

Abstract
Polycomb Group (PcG) proteins are essential regulators of development that maintain gene silencing in Drosophila and mammals through alterations of chromatin structure. One key PcG protein, Posterior Sex Combs (PSC), is part of at least two complexes: Polycomb Repressive Complex 1 (PRC1) and dRING Associated Factors (dRAF). PRC1-class complexes compact chromatin and inhibit chromatin remodeling, while dRAF has E3 ligase activity for ubiquitylation of histone H2A; activities of both complexes can inhibit transcription. The noncovalent effects of PRC1-class complexes on chromatin can be recapitulated by PSC alone, and the region of PSC required for these activities is essential for PSC function in vivo. To understand how PSC interacts with chromatin to exert its repressive effects, we compared the ability of PSC to bind to and inhibit remodeling of various nucleosomal templates, and determined which regions of PSC are required for mononucleosome binding and inhibition of chromatin remodeling. We find that PSC binds mononucleosome templates but inhibits their remodeling poorly. Addition of linker DNA to mononucleosomes allows their remodeling to be inhibited, although higher concentrations of PSC are required than for inhibition of multi-nucleosome templates. The C-terminal region of PSC (aa 456-1603) is important for inhibition of chromatin remodeling, and we identified aa 456-909 as sufficient for stable nucleosome binding but not for inhibition of chromatin remodeling. Our data suggest distinct mechanistic steps between nucleosome binding and inhibition of chromatin remodeling.
Keywords: chromatin, Polycomb Group, nucleosome, chromatin remodeling
PcG genes were initially identified in Drosophila [1, 2] and are conserved throughout metazoans [3]. They play key roles in Hox gene expression during development [4] and other cellular processes such as X-inactivation [5], genomic imprinting [6], cell cycle progression, and self-renewal of embryonic and adult stem cells [7]. PcG proteins form several multiprotein complexes that are thought to maintain gene silencing through interaction with chromatin (reviewed in [8]), including covalent modifications of histone proteins [7] and non-covalent changes of chromatin [9-12] and DNA [13] structures.
PRC1 was the first PcG complex to be purified from Drosophila [11, 14] and contains 4 main PcG proteins: Posterior Sex Combs (PSC), Polyhomeotic (Ph), dRING, and Polycomb (Pc), as well as substoichiometric levels of Sex Comb on Midleg (SCM) [15]. PRC1 inhibits chromatin remodeling by the human Swi/Snf (hSwi/Snf) ATP-dependent chromatin remodeling complex [11, 14] and also inhibits in vitro transcription [10]. Subsequent studies in mammalian cells identified a PRC1-like complex with a similar constellation of PcG proteins and activities [16]. Reconstitution of Drosophila PRC1 indicated that the four stoichiometric PcG proteins in this complex (PRC1 Core Complex, PCC), or a subcomplex of PSC, Pc, and dRING, are sufficient for its activities towards chromatin [14, 17]. PSC alone can recapitulate most of the activities of PRC1-class complexes, although it is slightly less efficient than PCC [14].
The importance of PSC interaction with chromatin in vivo was evidenced by the analysis of a series of Psc alleles that encode C-terminal truncations [18]. Proteins encoded by alleles that produce severe phenotypes in vivo assemble into PCC, but do not inhibit chromatin remodeling or transcription in vitro [18]. PSC truncations were also tested for their ability to compact chromatin, and the same sequences required for inhibition of chromatin remodeling and transcription are required for chromatin compaction [9]. Thus, chromatin compaction mediated by the C-terminal region of PSC (aa 456-1603) is likely responsible for inhibition of chromatin remodeling and transcription. This region is predicted to be unstructured [19], and the mechanism by which it interacts with chromatin is unknown. Other unstructured proteins function by assuming structured conformations when they contact their ligand(s); it is possible that the C-terminal region of PSC assumes a structured conformation when bound to chromatin. A homolog of PSC, Suppressor 2 of Zeste [Su(Z)2], resides adjacent to PSC in the Drosophila genome and can functionally substitute for some PSC activities in vivo [20, 21]. Su(Z)2 has similar in vitro activities as PSC [22], but the C-terminal region of Su(Z)2, which mediates its inhibition of chromatin remodeling activity, is not homologous to that of PSC. Instead, the two C-terminal regions share a unique amino acid composition [23]. Thus, the precise ordering of the primary sequence may not be important for the function of this region, but further work is needed to understand how it exerts its effects on chromatin. For example, it is not known whether different parts of the large C-terminal region of PSC (aa 456-1603) have different activities.
