

Abstract
Neurons have many specialized membrane domains with diverse functions responsible for receiving, integrating, and transmitting electrical signals between cells in a circuit. Both the locations and protein compositions of these domains defines their functions. In axons, two of the most important membrane domains are the axon initial segment and the nodes of Ranvier. Proper assembly and maintenance of these domains is necessary for action potential generation and propagation, and the overall function of the neuron.
Keywords: action potential, neuron-glia interaction, cell adhesion molecule
1. Introduction
To facilitate rapid action potential conduction, mammalian neurons have evolved a remarkable set of ion channel clustering mechanisms [1], some of which are just now being discovered. Clustered ion channels at the most proximal part of the axon, or the axon initial segment (AIS; Fig. 1A), function to integrate synaptic input into a single all-or-none response called the action potential (AP). Once generated, the AP must propagate rapidly and efficiently over very long distances; in mammalian myelinated axons, Na+ channels clustered at regularly spaced gaps in the myelin sheath called nodes of Ranvier (Fig. 1B, green) perform this function. Nodal channels are responsible for regenerating the inward Na+ currents that underlie saltatory AP conduction. In addition to Na+ channels, K+ channels are also highly clustered at discrete locations along axons where they modulate AP amplitude, duration, and frequency of firing [2]. These axonal membrane domains have been the focus of renewed attention as it has become clear their molecular compositions and physiologies are more diverse and plastic than originally thought, and because these domains represent a remarkable example of how neurons and their associated glial cells can regulate their membrane properties and protein localization to reflect their special functions. Here, I discuss the protein components that have been identified at the AIS and nodes of Ranvier, and describe how these domains are assembled during development. Finally, diseases and injuries can alter the functional organization of axons by disrupting AIS and nodes of Ranvier. I will discuss the evidence to support this previously unappreciated consequence of injury. Due to space limitations, I will not provide an exhaustive description of each molecule and domain or physiologies, but I will highlight key ideas that reflect principles of organization and function.
Figure 1.

Ion channels are clustered in high density at multiple sites along axons. A, Axon initial segment (AIS) of a cultured hippocampal neuron immunolabeled using antibodies against Nav channels (red), ankG (green), and the microtubule associated protein 2 (MAP2) to define the somatodendritic domain (blue). B, Central nervous system nodes of Ranvier immunolabled using antibodies against Nav channels (green), Caspr (red), and Kv1.2 K+ channels (blue). Scale bars, A = 10 μm; B = 5 μm.
2. Composition of the AIS and nodes of Ranvier
2.1 ion channels
The excitable domains along axons share a common molecular organization consisting of ion channels, cell adhesion molecules, cytoskeletal and scaffolding proteins, and extracellular matrix molecules. In some cases, two different domains can have virtually identical compositions! Of course, the main characteristic of the membrane domains discussed here is that they are highly enriched with voltage-gated ion channels. Both AIS and nodes of Ranvier have high densities of voltage-gated Na+ (Nav) channels (Fig. 1) [3] that are responsible for initiation and propagation of APs, respectively [4]. In mature nodes these channels consist mainly of Nav1.6 channels[5], although Nav1.1 can also be found in many mature nodes[6], while at developing nodes, especially in the CNS, Nav1.2 channels are first clustered and then replaced by Nav1.6 [7]. In the PNS both Nav1.6 and Nav1.2 can be found at developing nodes, but Nav1.6 is the predominant channel. This may be related to the fact that PNS myelination proceeds much more quickly and occurs earlier than CNS myelination [8]. The reason for the switch in Nav channel subtype expression during myelination is unknown, but may be related to the switch from continuous to saltatory conduction upon myelination.
In contrast to nodes, both Nav1.2 and Nav1.6 are often found in the mature AIS, albeit in different subdomains: Nav1.2 channels are found in the proximal AIS, while Nav1.6 channels are found in the distal AIS [9, 10]. The basis of this differential localization is unknown, but may reflect contributions from unique Na+ channel interacting proteins such as their accessory β-subunits, which can also be found at the AIS and nodes [11]. The function of the distal Nav1.6 has been proposed to be action potential initiation, while the proximal Nav1.2 channels facilitate backpropagation into the soma[9]. In the AIS of parvalbumin-positive GABAergic interneurons, the primary Nav channel subtype is Nav1.1 [12]. Consistent with the idea that these channels play key roles in regulating brain excitability, mutations in Nav1.1 lead to epilepsy, presumably due to impaired inhibition of excitatory neurons[13].
Besides Nav channels, the voltage-gated K+ channels KCNQ2 and KCNQ3 (also known as Kv7.2 and Kv7.3) are also clustered in high densities at nodes and the AIS where they modulate axonal excitability [14-16]. The interested reader is referred to the more detailed discussion of these ion channels and their functions in axons found elsewhere in this volume (Cooper, 2011).
