activated kinase (Oelze and et al., 2000; Blume and et al., 2007). We recently demonstrated that VASP phosphorylation at serine239 is involved in the growth inhibitory effects of NO on SMCs (Chen and et al., 2004). In an ex-vivo model, Schäfer et al. observed a correlation between VASP phosphorylation and vascular relaxation (Schafer and et al., 2003). Whether VASP phosphorylation at serine239 mediates the effect of NO on SMC relaxation is not clear. In addition, the role of NO as a modulator of SMC motility in vitro is also controversial. Several studies have shown that NO has the capacity to decrease SMC motility in culture (Sarkar and et al., 1996; Hassid and et al., 1999; Sreejayan and et al., 2002; Schafer and et al., 2003), while others demonstrated NO increases cell motility (Brown and et al., 1999; Brown and et al., 2001).
In this study, we tested the hypothesis that VASP phosphorylation at residue S239 mediates the NO-driven regulation of vascular SMC invasion, adhesion, and matrix contraction. To study the effects of phosphorylation on serine239, we used retroviral-mediated gene transfer to introduce either wild-type VASP (wt-VASP), a non-phosphorylatable VASP (S239A-VASP), or a phosphorylation mimicking VASP mutant (S239D-VASP) into cultured vascular SMCs isolated from VASP-/- mice.
Materials and Methods
Materials
Cell culture medium (DMEM), penicillin, and streptomycin were purchased from Invitrogen-Gibco Life Technologies. Fetal bovine serum (FBS) was purchased from Atlantic Biological. Type I collagen (Vitrogen) was purchased from Collagen Corporation. Rp-8-pCPT-cGMPS was purchased from BioLog. Deta NONOate was purchased from Cayman chemical. G418 was purchased from Calbiochem. Image-iT™ FX signal enhancer was purchased from Invitrogen. Rabbit anti-VASP antibody (M4, affinity purified) was purchased from Alexis and mouse anti-β-smooth muscle actin was purchased from Sigma. Rhodamine-phalloidin was purchased from Cytoskeleton. Alexa-568 conjugated goat anti-rabbit antibody was purchased from Molecular Probes.
Transfection of SMCs from VASP-/- mice with retroviral vectors encoding wt-VASP and VASP mutants
Retroviral constructs encoding either human wt-VASP or the VASP mutants (S239A-VASP and S239D-VASP), in which serine239 was replaced by alanine or aspartic acid respectively, were generated by insertion of the gene into the retroviral vector LXSN (kindly provided by A.D. Miller) (Miller AD, Rosman, 1989). Phosphomimetic VASP mutants were described and characterized previously (Benz and et al., 2008). The packaging cells were transfected with the constructs and selected (Miller AD, Rosman, 1989). Aortic SMCs, isolated from 2 month old VASP-/- mice on a C57BL/6X129sv background (Massberg and et al., 2004), were infected with LXSN (LXSN-SMCs), wt-VASP (wt-VASP SMCs), S239A-VASP (S239A-VASP SMCs), or S239D-VASP (S239D-VASP SMCs) virus. Multiple clones of each were selected, propagated and maintained in the presence of G418 (0.6mg/ml). The expression levels of VASP in various clones were assessed by Western blotting and clones with mutant VASP expressing levels similar to endogenous protein in wt-C57Bl/6 SMCs were selected for further study.
Collagen Gel Contraction Assay
All cell types were maintained in 15% serum and mixed with type I collagen at a final concentration of 3×105 cell/ml and 1mg collagen/ml. When indicated, SMCs were pretreated with DMEM in the presence or absence of deta NONOate (100 μM) or Rp-8-pCPT-cGMPS (50 μM) for 30 minutes at room temperature. The mixture of cells and collagen polymerized in 24-well tissue culture plates (1ml per well) that were precoated with 1% agarose at 37°C in a 5% CO2 incubator for 1h. Then, 1 ml of DMEM containing 15% FBS with or without deta NONOate (100μM) or Rp-8-pCPT-cGMPS (50 μM) was added. Gel contraction was measured at different times as indicated by scanning the plates with a transmission flat bed scanner and determining the area covered by the gel using ImageQuant (Molecular Dynamics). The area of an empty well (2 cm2) was used as a reference. Assays were performed in triplicate.
Cell Invasion Assays
All cell types were trypsinized and resuspended in DMEM containing 15% FBS, followed by three washes in serum-free media. When indicated, SMCs were pretreated with 100 μM deta NONOate in DMEM for 30 minutes at room temperature. The invasion assay was performed using Transwell (Costar) 24-well tissue-culture plates composed of a polycarbonate membrane containing 5 μm pores as described by Kanda et al (Kanda and et al., 2000). Briefly, twenty microliters of neutralized collagen solution (1.0 mg/ml) was added to the top chamber of the Transwell and polymerized at 37°C for 6 hours to form a thin collagen gel on top of the membrane. Then, 106 SMCs in 100 μl of DMEM were seeded into the top chamber with or without 100 μM deta NONOate. DMEM alone or with 10 ng/ml PDGF-BB, each with or without 100 μM deta NONOate, was added to the bottom chambers and the chambers were incubated at 37°C with 5% CO2 for 12 hours (Kanda and et al., 2000). Membranes were removed and the cells on the top side were scraped off. Remaining cells on the bottom side were fixed, stained using Dif-Quick and counted. Assays were performed in quadruplicate.
Cell Adhesion Assay
Adherent cells were washed with versene CA EDTA (1 mM EDTA in PBS), suspended with trypsin, washed twice with soybean trypsin inhibitor (STI, 0.5 mg/ml in PBS), and resuspended in DMEM containing 1 mg/ml BSA. When indicated, SMCs were pretreated with DMEM containing 1 mg/ml BSA in the presence or absence of 100 μM deta NONOate for 30 minutes at room temperature. Cells (3×104 cells/well) were plated in the absence or presence of 100 μM deta NONOate in 96-well plates coated with 100 μg/ml of polymeric type I collagen. After 1 hour incubation at 37°C, the wells were rinsed with warm PBS to remove non-attached cells, and the remaining cells were stained for 15 minutes with 0.2% crystal violet in 20% methanol. After five washes with water, the cell-bound dye was solubilized in 0.1M sodium citrate/ 50% ethanol (pH 4.2) and quantified by measuring absorbance at 550nm using an ELISA plate reader. Assays were performed in triplicate.
Fluorescent Microscopy
SMCs (3×105 cells/ml) cultured within collagen gels (1mg collagen/ml) for various times were fixed with 4% paraformaldehyde in PBS for 15 minutes and permeabilized with 0.5% Triton X-100 in PBS for 10 min. Image-iT FX signal enhancer was added for 30 minutes followed by addition of TRITC-labeled phalloidin (10μg/ml) for 1 hour. Fluorescence was analyzed with a Zeiss confocal microscope and the images were acquired using Zeiss LSM Aim examiner software.
Morphology Assay
All cell types were maintained in 15% serum. Slides were coated with polymeric Type 1 collagen (100 μg/ml in 1 X PBS) and washed with DMEM 3X prior to plating. When indicated, cells were treated with DMEM in the presence or absence of deta NONOate (100 μM) 30 minutes prior to trypsinization and again upon plating. Cells were trypsinized, resuspended in DMEM, plated (3×105 cells/well for 3h and 6h, 6×104 cells/well for 24h), and placed at 37°C until fixation. Cells were fixed in 4% paraformaldehyde in 1XPBS for 15 minutes at 3h, 6h and 24h after plating, incubated with anti-VASP antibody (M4, affinity purified, 1:500, NanoTools) and stained for α-smooth muscle actin and F-actin with anti-α-smooth muscle actin (Sigma, 1:200) or Rhodamine-Phalloidin (Cytoskeleton, 1:70), respectively. Images were acquired as described above.