In addition to PRC1, PSC is part of a second complex in Drosophila: dRING associated factors (dRAF) [24]. dRAF contains both PSC and dRING, as well as other subunits not found in PRC1, but not PRC1 components Pc or Ph. dRAF functions as an E3 ligase for ubiquitylation of histone H2A, an activity which also seems to be essential for PcG silencing but cannot be recapitulated by PSC alone and is carried out poorly by PCC [24]. Therefore, it seems that PSC contributes two key functions to PcG silencing: non-covalent modification of chromatin structure which requires its C-terminal, unstructured region [19], and ubiquitylation of histone H2A which involves the N-terminal RING domain [25-27] and other conserved sequences. The N-terminal region is also involved in assembly of PSC into both complexes.
Although extensive data support the significance of non-covalent modification of chromatin structure by PSC, exactly how this protein interacts with chromatin is not yet known. Initial work indicated that PSC inhibits remodeling of multinucleosome arrays but not of mononucleosomes [14]; however, it is unclear if this substrate discrimination is at the level of binding or a subsequent step. To understand the mechanism of PSC, the substrate and protein requirements for PSC binding to nucleosomes and inhibiting chromatin remodeling were investigated.
MATERIALS AND METHODS
Proteins and templates
FLAG epitope-tagged PSC and truncations were expressed in Sf9 cells using the Bac-to-Bac Baculovirus Expression System (Invitrogen) and purified as described [14]. hSwi/Snf, was purified from nuclear extracts from HeLa cells expressing FLAG-Ini-1 as described [28]. Active fractions of PSC and truncation preparations were determined using DNA in excess of protein [14] and were typically between 20%-30%, except for PSC1-572, which was <1% active. All stated concentrations refer to active concentrations unless otherwise indicated.
DNA templates contain one or tandem repeats of the Xenopus laevis 5S nucleosome positioning sequence [29] or the 601 nucleosome positioning sequence [30]. Plasmids and details are available on request. Different linker orientations were achieved by placing the positioning sequence in different locations relative to the DNA ends. Mononucleosome and dinucleosome (1N and 2N, where N stands for nucleosome) DNA templates were prepared by PCR followed by gel purification on acrylamide gels, while 6N and 12N DNAs were prepared by restriction enzyme digestion of plasmids followed by purification on a Sephacryl S-1000 gel filtration column (170×1.5 cm). In some cases, 1N and 2N templates were prepared with Cy5- or Cy3-labeled primers for visualization. Mononucleosome templates were assembled from purified 5S fragment and HeLa histone octamers by step dialysis as described [31, 32], followed by purification by sedimentation through 10-30% glycerol gradients. Nucleosomal templates greater than 1N were assembled by salt gradient dialysis with HeLa histone octamers as described [32]. Nucleosome assembly was tested by electrophoretic mobility shift assay (EMSA) or, for multinucleosome templates, by EcoRI digestion (EcoRI cuts between each 5S repeat) followed by EMSA [33]. Mononucleosomes were heated at 50 degrees for at least 1 hour prior to using for REAs in order to position the nucleosomes. All experiments were carried out with at least two preparations of mononucleosomes except for the 1N-100 template which was from a single nucleosome assembly.
To confirm mononucleosome positioning, nucleosomes were first analyzed on native acrylamide gels. End positioned nucleosomes migrate more rapidly than middle positioned ones (sFigure 1A). Mononucleosome positions were further confirmed by micrococcal nuclease digestion followed by purification of the protected, 150 base pair DNA fragment (carried out by adding SDS to 0.1% and PCR cleanup kit (Machery-Nagel)), and digestion with HhaI. If nucleosomes are positioned over the positioning sequence, then the restriction digest of the protected fragments should be identical, irrespective of the length or configuration of the template (sFigure 1B, C, D). This was observed for 601 (sFigure 1D) but not for 5S templates (sFigure 1C).