Kv1.1, Kv1.2, and Kv1.4 K+ channels are also clustered along axons[17]. Intriguingly, while these channels are enriched at the distal part of the AIS where they colocalize with Nav1.6 channels [10, 18], they are excluded from mammalian nodes of Ranvier. Instead clusters of Kv1 channels flank nodes beneath the myelin sheath in regions called juxtaparanodes [19]. The function of juxtaparanodal K+ channels is unclear, but they have been proposed to play important roles in modulating action potential propagation and to inhibit repetitive firing of injured and developing myelinated axons [20]. In contrast to mammals, amphibians have Kv1 channels enriched at nodes[21]. The reason for the exclusion of Kv1 channels from mammalian nodes is unknown, but may be related to the different temperatures at which action potential conduction occurs. The function of AIS Kv1 channels is better understood and they have been shown to play important roles in regulating AIS excitability and duration of the action potential [22]. In fast-spiking GABAergic interneurons these channels also function to reduce excitability [23].
2.2 Scaffolds
Clustering of Nav and KCNQ2/Q3 channels depends on interactions with the cytoskeletal scaffolding and adaptor protein ankyrinG (ankG); both Nav and KCNQ2/3 channels have a similar AIS targeting motif that mediates channel binding to ankG [24, 25]. One remarkable feature of this interaction is that phosphorylation of the Nav channel AIS targeting motif by protein kinase CK2 dramatically increases the affinity of Nav channels for ankG [26]. Whether KCNQ2/3 channels are also phoshorylated to promote their interaction with ankG is unknown. It will be important to determine if phosphorylation of AIS Nav and KCNQ2/3 channels results in dynamic regulation of channel densities at the AIS under different conditions of activity or disease. A more detailed discussion of the interactions that occur in neurons between Nav and KCNQ2 channels and the scaffold ankG can be found in the reviews by Leterrier et al. (2011) and Cooper (2011). AnkG links AIS membrane proteins to the underlying actin cytoskeleton through βIV spectrin [27, 28]. While disruption of βIV spectrin results in reduced densities of AIS Nav channels [29, 30], loss of ankG totally blocks ion channel clustering and the assembly of the AIS [25, 31, 32]. In vitro studies of myelinating dorsal root ganglion and Schwann cell co-cultures also implicate ankG as being essential for ion channel clustering at nodes. Specifically, shRNA was used to silence expression of ankG in cultured DRG neurons, resulting in loss of Nav channels from nodes [33]. Thus, ankG is a key organizer of the AIS and nodal protein complex.
The scaffolding proteins PSD-93 and PSD-95 are highly enriched at juxtaparanodes, but genetic ablation of one or both of these proteins does not impact Kv1 channel clustering [34, 35]. The cytoskeletal scaffolding protein 4.1B is also found at juxtaparanodes, and links juxtaparanodal CAMs (see below) to the underlying spectrin-based cytoskeleton [36, 37]. Loss of 4.1B results in disrupted clustering of Kv1 channels [38], although it is not yet clear whether this is due to failure to initiate clustering, or failure to maintain channel clusters along axons. In contrast, only PSD-93 can be detected at the AIS. Using cultured hippocampal neurons, Ogawa et al. [17] demonstrated that acute silencing of PSD-93 could inhibit the clustering of AIS Kv1 channels. However, the mechanisms responsible for PSD-93 clustering at the AIS remain unknown, and PSD-93-null mice still have clustered Kv1 channels at the AIS [39], indicating that there exist other, redundant mechanisms in vivo that can compensate for the chronic loss of PSD-93.
2.3 Cell Adhesion Molecules
AIS, nodes, and juxtaparanodes are also enriched with a variety of cell adhesion molecules (CAMs). The two main CAMs at nodes and the AIS are neurofascin (NF)-186 and NrCAM. These CAMs are both members of the L1 family of CAMs, and require their interaction with ankG for proper AIS localization [40, 41]. The functions of the L1 CAMs at the AIS are not well-understood, but silencing expression of NF-186 impairs the clustering of gephyrin, a postsynaptic cytoskeletal scaffold necessary for the recruitment of GABA receptors to the chandelier cell inputs found at the AIS of cortical pyramidal neurons [42]. Furthermore, disruption of NF-186 localization in Purkinje neurons leads to aberrant GABAergic basket cell innervation [43]. Thus, the CAM NF-186 appears to play important roles in regulating assembly of GABAergic inputs to the AIS of many neurons.