Protein Extraction and Western Blotting
Cellular proteins were extracted in HEPES extraction buffer (25mM Hepes, pH 7.5; 5mM EDTA; 5mM EGTA; 150mM NaCl; 100mM Na4P2O7; 50mM NaF; 1mM benzamidine; 1% Triton X-100; 10% glycerol; 0.1% β-mercaptoethanol; 1μg/ml pepstain-A; 5 μg/ml leupeptin and 5 μg/ml aprotinin) (Chen and et al, 1998), separated by SDS-polyacrylamide electrophoresis and analyzed by Western blotting as described (Chen and et al, 1998).
Statistical Analysis
Results are given as mean ± S.E.M. of n independent experiments. Comparisons among the groups were made using one-way ANOVA followed by Bonferroni multiple comparison test. P≤0.05 was considered significant.
Results
Collagen Gel Contraction
To study the role of VASP phosphorylation at serine239 in SMC contraction of collagen gels, we used SMCs derived from VASP-/- mice expressing similar levels of wt-VASP, nonphosphorylatable VASP (S239A-VASP), or phospho-mimicking VASP (S239D-VASP) (Figure 1A). As expected, ectopically expressed wt-VASP was phosphorylated at serine239 in response to NO, while no phosphor-serine239 was detected in ectopically expressed S239A-VASP or S239D-VASP (Supplemental Figure 1A). Treatment with a specific inhibitor of cGMP-dependent protein kinase (PKG), Rp-8-pCPT-cGMPS, completely prevented phosphorylation at serine239 of wt-VASP in response to NO (Supplemental Figure 1B).
Figure 1.
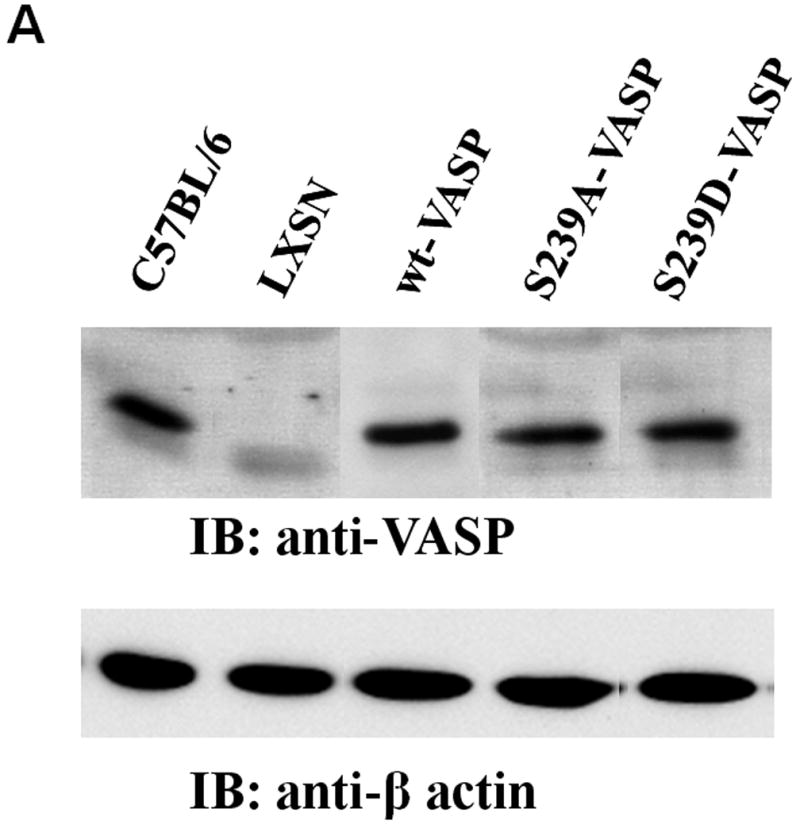
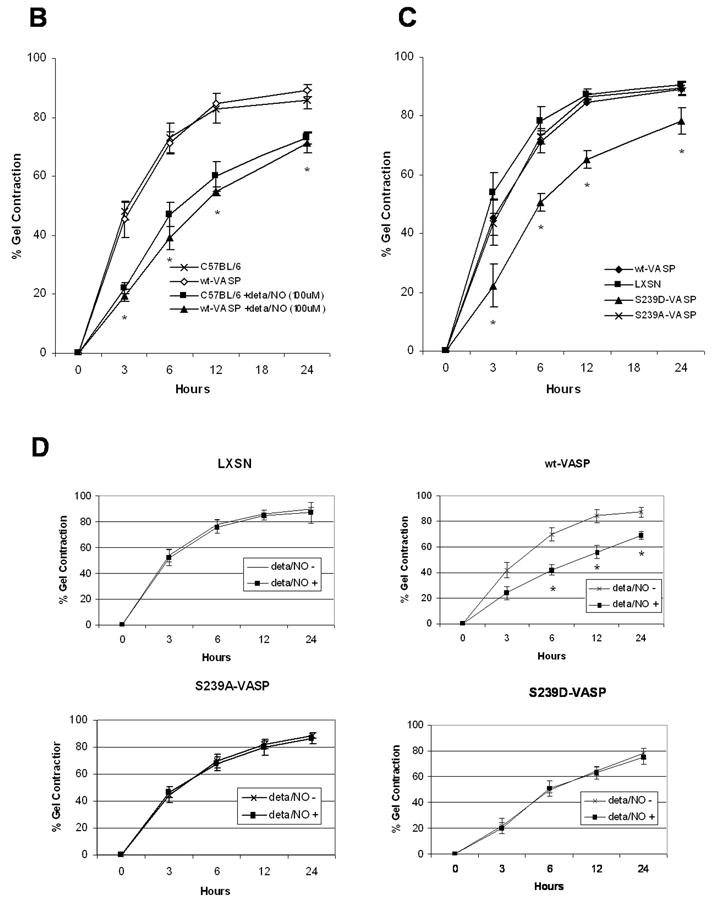
NO and expression of S239D-VASP in VASP-/- SMCs inhibit SMC-mediated collagen gel contraction. (A) Western blots of cell lysates of growing aortic SMCs (C57BL/6 wild-type SMCs and the following all derived from VASP-/- SMCs: LXSN SMCs, wt-VASP SMCs, S239A-VASP SMCs and S239D-VASP SMCs) were probed with antibodies against VASP or β-actin. (B) Collagen gel contraction by SMCs isolated from wild-type C57BL/6 mice or by wt-VASP SMCs was investigated in the absence or presence of a NO donor (100 μM deta NONOate). When indicated, SMCs were pretreated with deta NONOate for 30 minutes at room temperature. Data (mean±SEM; n=3-5 experiments) are expressed as percent of gel contraction. * With vs. without deta NONOate, P ≤ 0.05. (C) Collagen gel contraction by LXSN, wt-VASP, S239A-VASP or S239D-VASP SMCs. Data (mean±SEM; n=3-5 experiments) are expressed as the percent of gel contraction. * S239D-VASP SMCs vs. other cell types, P ≤ 0.05. (D) Collagen gel contraction by LXSN, wt-VASP, S239A-VASP or S239D-VASP SMCs in absence or presence of deta NONOate. Data (mean±SEM; n=3-5 experiments) are expressed as percent of gel contraction. * With vs. without deta NONOate, P ≤ 0.05.