Restriction Enzyme Accessibility (REA) Assay
REAs were carried out as described [14], with nucleosomes at 1nM. In some cases, mononucleosomes were also tested at 5nM. Concentration in all cases refers to the concentration of mononucleosome sized DNA (so that reactions with 12N or 1N templates contain the same amount of DNA). Standard reaction conditions are: 12mM Hepes, pH7.9, 0.12mM EDTA, 12% glycerol, 60mM KCl, 2mM ATP, 0.025% NP40 and either 4mM (multinucleosome templates) or 6mM MgCl2 (1N templates). PstI or HhaI (New England Biolabs), which is unique in each 1N, 2N, or 6N template and is covered by a nucleosome, was used for restriction digest of 5S templates. For 601 mononucleosomes, only HhaI is present so all experiments were carried out with HhaI. DNA was visualized with SYBR Gold or SYBR Green I stain (Invitrogen), or Cy5 or Cy3 fluorescence on a Typhoon Imager. hSwi/Snf was titrated to ensure that it was present at saturating concentrations for nucleosome remodeling with each template. % inhibition was calculated from the % uncut using the following equation:
Electrophoretic Mobility Shift Assay (EMSA)
Reactions were carried out in conditions described for REA assays except without ATP. Simple binding reactions contained 1 or 5 nM nucleosomes, and competition binding reactions contained 1 nM PSC, 0.5 nM nucleosomes of labeled substrates, and up to 10 nM nucleosomes of unlabeled competitors. Samples were resolved in 0.6% or 0.8% 0.5x-TBE agarose gels at 4 V/cm for 2-4 hours and visualized with SYBR Gold stain or by Cy3- or Cy5-fluorescence on a Typhoon Imager. All experiments were carried out at least three times with two different preparations of PSC. EMSAs were quantified in ImageQuant as indicated in sFigure 5.
Co-Immunoprecipitation
Nuclear extracts of Sf9 cells infected with viruses encoding FLAG-PSC, HA-PSC, or both were prepared as described [34] except that extracts were not dialyzed after the high salt extraction. M2 anti-FLAG agarose (Sigma) used for immunoprecipitation was blocked with 500 ng/μL BSA (NEB) in BC buffer (20mM Hepes, pH 7.9, 0.2mM EDTA) with 20% glycerol, 300 mM KCl, 0.05% NP40), and protease inhibitors for 30 min at 4°C. Nuclear extracts were incubated with anti-FLAG agarose at 4°C for 2 hours. Agarose beads were washed five times with blocking buffer and boiled in SDS-PAGE sample buffer. Samples were resolved by SDS-PAGE and immunoblotted with M5 anti-FLAG or HA-7 anti-HA antibodies (Sigma). Co-purification was carried out in two independent experiments.
Chemical Crosslinking
PSC (150-450 nM total protein) was diluted into a final reaction mix of 12mM Hepes, pH 7.4, 0.12 mM EDTA, 0.5mM PMSF, 2mM MgCl2 and allowed to equilibrate at 30°C for 15 min. followed by addition of up to 0.25 mM 1,8-bis-maleimidodiethyleneglycol (BM) or 2.5 mM 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (Thermo Fisher) at room temperature for 1-2 hours. Crosslinked products were resolved by SDS-PAGE and immunoblotted with M5 anti-FLAG antibody. Experiments were carried out at least three times with two different preparations of PSC.
Glycerol Gradient Sedimentation
5-35% glycerol gradients (280 μl) were created in BC buffer with 60-900mM KCl, 0.5mM DTT, 0.2mM PMSF, and 0.05% NP40. Proteins were adjusted to the appropriate KCl concentration and to 25-100mM total protein concentration in BC buffer with 0.5mM DTT, 0.2mM PMSF, and 0.05% NP40 and incubated 15 minutes at 30°C before loading onto gradients. Gradients were centrifuged for 3 hours at 55,000 rpm at 4°C in rotor TLS-55 (Optima mini-ultracentrifuge) and fractionated into 40 μl fractions by pipeting from the top. Fractions were analyzed by SDS-PAGE followed by Western blotting. Similar results were obtained on larger gradients (2.2 ml), in that PSC and PCC migrate as distinct peaks, but PSC migrates much further into the gradient than PCC.