The distal AIS and juxtaparanodes also contain the CAMs Caspr2, TAG-1, and ADAM22, but these CAMs associate with the Kv1 channel complex rather than ankG or Nav channels. The functions of these CAMs in the AIS is unknown, but mutations in Caspr2 lead to epilepsies and mental retardation, and have been linked to autism [44-46]. Caspr2, ADAM22, and Kv1 channels all co-immunoprecipitate with the cytoskeletal scaffolds PSD-93 and PSD-95, indicating that they form a larger macromolecular complex [35, 39]. Indeed, deletion of either Caspr2 or TAG-1 dramatically impairs juxtaparanodal Kv1 channel and ADAM22 clustering. In contrast, genetic deletion of the CAM ADAM22 blocks only the recruitment of PSD-93 and PSD-95 to juxtaparanodes, and does not affect the clustering of Kv1 channels or Caspr2 [17].
2.4 Extracellular matrix molecules
The AIS and nodes of Ranvier are also endowed with a specialized extracellular matrix. The chondroitin sulfate proteoglycans brevican and versican are both found at the AIS and CNS nodes of Ranvier [47-49]. Hedstrom et al. [32] demonstrated that NF-186 is required for the recruitment of brevican to the AIS, suggesting that assembly of the specialized ECM depends on nodal and AIS CAMs. In addition to brevican and versican, CNS nodal extracellular matrices also contain phosphacan, tenascin-R, and Bral1, although genetic loss of any single one of these ECM proteins does not affect the clustering of nodal Nav channels [49-51]. The function of AIS and nodal ECMs is unknown, but may include active clustering of channels at CNS nodes, stabilization of clusters, and even buffering of ions [50, 52].
Consistent with a role in assembly of Nav channel clusters, a unique nodal ECM exists in the PNS and by itself can induce clustering of channels along axons. This nodal, PNS ECM consists primarily of the Schwann cell-derived protein gliomedin and the L1 CAM NrCAM which is also cleaved from the Schwann cell surface and incorporated into the nodal ECM [53, 54]. These two proteins play important roles in the initial assembly of Nav channel clusters at PNS nodes (see below).
While many AIS and nodal ECM proteins have been identified, none have been reported at juxtaparanodes. Ogawa et al., [39] recently demonstrated the enrichment of ADAM22 at juxtaparanodes. Since ADAM22 interacts with members of the Leucine-rich Glioma Inactivated (LGI) family of ECM proteins [55], we performed a screen for LGI-proteins at juxtaparanodes and found that antibodies against LGI-3 labeled juxtaparanodes (unpublished results). Thus, one common feature of clustered axonal ion channels is that they associate with a complementary ECM.
2.5 AIS as both node and juxtaparanode
As described above, the juxtaparanodes and nodes of Ranvier occupy distinct axonal domains and do not share any proteins in common. In contrast, the AIS is comprised of the sum of the proteins found at nodes and juxtaparanodes, but the Kv1 channel protein complex is restricted to the distal part of the AIS, suggesting that it is the ‘juxtaparanodal’ part of the AIS. Nodes of Ranvier have been proposed to be evolutionary derivatives of the AIS that co-evolved with myelination [1]. However, there is no information on the evolutionary origin of the juxtaparanode. That the AIS has both properties of nodes and juxtaparanodes suggest that as with nodal Na+ channels, juxtaparanodes derive from the AIS and mechanisms arose concomitant with myelination that restricted Kv1 channels to juxtaparanodes.
3. Assembly of axonal domains
Although nodes, juxtaparanodes, and AIS share a common molecular organization, their mechanisms of assembly are distinct. The major difference is that AIS are instrinsically organized by the neuron, i.e. neurons contain all the necessary ‘ingredients’ to assemble an AIS. In contrast, despite having the necessary axonal ‘ingredients’, a node of Ranvier or a juxtaparanode will not form in the absence of glial-derived signals and neuron-glia interactions. Thus, AIS form from the inside-out, while nodes and juxtaparanodes are assembled from the outside-in. As described above, the key proteins necessary for AIS assembly are scaffolds. Thus, loss of ankG blocks clustering of all other AIS proteins [32, 41], while loss of PSD-93 from cultured neurons blocks clustering of Kv1 channels [17].
What are the key interactions that mediate assembly of nodes and juxtaparanodes? Recent experiments have revealed important details about the mechanisms underlying node of Ranvier formation. Intriguingly, there appear to be at least two overlapping mechanisms that exist to ensure nodes are properly assembled. The first of these mechanisms depends on ECMs secreted from myelinating glia. This route to nodal channel clustering has been described most thoroughly in the PNS [53, 54, 56]. Here, Schwann cells secrete gliomedin and NrCAM. These ECM proteins bind to axonal NF-186, which in turn functions as an attachment site for ankG. AnkG then assembles the ion channel and βIV spectrin protein complex necessary for saltatory conduction [33]. The second mechanism for channel clustering occurs when paranodal junctions form during myelination. These act as membrane diffusion barriers flanking nodes. They exclude nodal proteins from beneath the myelin sheath, and restrict them to nodes. In the CNS, the paranodal barrier mechanism appears to be the primary clustering mechanism [57]. While details of the ECM-dependent mechanism are an active area of investigation, it may depend on the chondroitin-sulfate proteoglycan brevican which has been shown to be a binding partner for NF-186 [32].