Treatment with the slow-releasing-long-lasting NO donor, Deta NONOate (100 μM) decreased collagen gel contraction by wild-type C57BL/6 SMCs as well as by wt-VASP reconstituted SMCs (Figure 1B). S239D-VASP SMCs contracted collagen gel significantly less at all time points compared to non-stimulated wt-VASP or S239A-VASP expressing SMCs (Figure 1C). Stimulation of S239D-VASP, S239A-VASP, or LXSN SMCs with deta NONOate did not alter gel contraction (Figure 1D). Furthermore, treatment with Rp-8-pCPT-cGMPS completely blocked the inhibitory effect of Deta NONOate on gel contraction in wt-VASP expressing SMCs (Table 1).
Table 1.
Percentage of Gel Contraction in SMCs at 12 Hours
| Cell Type | -deta/NO | +deta/NO | +deta/NO +Rp-8-pCPT-cGMPS (50μM) |
|---|---|---|---|
| wt-VASP | 84 ± 5 % | 56 ± 5 % # | 77 ± 7 % ≠ |
| LXSN | 86 ± 3 % | 85 ±4 % | 86 ± 4 % |
| S239A-VASP | 82 ± 3 % | 80 ± 6 % | 79 ± 4 % |
| S239D-VASP | 64 ± 3 % * | 63 ± 5 % | 60 ± 5 % |
P ≤ 0.02 S239D-VASP vs. other cell types.
P ≤ 0.03 deta/NO-treated wt-VASP vs. wt-VASP without deta/NO.
P ≤ 0.04 deta/NO-treated wt-VASP in the presence of Rp-8-pCPT-cGMPS vs. deta/NO-treated wt-VASP without Rp-8-pCPT-cGMPS.
These results suggest that VASP phosphorylation at serine239 is required for NO-mediated inhibition of collagen gel contraction by SMCs.
Cell Migration and Invasion
The role of VASP phosphorylation at serine239 in SMC migration through a collagen gel barrier (3D invasion assay) was investigated In the absence of PDGF-BB in the bottom chamber, SMCs did not invade the gel over a period of 12 hours (data not shown). While addition of PDGF-BB to the bottom chamber significantly stimulated invasion by all of the SMCs studied. Matrix invasion was higher by S239D-VASP SMCs compared to LXSN, wt-VASP or S239A-VASP SMCs (Figure 2). Consistent with this observation, deta NONOate (100μM) significantly increased wt-VASP SMC invasion, but had no effect on S239D-VASP, S239A-VASP, or LXSN SMC invasion (Figure 2).
Figure 2.
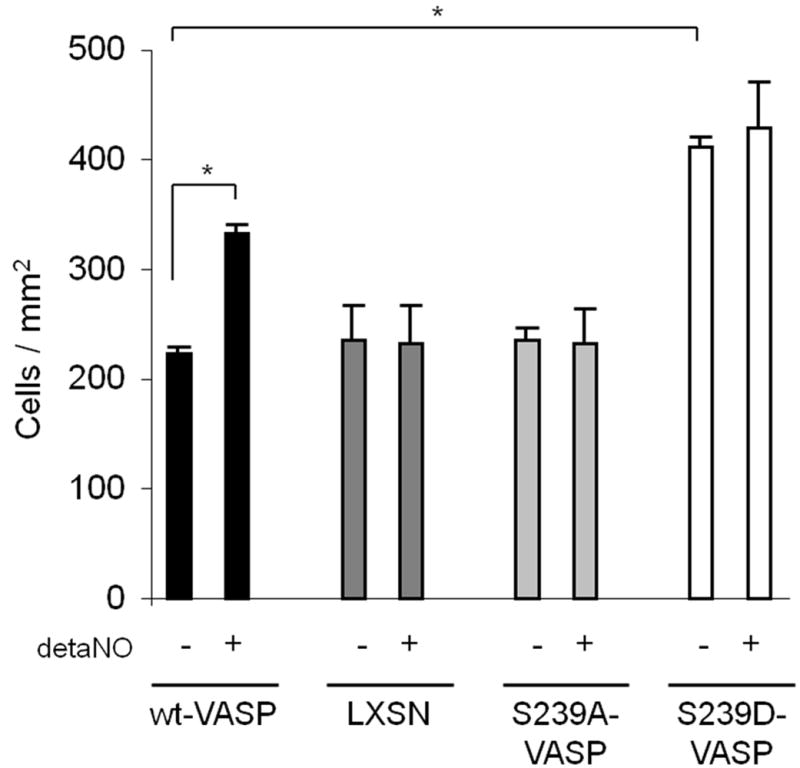
NO and expression of S239D-VASP enhance invasion through a collagen gel barrier. When indicated, SMCs were pretreated with detaNONOate (100 μM) for 30 minutes at room temperature. Invasion of SMCs through a thick collagen gel with PDGF-BB (10ng/mL) in the bottom chambers was investigated in the absence or presence of NO. Data (mean±SEM; n=3 experiments) are expressed as cells / mm2. * P ≤ 0.05.
Cell adhesion and spreading
Since SMCs need to attach to the matrix to gain traction for spreading, migration and gel contraction, we investigated the possibility that phosphorylation of serine239 might alter cell adhesion and spreading on polymeric collagen. LXSN, wt-VASP and S239A-VASP SMCs adhered to polymeric type I collagen to the same extent, whereas adhesion of S239D-VASP SMCs was significantly decreased. The addition of 100μM deta NONOate largely decreased adhesion of wt-VASP SMCs, but did not significantly alter adhesion of LXSN, S239A-VASP, or S239D-VASP SMCs (Figure 3).
Figure 3.
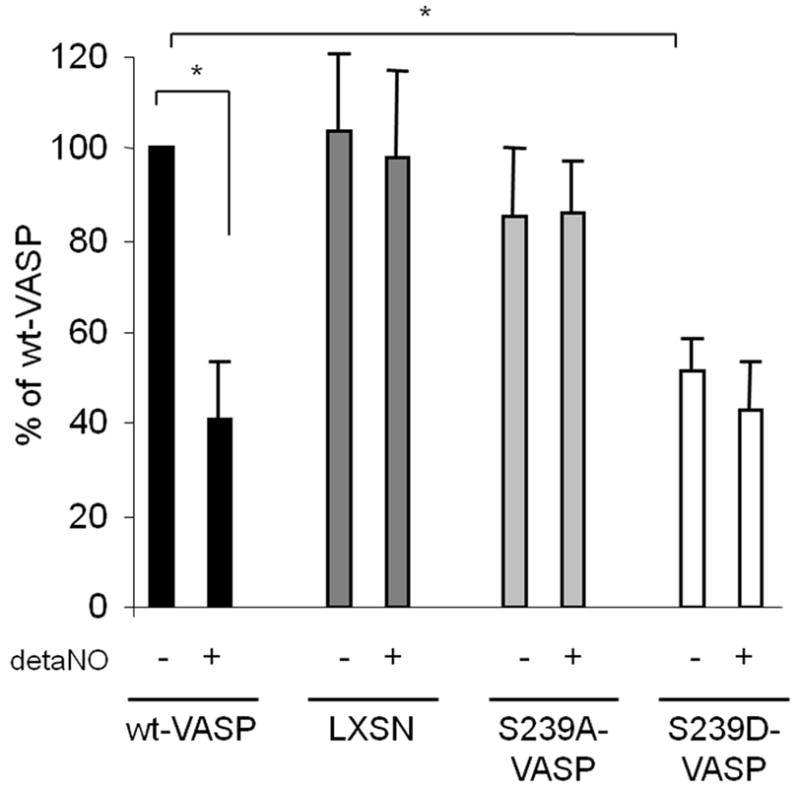
NO and expression of S239D-VASP inhibit adhesion. When indicated, SMCs were pretreated with 100 μM deta NONOate for 30 minutes at room temperature. Adhesion to polymeric collagen was investigated in the absence or presence of deta NONOate. Data (mean±SEM; n=3) are expressed as percent of adhesion of wt-VASP SMCS. *P ≤ 0.05.