RESULTS
PSC inhibits nucleosome remodeling of 2N substrates more effectively than 1N
To determine the chromatin substrate requirements for PSC, inhibition of remodeling was compared on templates with different numbers of nucleosomes. Remodeling of templates consisting of 2 or 6 nucleosomes (referred to as 2N or 6N) was 50% inhibited at concentrations between 0.5 and 1nM PSC, a ratio of 0.5-1 PSC: nucleosomes, and inhibition was saturated at 2nM PSC (Figure 1A-C). (All concentrations refer to the active concentration unless otherwise indicated; see Methods for explanation.) This is the same concentration range previously demonstrated for inhibition of remodeling of 12N templates [14]. In contrast, but consistent with previous work [14], remodeling of a 157-bp mononucleosome template (10-1N) is inhibited poorly (Figure 1D, F). Increasing the concentration of mononucleosomes to 5nM allowed some inhibition of remodeling although inhibition did not reach 50% at a 40-fold higher concentration than needed to reach 50% inhibition of nucleosomal arrays (Figure 1E, F).
Figure 1. PSC inhibits remodeling of mononucleosomes poorly.
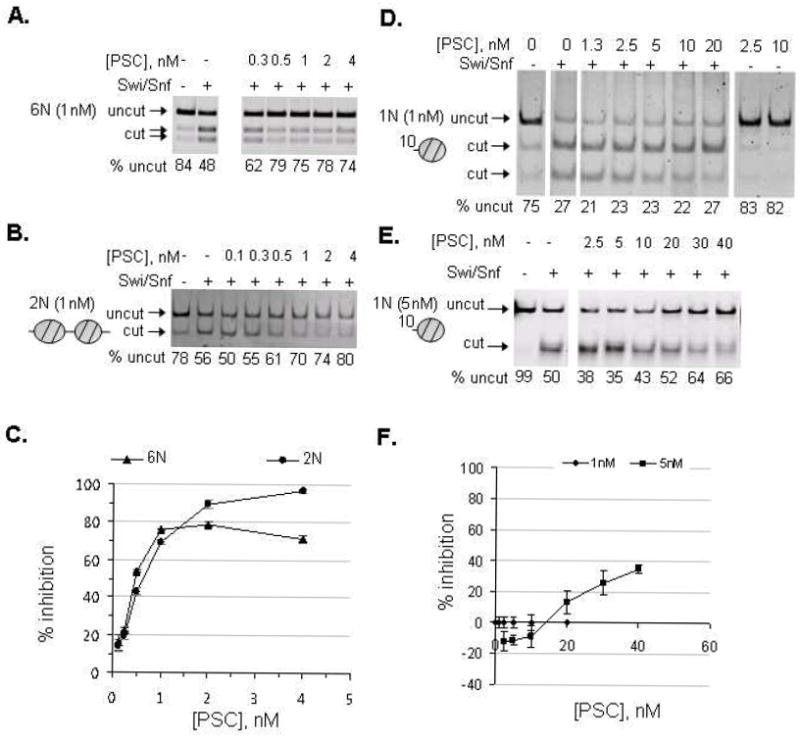
Restriction enzyme accessibility (REA) assays were carried out with PSC at the indicated concentrations and hSwi/Snf. A,B) Representative gels of REAs with 6N (A) and 2N (B) templates. (N refers to nucleosomes, so 6N is a 6-nucleosome template; all of these templates are composed of repeats of the 5S nucleosome positioning sequence.). C) Summary of REAs with 6N and 2N templates. D, E) Representative gel of REA with mononucleosomes assembled on a 157-bp (10-1N) 5S template at 1nM (D) or 5nM (E). Note that PstI was used for digestion in the experiment with 1nM nucleosomes, while HhaI was used with 5nM nucleosomes. Both enzymes have single digestion sites in the template, but produce slightly different digestion patterns (HhaI digests the template into 77 and 80 bp fragments, which are not resolved, while PstI digests it into 99 and 52 bp fragments). Both enzymes were used in these assays and they produce very similar results. D) Summary of REAs with mononucleosome template. Error bars in are SEM.