The idea that at least two mechanisms exists for channel clustering at nodes finds key experimental support in both the PNS and CNS. In the PNS, gliomedin-null and NrCAM-null mice still assemble nodes of Ranvier [54, 58]. Similarly, paranodal mutant mice lacking Caspr, a CAM necessary for the proper formation of paranodal junctions, also have normal nodal Nav channel clustering [59]. However, double-mutant mice lacking both gliomedin and Caspr have dramatically impaired node formation [54], suggesting that paranodal junctions and ECM-CAM interactions at nodes are overlapping mechanisms to facilitate Nav channel clustering. In the CNS, mice with disrupted paranodal junctions and lacking nodal neurofascin-186 fail to cluster Nav channels. However, if the paranodal junctions are rescued, nodes still form [60]. Together, these results strongly suggest that there exist both paranodal and ECM-dependent mechanisms to facilitate ion channel clustering at nodes of Ranvier[52].
Like nodes of Ranvier, the proper clustering of juxtaparanodal K+ channels also depends on at least two distinct neuron-glia interactions. As for nodes, paranodal junctions function to exclude Kv1 channels from paranodal regions and restrict their lateral diffusion in the axonal membrane. Thus, paranodal mutant mice have Kv1 channels that invade into regions immediately adjacent to nodal Nav channel clusters [59, 61, 62]. The second mechanism is also similar to that occurring at nodes. Specifically, the CAM TAG-1 found on the inner membrane of Schwann cells and axonal TAG-1/Caspr2 interact as a hetero-trimeric protein complex to initiate Kv1 channel clustering. Mice lacking either one of these CAMs have dramatically impaired Kv1 channel clustering in PNS myelinated axons [63, 64]. Surprisingly, although Kv1 channels and Caspr2 can co-immunoprecipitate one another, their interactions are indirect, and the mediator(s) of their interaction are unknown, although protein 4.1B may contribute since loss of this scaffold impairs Kv1 channel clustering [38].
Taken together, these results demonstrate that the assembly of membrane domains along myelinated axons follows a similar set of ‘rules’: axonal CAMs interact with glial ECM proteins or glial CAMs (Fig. 2). These glial proteins position or stabilize the CAMs along the axon in appropriate locations. These axonal CAMs then function to recruit scaffolds that can bind and stabilize ion channels. Finally, membrane barriers also exist in the axon to further restrict channels and other axonal proteins to distinct sites.
Figure 2.
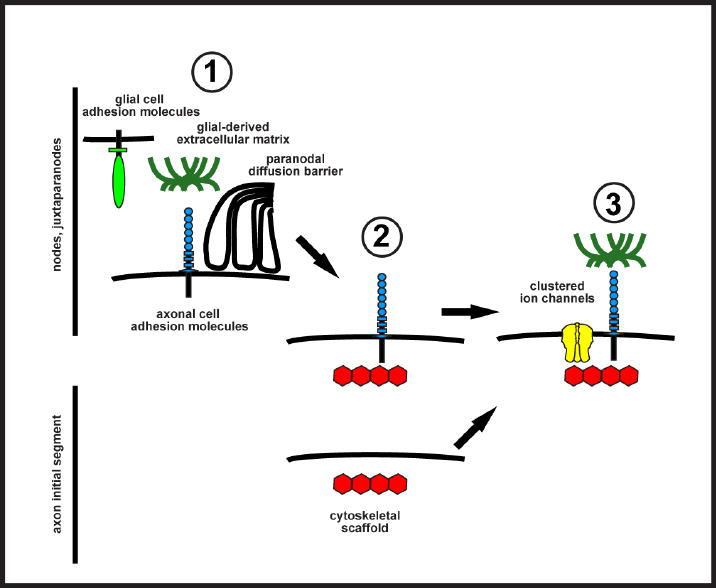
Cartoon illustrating domain assembly in axons. Nodes and juxtaparanodes are assembled from the ‘outside-in’ through neuron-glia interactions, but AIS are formed from the ‘inside-out’ without any dependence on other cell types. Three different neuron-glia interactions ( ) can contribute to the initiation of ion channel clustering in myelinated axons. Glial cell adhesion molecules, glia-derived ECM proteins, or paranodal junctions bind to and restrict the position of axonal cell adhesion molecules in the axolemma. These axonal CAMs function as attachment sites for cytoskeletal scaffolds (
) can contribute to the initiation of ion channel clustering in myelinated axons. Glial cell adhesion molecules, glia-derived ECM proteins, or paranodal junctions bind to and restrict the position of axonal cell adhesion molecules in the axolemma. These axonal CAMs function as attachment sites for cytoskeletal scaffolds ( ). At axon initial segments, the upstream mechanisms that recruit or restrict scaffolds to the AIS are unknown. Finally, ion channels become restricted to excitable domains by binding to scaffolds (
). At axon initial segments, the upstream mechanisms that recruit or restrict scaffolds to the AIS are unknown. Finally, ion channels become restricted to excitable domains by binding to scaffolds ( ). At AIS, ECM proteins are also recruited through binding to AIS CAMs.