In order to study cell spreading, LXSN, wt-VASP, S239A-VASP or S239D-VASP SMCs were plated in collagen gel, and spreading of the cells was analyzed by phase-contrast microscopy and by fluorescent microscopy following TRITC-phalloidin actin fiber staining. wt-VASP SMCs displayed a dendritic morphology characterized by the presence of long, branched membrane protrusions with the leading edge being rich in actin (Figure 4A-B). Spreading of LXSN, S239A-VASP, and wt-VASP expressing SMCs was similar. In the presence of 100μM deta NONOate, spreading of wt-VASP SMCs was significantly reduced and the number of cell protrusions decreased (Figure 4B and Table 2). S239D-VASP SMCs exhibited a significantly reduced number of membrane protrusions (Figure 4A-B and Table 2). Addition of deta NONOate did not alter spreading or the number of membrane protrusions of LXSN, S239A-VASP or S239D-VASP SMCs (Figure 4B and Table 2).
Figure 4.
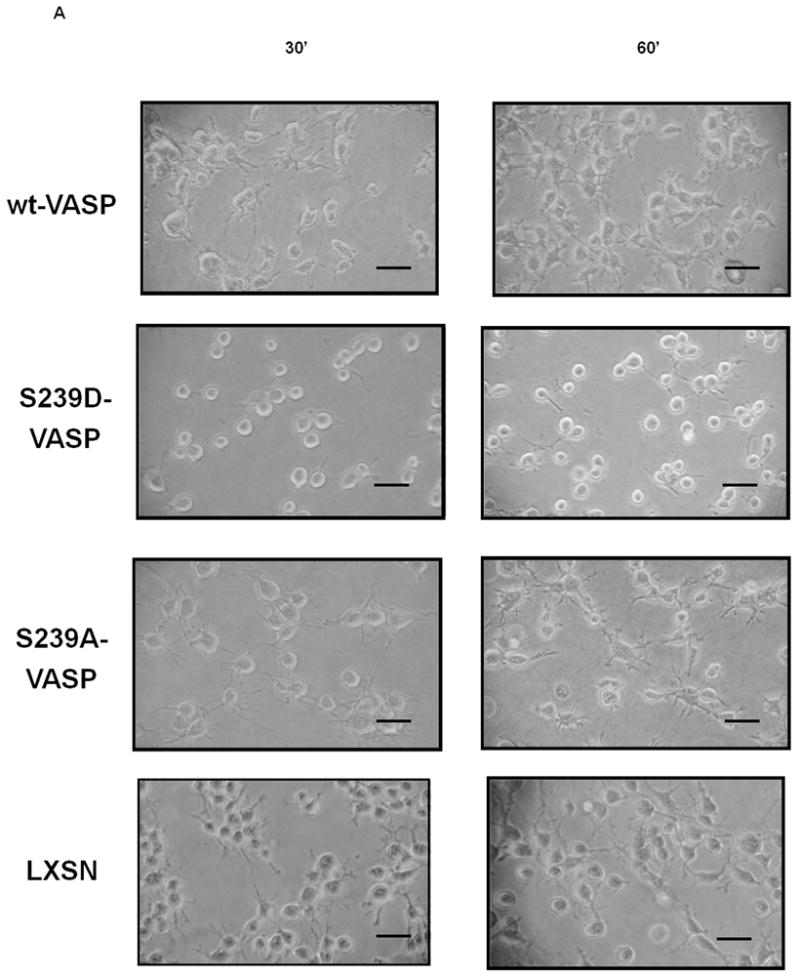
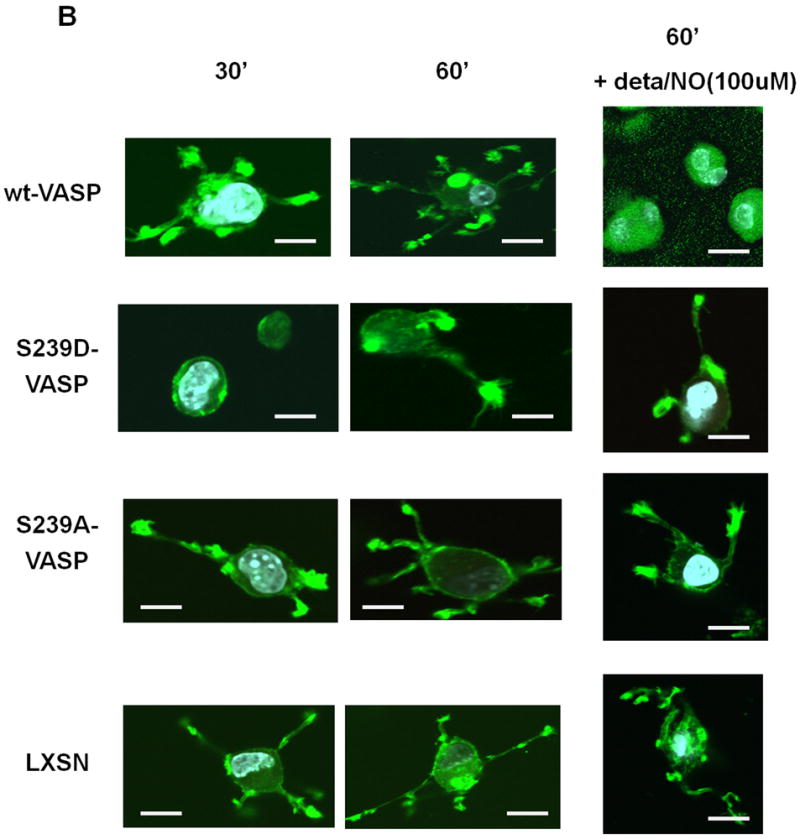
NO and expression of S239D-VASP inhibit membrane protrusion formation and SMC spreading within collagen. (A) Cell morphology was analyzed by phase contrast microscopy 30 and 60 minutes after the cells were seeded within collagen. Scale bar represents 10μm. (B) wt-VASP, LXSN, S239A-VASP or S239D-VASP SMCs were pretreated with or without 100 μM deta NONOate for 30 minutes at room temperature and then seeded within collagen gel in presence or absence of deta NONOate. After 30 and 60 minutes, the cells were analyzed by fluorescence microscopy after staining with TRITC-conjugated phalloidin. Scale bar represents 5μm. The photomicrographs are representative of three independent experiments.
Table 2.