Remodeling of a nucleosome with linker DNA is inhibited by PSC
2N templates differ from 1N in two ways: an additional nucleosome and the presence of linker DNA. Because PSC binds tightly to naked DNA (Kd = 0.2nM for 155-bp fragment [14]), linker DNA might be important for PSC interaction with mononucleosomes. A series of mononucleosomes was created with linker DNA on one or both sides, and the ability of PSC to inhibit Swi/Snf-mediated chromatin remodeling of them was tested. We refer to these templates according to the length of the linker DNA and its orientation. Thus, 50-1N-50 templates should have a nucleosome positioned in the middle of a 247 base pair fragment with 50 base pair linkers on each side, while 1N-100 has a 100 bp linker 3’ to the nucleosome positioning sequence. We initially prepared these templates using the 5S nucleosome positioning sequence for direct comparison with our polynucleosome template results. However, although the nucleosome positioning was sufficient to block restriction enzymes present in the 5S sequence (data not shown), detailed analysis of positioning indicated that the longer templates all had multiple nucleosome positions (sFigure 1 and data not shown). Thus, we prepared a second series of mononucleosome templates using the artificial 601 nucleosome positioning sequence [30] (sFigure 1). We were able to obtain well-positioned populations mononucleosomes after purification by glycerol gradient sedimentation. We note, however, that all 601 assemblies onto 247 base pair fragments produced two positions, one which the nucleosome is likely at one end of the template, and the other of which positions it in the middle of the fragment. These positions could be separated on glycerol gradients.
In order to compare inhibition of chromatin remodeling on different mononucleosome templates, it is important that all of the substrates can be remodeled similarly. We therefore characterized the kinetics of remodeling by hSwi/Snf on each of our mononucleosome templates (sFigure 2). We find that two different 5S templates (10-1N, 50-1N-50), and two 601 mononucleosomes (100-1N, 50-1N-50) are all remodeled with similar kinetics (sFigure 2A, B); the rate constants are also similar to those previously measured for mononucleosomes (between 0.07 and 0.14 min-1) [35]. In contrast, two of the 601 mononucleoosmes (10-1N, 1N-100) were remodeled 3-14 times slower than the other templates (rate constants 0.01-0.02 min-1) (sFigure 2C-E). We do not understand the basis of this effect. Previous studies have demonstrated that RNA polymerase II transcription is differentially affected in the forward and reverse orientation on 601 mononucleosomes [36], so it is possible that our data reflect these differences. However, the two mononucleosomes that are remodeled slowly have linkers in opposite orientation (for the 10-1N, the linker protrudes from the 5’ end of the 601 sequence, while for the 1N-100, it protrudes from the 3’ end), so that a simple orientation effect seems unlikely. Nevertheless, we chose to compare inhibition of remodeling on the rapidly remodeled nucleosomes (i.e. 5S 10-1N, 5S 50-1N-50, 601 100-1N, 601 50-1N-50), since we were concerned that the slow remodeling might enhance inhibition by PSC. The 5S 1N-10 mononucleosome is well positioned (sFigure 1A, C) so can be compared with the 601 templates with longer linkers.
Addition of 100 bp of linker DNA to one side, or 50 bp to both sides of 601 mononucleosomes enhanced inhibition of chromatin remodeling by PSC. 50% inhibition was reached at between 8 and 10nM PSC (Table I summarizes all of the templates used and the results of REAs). This is about a 10-fold higher concentration of PSC than required for inhibition on 2N templates (Figure 2, Table I). We also tested 5S mononucleosomes with 50 base pair linkers on both sides and find that their remodeling is also inhibited by PSC, but requires about two-fold higher concentrations than the 601 mononucleosomes. Since our initial mononucleosome experiments suggested that PSC can inhibit 10-1N templates when the templates are used at 5nM instead of 1nM, we repeated the analysis of the mononucleosomes with linkers using template at 5nM (sFigure 3, Table I). PSC inhibited remodeling more efficiently when then template was used at 5nM. 50% inhibition was reached between 7.5 and 15nM PSC for templates with two 50 bp linkers or one 100 bp linker. This is a ratio of 1.5-3 PSC per nucleosome, which is lower than the 8-10 PSC per nucleosome required for inhibition using 1nM substrate, but is still higher than the amount of PSC required for 50% inhibition of a 2N template (less than 1 PSC per nucleosome, Figure 1C, Table I). These experiments indicate that linker DNA contributes to PSC inhibition of chromatin remodeling but is not sufficient to explain the difference between 1N and 2N templates.
Table I.
Summary of REA results.
Figure 2. PSC inhibits mononucleosomes with one long linker or two linkers better than mononucleosomes with one short linker.