). At AIS, ECM proteins are also recruited through binding to AIS CAMs.
4. Maintenance of axonal domains
In addition to the membrane specializations found along axons, neurons are subdivided into two major polarized domains: the axonal and somatodendritic domains, each of which is populated by unique sets of molecules that are important for synaptic input and integration (somatodendritic domain) or action potential propagation (axonal domain). The junction between these two domains corresponds to the AIS, and proteins and lipids found here are remarkably stable with very long half-lives or dramatically reduced diffusion rates when compared with other regions of the cell [65-67]. Based on these observations, the AIS has been proposed to function as a kind of diffusion barrier, to regulate trafficking and restrict membrane protein distribution in axons. Direct evidence to support this idea has come from the analysis of neurons lacking ankG. For example, Hedstrom et al. [65] used a short-hairpin RNA (shRNA) to silence expression of ankG in mature, fully polarized hippocampal neurons. Upon loss of ankG, all other AIS protein components including ion channels, CAMs, and ECM proteins were also lost from the AIS, indicating that ankG is necessary for initial recruitment of these proteins and their long-term stability. Even more surprising, however, was the observation that upon loss of ankG the axon began to de-differentiate and revert to a dendritic phenotype with all the molecular and structural characteristics of a dendrite. Similar observations were made in vivo using a mouse that lacks ankG in Purkinje neurons [68]. One other way the AIS maintains neuronal polarity is by excluding distinct classes of vesicular cargoes from the axon, thereby contributing to the maintenance of neuronal polarity[69, 70]. A more thorough discussion of the important role the AIS plays in maintenance of neuronal polarity can be found in [71].
4.2 plasticity of axonal membrane domains
Although the AIS, nodes of Ranvier, and juxtaparanodes have traditionally been considered static structures that are responsible only for AP initiation and propagation, recent data suggests that the properties of these domains may in fact be plastic such that different types of neuronal activity can result in changes in the density, length, or position of ion channel clusters. For example, Grubb and Burrone [72] recently demonstrated that chronic depolarization or sustained patterns of activity could cause the physical translocation of the AIS to more distal sites along the axon, resulting in a neuron that is less excitable. In contrast, Kuba et al. [73] demonstrated that depriving auditory neurons of synaptic input resulted in the expansion of the AIS, with more Nav channels available to decrease the membrane depolarization needed to generate an AP. Together, these experiments support the general concept of AIS homeostatic plasticity, and point to the AIS as a previously unappreciated site that can dramatically influence a neuron’s input-output function. One important point to emphasize is that the changes occurring at the AIS happen over much longer time-scales than the kind of plasticity observed at synapses.
5. Axonal domains in disease
Since the clustering of Na+ and K+ channels at nodes and juxtaparanodes depends on neuron-glia interactions, it is not surprising that demyelination, dysmyelination, and altered neuron-glia interactions leads to loss of nodal and juxtaparanodal ion channel clusters and altered nerve conduction [74-77]. Demyelination and disrupted neuron-glia interactions also results in the upregulation of Nav1.2 in several CNS disease models [76, 78, 79], while demyelination in Trembler-J mice, a model of the peripheral demyelinating Charcot-Marie-Tooth disease, results in upregulation of the tetrodotoxin-resistant Nav channel Nav1.8 [80]. In contrast to these models of demyelination, some investigators have also examined the capacity of transplanted glial cells to reestablish proper ion channel clustering along axons during remyelination. For example, transplantation of human oligodendrocyte precursors cells into congenitally hypomyelinated Shiverer mice resulted in profound remyelination and functional recovery that included the proper clustering of Nav and Kv1 channels along axons [81]. Together, these studies highlight the important role that myelin plays in regulating axonal ion channel expression and localization.