Number of Protrusions per Cell in SMCs at 60 minutes (n=50)
| Cell Type | -deta/NO | +deta/NO |
|---|---|---|
| wt-VASP | 4.1 ± 1.3 | 1.8 ± 1.3 # |
| LXSN | 3.4 ± 1.6 | 3.6 ± 1.2 |
| S239A-VASP | 3.6 ± 1.8 | 4.0 ± 1.5 |
| S239D-VASP | 1.3 ± 1.2 % * | 1.4 ± 1.1 |
P ≤ 0.05 S239D-VASP vs. other cell types.
P ≤ 0.03 deta/NO-treated wt-VASP vs. wt-VASP without deta/NO.
Cell Morphology
To determine whether phospho- serine239 VASP colocalizes with F-actin, we stained SMCs plated on polymeric collagen coated glass slides for VASP and F-actin (Figure 5). In agreement with previous reports (Reinhard and et al., 2001; Krause and et al., 2003; Kwiatkowski and Gertler, 2003), we found VASP to be localized to the focal adhesions, the leading edge of lamellipodia and the cytosol. In comparison to wt-VASP SMCs, S239D-VASP SMCs were smaller, rounder, and had fewer protrusions at 3h (Figure 5) and 6h (Supplemental Figure 2B). At 24h, though the numbers of protrusions of the cells were the same in all cell types, S239D-VASP SMCs still appeared smaller in size and less spread than any of the other cell types (Supplemental Figure 2D). Staining of cells for filamentous actin with rhodamine-phalloidin and for VASP revealed colocalization of VASP and F-actin in wt-VASP SMCs and S239A-VASP SMCs, but not in S239D-VASP SMCs (Figure 5 and Supplemental Figure 2B). No colocalization was observed in any of the SMCs at 24h (Supplemental Figure 2D), suggesting that VASP plays a critical role during the earlier stages of F-actin polymerization, corresponding to the time when the cells are actively adhering and spreading. Cells were also stained for smooth muscle-alpha actin and VASP, and VASP colocalized with smooth muscle-α actin in both the wt-VASP and S239A-VASP SMCs (data not shown).
Figure 5.
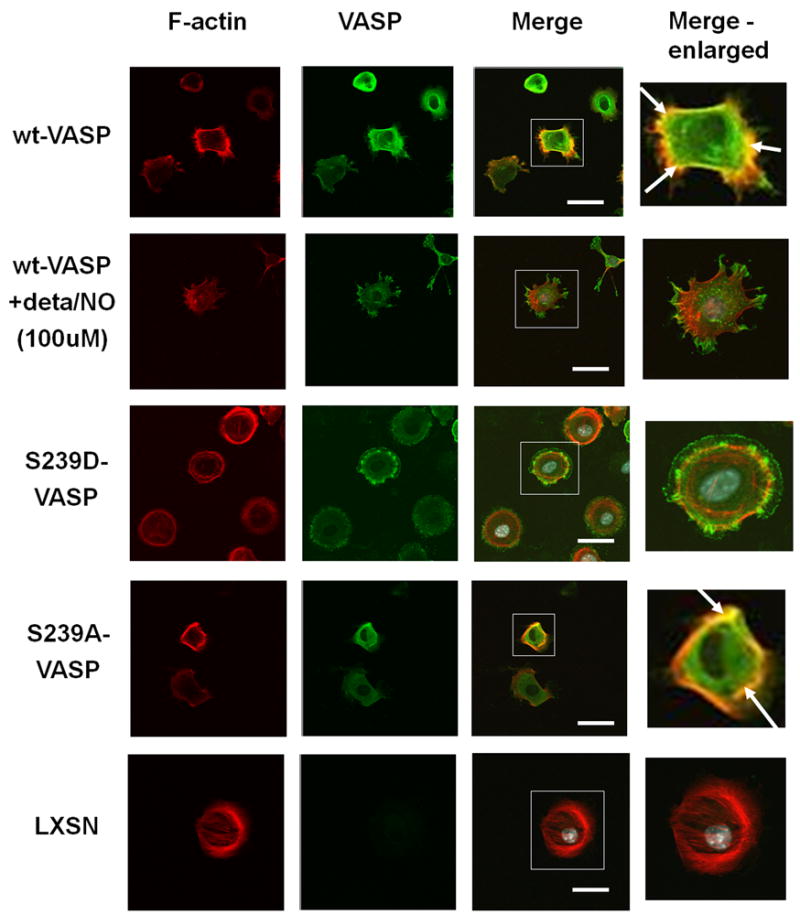
Colocalization of VASP and F-actin is inhibited by phosphorylation of VASP serine239. wt-VASP, LXSN, S239A-VASP or S239D-VASP SMCs were plated on polymeric collagen-coated glass slides, and wt-VASP SMCs were pretreated with 100 μM deta NONOate for 30 minutes at room temperature and then plated on polymeric collagen-coated glass slides in the presence of 100 μM deta NONOate. 3 hours after plating localization of VASP and F-actin was analyzed by staining with rhodamine-phalloidin (red) and VASP (green). Boxed regions are enlarged in insets. Arrows indicated where VASP and F-actin are colocalized (yellow). Scale bar represents 5μm.
Addition of 100μM deta NONOate inhibited the localization of VASP to focal adhesions in wt-VASP SMCs at 3 and 6 hours (Figure 5 and Supplemental and 2C), suggesting that phosphorylation impairs the binding of F-actin to VASP. The addition of NO had no effect on interaction between VASP and F-actin in S239D-VASP SMCs or in S239A-VASP SMCs (Figure 5 and Supplemental Figure 2A and 2C). At 24 hours, no effects of NO on VASP and actin could be detected in any of the cell types (Supplemental Figure 2E).
Discussion
Nitric oxide affects a wide variety of SMC functions such as contraction, proliferation, and migration through the activation of PKG. We recently showed that VASP phosphorylation at serine239 by PKG accounts, in part, for the growth inhibitory effect of NO (Chen and et al., 2004). In the present study, we have investigated the role of serine239 phosphorylation in SMC contraction, adhesion, spreading and invasion.
In agreement with previous reports that NO causes smooth muscle relaxation (Lincoln TM, Cornwell; 1993; Hofmann and et al., 2000), we have demonstrated that NO inhibits collagen gel contraction by SMCs and increases SMC invasion of collagen (Figures 1 and 2). We hypothesized that phosphorylation of VASP is required for the inhibitory effect of NO on SMC collagen contraction and enhancement of SMC invasion of collagen. We found that SMCs expressing the phospho-mimicking serine239 VASP mutant (S239D-VASP) exhibit less collagen gel contraction and increased invasion of collagen as compared to wt-VASP SMCs, and that treatment with a NO donor has no further effect. S239A-VASP SMCs, which express the non-phosphorylatable VASP mutant, contracted collagen gels and invaded collagen to the same extent as wt-VASP SMCs, but these processes were not blocked by NO. Furthermore a specific inhibitor of PKG, Rp-8-pCPT-cGMPS, completely blocked NO-mediated inhibition of gel contraction in wt-VASP SMCs (Table 1). These results suggest that NO inhibits SMC-mediated collagen gel contraction and increases SMC invasion of collagen by a mechanism that involves PKG-mediated VASP phosphorylation at serine239. Serine239 is not only critical for SMC morphology and actin based processed but NO-initiated and PKG-mediated phosphorylation at the second VASP phosphorylation state is critical for actin turnover in HEK cells and kerationocytes (Lindsay and et al., 2007), and in glioma cells (Zhuang and et al., 2004).