Nodes of Ranvier have also been proposed to be the direct targets of autoantibodies in some forms of PNS and CNS autoimmune diseases. For example, in acute motor axonal neuropathy (AMAN), Susuki et al. [82] showed that nodes of Ranvier are dismantled by the deposition of IgG, complement, and membrane attack complex. Intriguingly, multiple sclerosis patients have also been shown to have autoantibodies against the nodal CAM NF-186, suggesting that disruption of nodes themselves may be a contributor to the pathophysiology of MS [83].
AIS are also susceptible to disruption after injury. Recently, Schafer et al. [84] demonstrated that after ischemic injury the AIS cytoskeletal proteins ankG and βIV spectrin are rapidly proteolyzed by the calcium-dependent cysteine protease calpain. Since maintenance of ion channel clusters require AIS ankG, upon proteolysis of this scaffold Nav channel clusters could no longer be detected. Consistent with its important role in neuronal polarity, proteolysis of ankG also resulted in redistribution of MAP2, a microtubule assocated protein, into the axon. Thus, ischemic injury resulted in both loss of ion channel clusters, and loss of the neuronal polarity that is necessary for proper nervous system function. Other laboratories have reported that calpain can directly proteolyze Nav channels and affect ion channel function[85]. Besides ischemic injury, traumatic injury and nerve crush have also been reported to cause proteolysis of ankG and βIV spectrin [84, 86]. Together, these results suggest that disruption of ion channel clusters may be a common consequence of many types of nervous system injuries and diseases. Given the central role played by clustered ion channels in nervous system function, it will be important to understand the developmental mechanisms that lead to channel clustering when considering therapeutic strategies aimed at nervous system repair.
6. Conclusion
Clustering of ion channels along axons is essential to normal nervous system function. While some of the proteins that contribute to clustering have been identified, our knowledge of the composition of these domains remains rudimentary. This limitation has impaired our ability to determine the mechanisms underlying assembly of these important axonal domains. Key questions that remain unanswered include: how is ankG restricted to the AIS? Do ECM proteins contribute to Nav channel clustering in the CNS? How do Caspr2 and Kv1 channels interact? What are the functions of PSD-93, PSD-95, and ADAM22 at juxtaparanodes? These and many other questions will continue to keep neurobiologists and cell biologists busy for many years to come.
Acknowledgments
Supported by NIH grants NS044916 and NS069688. MNR is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hill AS, et al. Ion channel clustering at the axon initial segment and node of ranvier evolved sequentially in early chordates. PLoS Genet. 2008;4(12):e1000317. doi: 10.1371/journal.pgen.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hille B. Ionic Channels of Excitable Membranes. third edition. Sunderland, MA: Sinauer Associates, Inc; 2001. [Google Scholar]
- 3.Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;328(5980):906–9. doi: 10.1126/science.1187958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kole MH, et al. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11(2):178–86. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- 5.Caldwell JH, et al. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A. 2000;97(10):5616–20. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duflocq A, et al. Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol Cell Neurosci. 2008 doi: 10.1016/j.mcn.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Boiko T, et al. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30(1):91–104. doi: 10.1016/s0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- 8.Schafer DP, et al. Early events in node of Ranvier formation during myelination and remyelination in the PNS. Neuron Glia Biol. 2006;2(2):69–79. doi: 10.1017/S1740925X06000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu W, et al. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12(8):996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- 10.Van Wart A, Trimmer JS, Matthews G. Polarized distribution of ion channels within microdomains of the axon initial segment. J Comp Neurol. 2007;500(2):339–52. doi: 10.1002/cne.21173. [DOI] [PubMed] [Google Scholar]
- 11.Brackenbury WJ, et al. Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc Natl Acad Sci U S A. 2010;107(5):2283–8. doi: 10.1073/pnas.0909434107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28(53):14329–40. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogiwara I, et al. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27(22):5903–14. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devaux JJ, et al. KCNQ2 Is a Nodal K+ Channel. J Neurosci. 2004;24(5):1236–44. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156(8):1185–95. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz JR, et al. KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J Physiol. 2006;573(Pt 1):17–34. doi: 10.1113/jphysiol.2006.106815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogawa Y, et al. Postsynaptic density-93 clusters Kv1 channels at axon initial segments independently of Caspr2. J Neurosci. 2008;28(22):5731–9. doi: 10.1523/JNEUROSCI.4431-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inda MC, Defelipe J, Munoz A. Voltage-gated ion channels in the axon initial segment of human cortical pyramidal cells and their relationship with chandelier cells. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0511197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, et al. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature. 1993;365(6441):75–9. doi: 10.1038/365075a0. [DOI] [PubMed] [Google Scholar]
- 20.Vabnick I, et al. Dynamic potassium channel distributions during axonal development prevent aberrant firing patterns. J Neurosci. 1999;19(2):747–58. doi: 10.1523/JNEUROSCI.19-02-00747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasband MN. It’s ‘juxta’ potassium channel. J Neurosci Res. 2004;76:749–757. doi: 10.1002/jnr.20073. [DOI] [PubMed] [Google Scholar]
- 22.Kole MH, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55:633–647. doi: 10.1016/j.neuron.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 23.Goldberg EM, et al. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58(3):387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garrido JJ, et al. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003;300(5628):2091–4. doi: 10.1126/science.1085167. [DOI] [PubMed] [Google Scholar]
- 25.Pan Z, et al. A common ankyrin-G-based mechanism retains KCNQ and Nav channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bréchet A, et al. Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interaction with ankyrin G. J Cell Biol. 2008;183:1101–14. doi: 10.1083/jcb.200805169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berghs S, et al. betaIV spectrin, a new spectrin localized at axon initial segments and nodes of ranvier in the central and peripheral nervous system. J Cell Biol. 2000;151(5):985–1002. doi: 10.1083/jcb.151.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y, et al. {beta}IV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol. 2007;176(4):509–19. doi: 10.1083/jcb.200610128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komada M, Soriano P. [Beta]IV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol. 2002;156(2):337–48. doi: 10.1083/jcb.200110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacas-Gervais S, et al. BetaIVSigma1 spectrin stabilizes the nodes of Ranvier and axon initial segments. J Cell Biol. 2004;166(7):983–90. doi: 10.1083/jcb.200408007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou D, et al. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143(5):1295–304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hedstrom KL, et al. Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol. 2007;178(5):875–86. doi: 10.1083/jcb.200705119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dzhashiashvili Y, et al. Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J Cell Biol. 2007;177(5):857–70. doi: 10.1083/jcb.200612012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rasband MN, et al. Clustering of neuronal potassium channels is independent of their interaction with PSD-95. J Cell Biol. 2002 doi: 10.1083/jcb.200206024. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horresh I, et al. Multiple molecular interactions determine the clustering of Caspr2 and Kv1 channels in myelinated axons. J Neurosci. 2008;28(52):14213–22. doi: 10.1523/JNEUROSCI.3398-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poliak S, et al. Localization of Caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J Neurosci. 2001;21(19):7568–75. doi: 10.1523/JNEUROSCI.21-19-07568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogawa Y, et al. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J Neurosci. 2006;26(19):5230–9. doi: 10.1523/JNEUROSCI.0425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horresh I, et al. Organization of myelinated axons by Caspr and Caspr2 requires the cytoskeletal adapter protein 4.1B. J Neurosci. 2010;30(7):2480–9. doi: 10.1523/JNEUROSCI.5225-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogawa Y, et al. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J Neurosci. 2010 doi: 10.1523/JNEUROSCI.4661-09.2010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J Biol Chem. 1994;269(44):27163–6. [PubMed] [Google Scholar]
- 41.Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155(5):739–46. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burkarth N, et al. Neurofascin regulates the formation of gephyrin clusters and their subsequent translocation to the axon hillock of hippocampal neurons. Mol Cell Neurosci. 2007;36(1):59–70. doi: 10.1016/j.mcn.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Ango F, et al. Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at purkinje axon initial segment. Cell. 2004;119(2):257–72. doi: 10.1016/j.cell.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Zweier C, et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. 2009;85(5):655–66. doi: 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strauss KA, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354(13):1370–7. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 46.Alarcon M, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82(1):150–9. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.John N, et al. Brevican-containing perineuronal nets of extracellular matrix in dissociated hippocampal primary cultures. Mol Cell Neurosci. 2006 doi: 10.1016/j.mcn.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 48.Bekku Y, et al. Brevican distinctively assembles extracellular components at the large diameter nodes of Ranvier in the CNS. J Neurochem. 2009;108(5):1266–76. doi: 10.1111/j.1471-4159.2009.05873.x. [DOI] [PubMed] [Google Scholar]
- 49.Dours-Zimmerman MT, et al. Versican V2 assembles the extracellular matrix surrounding the nodes of Ranvier in the central nervous system. J Neurosci. 2009 doi: 10.1523/JNEUROSCI.4158-08.2009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bekku Y, et al. Bral1: its role in diffusion barrier formation and conduction velocity in the CNS. J Neurosci. 2010;30(8):3113–23. doi: 10.1523/JNEUROSCI.5598-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weber P, et al. Mice deficient for tenascin-R display alterations of the extracellular matrix and decreased axonal conduction velocities in the CNS. J Neurosci. 1999;19(11):4245–62. doi: 10.1523/JNEUROSCI.19-11-04245.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Susuki K, Rasband MN. Molecular mechanisms of node of Ranvier formation. Curr Opin Cell Biol. 2008;20(6):616–23. doi: 10.1016/j.ceb.