Because attachment and spreading of SMCs within the collagen matrix are required for gel contraction and invasion, we investigated the possibility that they are affected by phosphorylation at serine239. We found that NO significantly reduced adhesion and spreading of wt-VASP SMCs (Figures 3 and 4 and Table 2). Attachment and spreading of S239D-VASP SMCs were reduced compared to cells expressing wild-type VASP, but were not altered by NO treatment. Finally, attachment and spreading of S239A-VASP SMCs were similar to wt-VASP SMCs; NO had no effect. These data suggest that phosphorylation of S239 is required for the effect of NO on attachment and spreading. In addition, the correlation between the inhibition of gel contraction, and the enhancement of invasion of collagen and the inhibition of cell spreading supports the concept that cell traction is proportional to the degree of cell spreading (Tolic-Norrelykke and Wang, 2005).
In this study, we showed that S239D-VASP SMCs in a collagen matrix as well as wt-VASP SMCs in the presence of a NO donor, have fewer membrane protrusions compared to wt-VASP SMCs in the absence of NO (Figure 4 and Table 2). This result is in agreement with another study in epithelial cells which showed that NO-dependent phosphorylation of VASP serine239 causes rapid retraction of lamellipodia and loss of protrusions resulting in cell rounding (Lindsay and et al., 2007). In addition, we also demonstrated that the S239D-VASP SMCs and NO-treated wt-VASP SMCs adhere less to polymeric collagen than wt-VASP SMCs in the absence of a NO donor (Figure 3). Therefore, the enhanced invasion through a collagen gel barrier by S239D-VASP SMCs or by wt-VASP SMCs in the presence of a NO donor may be explained, at least in part, by decreased adhesion to the extracellular matrix.
The molecular mechanisms that lead to an orchestrated interplay between cell spreading, adhesion, and contraction are poorly understood. All of these cellular functions are controlled by the actin cytoskeleton, its assembly, and its remodeling. There are several possible mechanisms by which VASP could mediate the effects of NO on SMC adhesion, spreading, contraction and invasion. First, VASP may exert a direct effect on actin filament formation, dynamics, and molecular organization. We observed a decrease in the number of membrane protrusions in NO-treated wild-type VASP SMCs and in S239D-VASP SMCs but not in S239A-VASP SMCs. These results are consistent with previously published data (Butt and et al., 1998; Zhuang and et al., 2004; Bundschu and et al., 2006; Blume and et al., 2007) demonstrating that phosphorylation of VASP at serine239 within the EVH2 domain but not at serine 157 within the proline-rich region correlates with inhibition of F-actin bundling by VASP. Phosphorylation of threonine278 by AMP-activated kinase also inhibits F-actin bundling by VASP but this site is not phosphorylated in response to NO. One fact worth noting is that S239D-VASP SMCs as well as NO-treated wt-VASP SMCs are able to form protrusions, which suggests that phosphorylation of VASP at serine239 is not critical for the initial events in cell protrusion. This is comsistent with the conclusions of other researchers that VASP facilitates actin polymerization (Krause and et al., 2003; Kwiatkowski and et al., 2003).
Previous studies using Ena/VASP-deficient fibroblasts and cells overexpressing Mena mutants, another member of the Ena/VASP family, show that membrane protrusions and subcortical actin network organization are regulated by the actin binding EVH2 domain of Ena/VASP proteins (Loureiro and et al., 2002). It has been suggested that basic stretches within the EVH2 domain, that contains the S239 phosphorylation site, mediate VASP binding to F-actin via electrostatic forces (Huttelmaier and et al., 1999; Bachmann and et al., 1999). In addition, Chereau and Dominguez suggest that a subdomain of the EVH2 domain binds profilin-G-actin and mediates the incorporation of the actin monomer onto the barbed end of the growing actin filament (Chereau and Dominguez, 2006). Using an identical approach to ours (expression of a mutant VASP mimicking phosphorylation), Harbeck et al. (2000) were able to show that the introduction of negative charges by phosphorylation weakens the VASP-F-actin interaction resulting in less actin polymerization. There was a more pronounced effect when serine235 (equivalent to serine239 in human cells) was phosphorylated. The serine239 site is located immediately adjacent to the G-actin-binding (GAB) site, which functions by binding actin monomers that are to be incorporated onto the barbed ends of elongating filaments. A recent study demonstrated that both the poly-Pro domain and the GAB domain have increased affinity for profilin-actin complexes than for profilin or actin monomers alone (Ferron and et al., 2007). They suggested a mechanism by which profilin-actin complexes are transferred from the poly-Pro to the GAB domain. The actin monomers are transferred to the F-actin-binding (FAB) domain, where they bind to the barbed end of growing filaments as they dissociate from profilins (Ferron and et al., 2007). Recruitment of profilin-actin complexes increases the rate of elongation. The subcellular distribution of VASP may be important for reorganizing actin during cell adhesion and spreading (Drees and et al., 2000; Smolenski, and et al., 2000), although VASP phosphorylation does not appear to alter VASP interaction with focal adhesion proteins (e.g. vinculin and zyxin) or its subcellular distribution (Smolenski and et al., 1998; Harbeck and et al., 2000). In this study, we investigated the possibility that phosphorylation at VASP serine239 interferes with localization of VASP and its interaction with actin. VASP localized to the focal adhesions in the different VASP variants, and its localization was unaffected by the addition of a NO donor. No significant difference was observed regarding complex formation with other focal adhesion proteins like zyxin, vinculin or profilins (data not shown). However, colocalization between VASP and filamentous actin was observed at the earlier time points in wt-VASP SMCs, but not in S239D-VASP SMCs or in NO-treated wt-VASP SMCs. The absence of colocalization in S239D-VASP SMCs suggests that the mutation interferes with VASP’s ability to associate with F-actin, providing further evidence for the role of phosphorylation in the regulation of actin polymerization.
Another potential mechanism for VASP in SMC contraction is that VASP modulates a signal transduction pathway that affects the actin cytoskeleton. Reports using VASP-/- fibroblasts have shown that VASP is involved in the regulation of myosin light chain phosphorylation as well as in the activation of Rac and PAK (Garcia-Arguinzonis and et al., 2002; Galler and et al., 2006). Recently, we have shown that VASP phosphorylation at serine157 is required for Rac1 activation (Chen and Clowes, unpublished data), but NO-mediated VASP phosphorylation at serine239 has no effect on the activation of the small GTPases RhoA, Rac1 and Cdc42 by serum (Supplementary Figure 3). MAPK signaling is also closely linked to the regulation of cellular functions depending on actin cytoskeleton reorganization such as contractility, spreading, adhesion and migration. However, we did not find significant differences in the expression levels or activities of ERK, p38 or JNK in wt-VASP or S239D-VASP SMCs during adhesion (Supplementary Figure 4). Also, while Ena/VASP proteins have been shown to modulate FAK phosphorylation (Kragtorp and Miller JR, 2006), we found no difference in activation of FAK between wt-VASP and S239D-VASP SMCs during adhesion (Supplementary Figure 4). Therefore, it is unlikely that the effects of VASP phosphorylation at serine239 on cell adhesion, spreading and contraction are mediated by the modulation of these transduction pathways.
In conclusion, our study identifies phosphorylation of VASP at serine239 as a critical mediator for the inhibitory effect of NO on SMC matrixcontraction, spreading and adhesion. SMC invasion is stimulated by NO. The clinical relevance of our results is that SMC contraction, adhesion and spreading as well as invasion are all involved in important arterial pathology such as restenosis after angioplasty. The control of the phosphorylation status of VASP may form the basis of a novel treatment that may minimize the effect of restenosis.
Supplementary Material
Acknowledgments
The study was supported by two grants from the National Institute of Health (HL-52459, A.W.C.; T32 GM07037, O.D.D.), grants from the Deutsche Forschungsgemeinschaft (DFG) SFB688 (A3 to T.R. and A1 to U.W.), and a fellowship from the Belgian American Educational Foundation (O.D.D.).
References
- Bachmann C, Fischer L, Walter U, Reinhard M. The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding, and actin bundle formation. J Biol Chem. 1999;274(33):23549–23557. doi: 10.1074/jbc.274.33.23549. [DOI] [PubMed] [Google Scholar]
- Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, Schafer DA. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem. 2005;280(31):28653–28662. doi: 10.1074/jbc.M503957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109(4):509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Benz PM, Blume C, Moebius J, Oschatz C, Schuh K, Sickmann A, Walter U, Feller SM, Renné T. Cytoskeleton assembly at endothelial cell-cell contacts is regulated by αII-spectrin-VASP complexes. J Cell Biol. 2008;180(1):205–219. doi: 10.1083/jcb.200709181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume C, Benz PM, Walter U, Ha J, Kemp BE, Renné T. AMP-activated protein kinase impairs endothelial actin cytoskeleton assembly by phosphorylating vasodilator-stimulated phosphoprotein. J Biol Chem. 2007;282(7):4601–4612. doi: 10.1074/jbc.M608866200. [DOI] [PubMed] [Google Scholar]
- Brown C, Pan X, Hassid A. Nitric oxide and C-type atrial natriuretic peptide stimulate primary aortic smooth muscle cell migration via a cGMP-dependent mechanism: relationship to microfilament dissociation and altered cell morphology. Circ Res. 1999;84:655–667. doi: 10.1161/01.res.84.6.655. [DOI] [PubMed] [Google Scholar]
- Brown C, Lin Y, Hassid A. Requirement of protein tyrosine phosphatase SHP2 for NO-stimulated vascular smooth muscle cell motility. Am J Physiol. 2001;281:H1598–H1605. doi: 10.1152/ajpheart.2001.281.4.H1598. [DOI] [PubMed] [Google Scholar]
- Bundschu K, Walter U, Schuh K. The VASP-Spred-Sprouty domain puzzle. J Biol Chem. 2006;281(48):36477–36481. doi: 10.1074/jbc.R600023200. [DOI] [PubMed] [Google Scholar]
- Butt E, Abel K, Krieger M, Palm D, Hoppe V, Hoppe J, Walter U. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J Biol Chem. 1994;269(20):14509–14517. [PubMed] [Google Scholar]
- Chang Y, Ceacareanu B, Dixit M, Sreejayan N, Hassid A. Nitric oxide-induced motility in aortic smooth muscle cells: role of protein tyrosine phosphatase SHP-2 and GTP-binding protein Rho. Circ Res. 2002;91(5):390–397. doi: 10.1161/01.res.0000033524.92083.64. [DOI] [PubMed] [Google Scholar]
- Chen L, Daum G, Forough R, Clowes M, Walter U, Clowes AW. Overexpression of human endothelial nitric oxide synthase in rat vascular smooth muscle cells and in balloon-injured carotid artery. Circ Res. 1998;82(8):862–870. doi: 10.1161/01.res.82.8.862. [DOI] [PubMed] [Google Scholar]
- Chen L, Daum G, Chitaley K, Coats SA, Bowen-Pope DF, Eigenthaler M, Thumati NR, Walter U, Clowes AW. Vasodilator-stimulated phosphoprotein regulates proliferation and growth inhibition by nitric oxide in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24(8):1403–1408. doi: 10.1161/01.ATV.0000134705.39654.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereau D, Dominguez R. Understanding the role of the G-actin-binding domain of Ena/VASP in actin assembly. J Struct Biol. 2006;155(2):195–201. doi: 10.1016/j.jsb.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Chitaley K, Chen L, Galler A, Walter U, Daum G, Clowes AW. Vasodilator-stimulated phosphoprotein is a substrate for protein kinase C. FEBS Lett. 2004;556(1-3):211–215. doi: 10.1016/s0014-5793(03)01435-2. [DOI] [PubMed] [Google Scholar]
- Cohen RA. The role of nitric oxide and other endothelium-derived vasoactive substances in vascular disease. Prog Cardiovasc Dis. 1995;38(2):105–128. doi: 10.1016/s0033-0620(05)80002-7. [DOI] [PubMed] [Google Scholar]
- Drees B, Friederich E, Fradelizi J, Louvard D, Beckerle MC, Golsteyn RM. Characterization of the interaction between zyxin and members of the Ena/vasodilator-stimulated phosphoprotein family of proteins. J Biol Chem. 2000;275(29):22503–22511. doi: 10.1074/jbc.M001698200. [DOI] [PubMed] [Google Scholar]
- Dubey RK, Jackson EK, Luscher TF. Nitric oxide inhibits angiotensin II-induced migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. J Clin Invest. 1995;96(1):141–149. doi: 10.1172/JCI118014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron F, Rebowski G, Lee SH, Dominguez R. Structural basis for the recruitment of profilin—actin complexes during filament elongation by Ena/VASP. EMBO J. 2007;26:4597–4606. doi: 10.1038/sj.emboj.7601874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galler AB, Garcia Arguinzonis MI, Baumgartner W, Kuhn M, Smolenski A, Simm A, Reinhard M. VASP-dependent regulation of actin cytoskeleton rigidity, cell adhesion, and detachment. Histochem Cell Biol. 2006;125(5):457–474. doi: 10.1007/s00418-005-0091-z. [DOI] [PubMed] [Google Scholar]
- Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83(5):1774–1777. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbeck B, Huttelmaier S, Schluter K, Jockusch BM, Illenberger S. Phosphorylation of the vasodilator-stimulated phosphoprotein regulates its interaction with actin. J Biol Chem. 2000;275(40):30817–30825. doi: 10.1074/jbc.M005066200. [DOI] [PubMed] [Google Scholar]
- Hassid A, Yao J, Huang S. NO alters cell shape and motility in aortic smooth muscle cells via protein tyrosine phosphatase 1B activation. Am J Physiol. 1999;277:H1014–1026. doi: 10.1152/ajpheart.1999.277.3.H1014. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Ammendola A, Schlossmann J. Rising behind NO: cGMP-dependent protein kinases. J Cell Sci. 2000;113:1671–1676. doi: 10.1242/jcs.113.10.1671. [DOI] [PubMed] [Google Scholar]
- Huttelmaier S, Harbeck B, Steffens O, Messerschmidt T, Illenberger S, Jockusch BM. Characterization of the actin binding properties of the vasodilator-stimulated phosphoprotein VASP. FEBS Lett. 1999;451(1):68–74. doi: 10.1016/s0014-5793(99)00546-3. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G, del Soldato P. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A. 2001;98(7):4202–4208. doi: 10.1073/pnas.071054698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda S, Kuzuya M, Ramos MA, Koike T, Yoshino K, Ikeda S, Iguchi A. Matrix metalloproteinase and alphavbeta3 integrin-dependent vascular smooth muscle cell invasion through a type I collagen lattice. Arterioscler Thromb Vasc Biol. 2000;20(4):998–1005. doi: 10.1161/01.atv.20.4.998. [DOI] [PubMed] [Google Scholar]