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eshed Y, et al. Secreted gliomedin is a perinodal matrix component of peripheral nerves. J Cell Biol. 2007;177(3):551–62. doi: 10.1083/jcb.200612139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feinberg K, et al. A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron. 2010;65(4):490–502. doi: 10.1016/j.neuron.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sagane K, Ishihama Y, Sugimoto H. LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int J Biol Sci. 2008;4(6):387–96. doi: 10.7150/ijbs.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eshed Y, et al. Gliomedin mediates schwann cell-axon interaction and the molecular assembly of the nodes of ranvier. Neuron. 2005;47(2):215–29. doi: 10.1016/j.neuron.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 57.Rasband MN, et al. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci. 1999;19(17):7516–28. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Custer AW, et al. The role of the ankyrin-binding protein NrCAM in node of Ranvier formation. J Neurosci. 2003;23(31):10032–9. doi: 10.1523/JNEUROSCI.23-31-10032.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhat MA, et al. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30(2):369–83. doi: 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- 60.Zonta B, et al. Glial and neuronal isoforms of Neurofascin have distinct roles in the assembly of nodes of Ranvier in the central nervous system. J Cell Biol. 2008;181(7):1169–77. doi: 10.1083/jcb.200712154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dupree JL, Girault J-A, Popko B. Axo-glial interactions regulate the localization of axonal paranodal proteins. J Cell Biol. 1999;147(6):1145–1151. doi: 10.1083/jcb.147.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boyle ME, et al. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30(2):385–97. doi: 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- 63.Poliak S, et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162(6):1149–60. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Traka M, et al. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J Cell Biol. 2003;162(6):1161–72. doi: 10.1083/jcb.200305078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183(4):635–40. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakada C, et al. Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat Cell Biol. 2003;5(7):626–32. doi: 10.1038/ncb1009. [DOI] [PubMed] [Google Scholar]
- 67.Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999;397(6721):698–701. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- 68.Sobotzik JM, et al. AnkyrinG is required to maintain axo-dendritic polarity in vivo. Proc Natl Acad Sci U S A. 2009;106(41):17564–9. doi: 10.1073/pnas.0909267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Song AH, et al. A selective filter for cytoplasmic transport at the axon initial segment. Cell. 2009;136(6):1148–60. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 70.Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6(3):201–14. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- 71.Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010 doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- 72.Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010 doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuba H, Oichi Y, Ohmori H. Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature. 2010 doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- 74.Dugandzija-Novakovic S, et al. Clustering of Na+ channels and node of Ranvier formation in remyelinating axons. J Neurosci. 1995;15(1 Pt 2):492–503. doi: 10.1523/JNEUROSCI.15-01-00492.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rasband MN, et al. Potassium channel distribution, clustering, and function in remyelinating rat axons. J Neurosci. 1998;18(1):36–47. doi: 10.1523/JNEUROSCI.18-01-00036.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rios JC, et al. Paranodal interactions regulate expression of sodium channel subtypes and provide a diffusion barrier for the node of Ranvier. J Neurosci. 2003;23(18):7001–11. doi: 10.1523/JNEUROSCI.23-18-07001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coman I, et al. Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain. 2006;129(Pt 12):3186–95. doi: 10.1093/brain/awl144. [DOI] [PubMed] [Google Scholar]
- 78.Craner MJ, et al. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. 2004;101(21):8168–73. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rasband MN, et al. Dysregulation of axonal sodium channel isoforms after adult-onset chronic demyelination. J Neurosci Res. 2003;73(4):465–70. doi: 10.1002/jnr.10675. [DOI] [PubMed] [Google Scholar]
- 80.Devaux JJ, Scherer SS. Altered ion channels in an animal model of Charcot-Marie-Tooth disease type IA. J Neurosci. 2005;25(6):1470–80. doi: 10.1523/JNEUROSCI.3328-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Windrem MS, et al. Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell. 2008;2(6):553–65. doi: 10.1016/j.stem.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Susuki K, et al. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J Neurosci. 2007;27(15):3956–67. doi: 10.1523/JNEUROSCI.4401-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mathey EK, et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med. 2007;204(10):2363–72. doi: 10.1084/jem.20071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schafer DP, et al. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci. 2009;29(42):13242–54. doi: 10.1523/JNEUROSCI.3376-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.von Reyn CR, et al. Calpain mediates proteolysis of the voltage-gated sodium channel alpha-subunit. J Neurosci. 2009;29(33):10350–6. doi: 10.1523/JNEUROSCI.2339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reeves TM, et al. Proteolysis of Submembrane Cytoskeletal Proteins Ankyrin-G and alphaII-Spectrin Following Diffuse Brain Injury: A Role in White Matter Vulnerability at Nodes of Ranvier. Brain Pathol. 2010 doi: 10.1111/j.1750-3639.2010.00412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]