- Kragtorp KA, Miller JR. Regulation of somitogenesis by Ena/VASP proteins and FAK during Xenopus development. Development. 2006;133(4):685–695. doi: 10.1242/dev.02230. [DOI] [PubMed] [Google Scholar]
- Garcia-Arguinzonis MI, Galler AB, Walter U, Reinhard M, Simm A. Increased spreading, Rac/p21-activated kinase (PAK) activity, and compromised cell motility in cells deficient in vasodilator-stimulated phosphoprotein (VASP) J Biol Chem. 2002;277(47):45604–45610. doi: 10.1074/jbc.M202873200. [DOI] [PubMed] [Google Scholar]
- Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- Kühnel K, Jarchau T, Wolf E, Schlichting I, Walter U, Wittinghofer A, Strelkov SV. The VASP tetramerization domain is a right-handed coiled coil based on a 15-residue repeat. Proc Natl Acad Sci U S A. 2004;101(49):17027–17032. doi: 10.1073/pnas.0403069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski AV, Gertler FB, Loureiro JJ. Function and regulation of Ena/VASP proteins. Trends Cell Biol. 2003;13(7):386–392. doi: 10.1016/s0962-8924(03)00130-2. [DOI] [PubMed] [Google Scholar]
- Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. FASEB J. 1993;7(2):328–338. doi: 10.1096/fasebj.7.2.7680013. [DOI] [PubMed] [Google Scholar]
- Lindsay SL, Ramsey S, Aitchison M, Renné T, Evans TJ. Modulation of lamellipodial structure and dynamics by NO-dependent phosphorylation of VASP Ser239. J Cell Sci. 2007;120:3011–3021. doi: 10.1242/jcs.003061. [DOI] [PubMed] [Google Scholar]
- Loscalzo J, Welch G. Nitric oxide and its role in the cardiovascular system. Prog Cardiovasc Dis. 1995;38(2):87–104. doi: 10.1016/s0033-0620(05)80001-5. [DOI] [PubMed] [Google Scholar]
- Loureiro JJ, Rubinson DA, Bear JE, Baltus GA, Kwiatkowski AV, Gertler FB. Critical roles of phosphorylation and actin binding motifs, but not the central proline-rich region, for Ena/vasodilator-stimulated phosphoprotein (VASP) function during cell migration. Mol Biol Cell. 2002;13(7):2533–2546. doi: 10.1091/mbc.E01-10-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massberg S, Gruner S, Konrad I, Garcia Arguinzonis MI, Eigenthaler M, Hemler K, Kersting J, Schulz C, Muller I, Besta F, Nieswandt B, Heinzmann U, Walter U, Gawaz M. Enhanced in vivo platelet adhesion in vasodilator-stimulated phosphoprotein (VASP)-deficient mice. Blood. 2004;103(1):136–142. doi: 10.1182/blood-2002-11-3417. [DOI] [PubMed] [Google Scholar]
- Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- Oelze M, Mollnau H, Hoffmann N, Warnholtz A, Bodenschatz M, Smolenski A, Walter U, Skatchkov M, Meinertz T, Munzel T. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ Res. 2000;87(11):999–1005. doi: 10.1161/01.res.87.11.999. [DOI] [PubMed] [Google Scholar]
- Pelletier S, Julien C, Popoff MR, Lamarche-Vane N, Meloche S. Cyclic AMP induces morphological changes of vascular smooth muscle cells by inhibiting a Rac-dependent signaling pathway. J Cell Physiol. 2005;204(2):412–422. doi: 10.1002/jcp.20308. [DOI] [PubMed] [Google Scholar]
- Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch BM, Walter U. The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilins. EMBO J. 1995;14(8):1583–1589. doi: 10.1002/j.1460-2075.1995.tb07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M, Jarchau T, Walter U. Actin-based motility: stop and go with Ena/VASP proteins. Trends Biochem Sci. 2001;26(4):243–249. doi: 10.1016/s0968-0004(00)01785-0. [DOI] [PubMed] [Google Scholar]
- Samarin S, Romero S, Kocks C, Didry D, Pantaloni D, Carlier MF. How VASP enhances actin-based motility. J Cell Biol. 2003;163(1):131–142. doi: 10.1083/jcb.200303191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar R, Meinberg EG, Stanley JC, Gordon D, Webb RC. Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ Res. 1996;78(2):225–230. doi: 10.1161/01.res.78.2.225. [DOI] [PubMed] [Google Scholar]
- Schafer A, Burkhardt M, Vollkommer T, Bauersachs J, Munzel T, Walter U, Smolenski A. Endothelium-dependent and -independent relaxation and VASP serines 157/239 phosphorylation by cyclic nucleotide-elevating vasodilators in rat aorta. Biochem Pharmacol. 2003;65(3):397–405. doi: 10.1016/s0006-2952(02)01523-x. [DOI] [PubMed] [Google Scholar]
- Schirenbeck A, Arasada R, Bretschneider T, Stradal TE, Schleicher M, Faix J. The bundling activity of vasodilator-stimulated phosphoprotein is required for filopodium formation. Proc Natl Acad Sci U S A. 2006;103(20):7694–7699. doi: 10.1073/pnas.0511243103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolenski A, Bachmann C, Reinhard K, Honig-Liedl P, Jarchau T, Hoschuetzky H, Walter U. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J Biol Chem. 1998;273(32):20029–20035. doi: 10.1074/jbc.273.32.20029. [DOI] [PubMed] [Google Scholar]
- Smolenski A, Poller W, Walter U, Lohmann SM. Regulation of human endothelial cell focal adhesion sites and migration by cGMP-dependent protein kinase I. J Biol Chem. 2000;275(33):25723–25732. doi: 10.1074/jbc.M909632199. [DOI] [PubMed] [Google Scholar]
- Sreejayan N, Lin Y, Hassid A. NO attenuates insulin signaling and motility in aortic smooth muscle cells via a protein tyrosine phosphatase 1B-mediated mechanism. Arterioscler Thromb Vasc Biol. 2002;22:1086–1092. doi: 10.1161/01.atv.0000020550.65963.e9. [DOI] [PubMed] [Google Scholar]
- Tolic-Norrelykke IM, Wang N. Traction in smooth muscle cells varies with cell spreading. J Biomech. 2005;38(7):1405–1412. doi: 10.1016/j.jbiomech.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Umans JG, Levi R. Nitric oxide in the regulation of blood flow and arterial pressure. Annu Rev Physiol. 1995;57:771–790. doi: 10.1146/annurev.ph.57.030195.004011. [DOI] [PubMed] [Google Scholar]
- Walders-Harbeck B, Khaitlina SY, Hinssen H, Jockusch BM, Illenberger S. The vasodilator-stimulated phosphoprotein promotes actin polymerisation through direct binding to monomeric actin. FEBS Lett. 2002;529(2-3):275–280. doi: 10.1016/s0014-5793(02)03356-2. [DOI] [PubMed] [Google Scholar]
- Zhuang S, Nguyen GT, Chen Y, Gudi T, Eigenthaler M, Jarchau T, Walter U, Boss GR, Pilz RB. Vasodilator-stimulated phosphoprotein activation of serum-response element-dependent transcription occurs downstream of RhoA and is inhibited by cGMP-dependent protein kinase phosphorylation. J Biol Chem. 2004;279(11):10397–10407. doi: 10.1074/jbc.M313048200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.