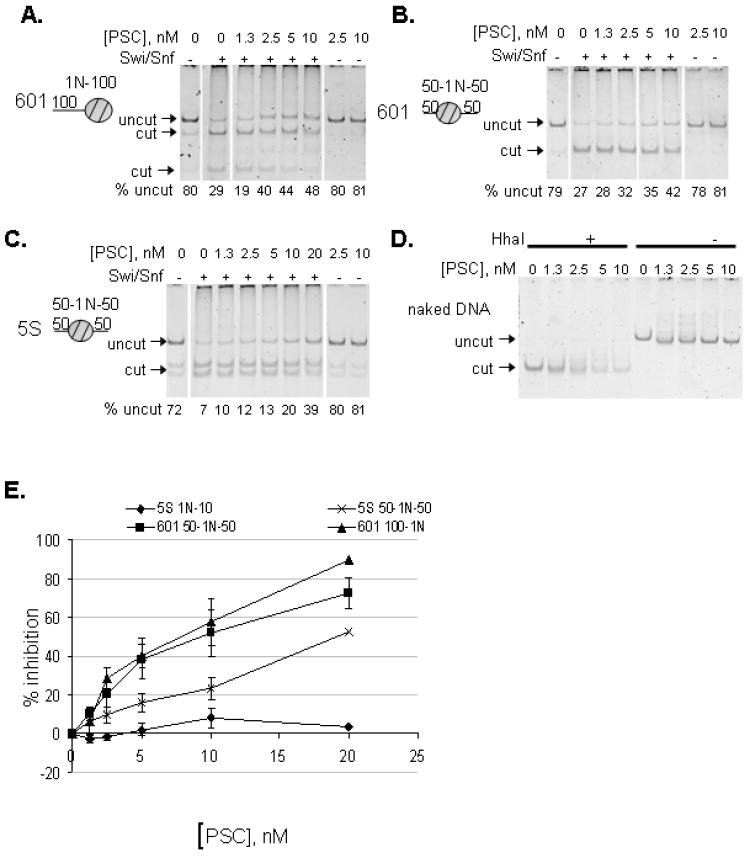
A-C) Representative REAs on a series of mononucleosome templates. The three templates are all 247-bp long. D) PSC does not inhibit restriction enzyme digestion. Naked DNA at 1nM was incubated with PSC under identical conditions as in the remodeling assays either with or without the restriction enzyme HhaI. E) Summary of REAs on the various mononucleosome templates. Note that even though remodeling of templates with linkers can be inhibited by PSC, higher concentrations are required than for 2N templates (compare with Figure 1C). Error bars are SEM.
Previously, we found that PSC does not inhibit background restriction enzyme accessibility of polynucleosomal templates [14]. However, because the mononucleosome experiments involve using high concentrations of PSC, we repeated these experiments with mononucleosome templates. We find that even low concentrations of PSC inhibit background restriction enzyme access (Figure 1D, Figure 2). This does not seem likely to account for inhibition of hSwi/Snf induced chromatin remodeling since it occurs even at low concentrations of PSC that do not block hSwi/Snf induced restriction enzyme accessibility. To further test the effect of PSC on restriction enzyme access, we incubated PSC with naked DNA under the same conditions as used for REA. Under these conditions, PSC does not inhibit restriction enzyme access, although some template loss was observed at higher concentrations of PSC (Figure 2D, sFigure 3D). Thus, inhibition of restriction enzyme activity seems unlikely to account for the effect on accessibility observed in chromatin remodeling reactions.
Mononucleosome experiments were carried out with 4mM free MgCl2, while experiments with polynucleosome templates, which can be compacted by MgCl2, were carried out with 2mM free MgCl2. To confirm that this difference in reaction conditions did not explain less efficient inhibition of remodeling of mononucleosomes, a small number of experiments were carried out with mononucleosome templates with 2mM free MgCl2 (sFigure 4). Results from these experiments were very similar to those with 4mM free MgCl2 in that remodeling of the 10-1N template was inhibited poorly by PSC, while remodeling of the 50-1N-50 was inhibited by high concentrations of PSC.
PSC binds mononucleosomes under conditions where it does not inhibit their remodeling
One explanation for the poor ability of PSC to inhibit mononucleosome templates without linkers is that it binds these templates poorly. This would also be consistent with the observation that increasing the template concentration by five fold improves inhibition of remodeling (compare Figure 2 vs. sFigure 3). To test this idea, EMSAs were carried out using mononucleosome templates. PSC shifts the mobility of all of the mononucleosomes tested, indicating that it binds to them (Figure 3A, top panel). Quantification of EMSAs (see sFigure 5 for explanation of quantification) indicates that 50% binding occurs between 0.9 and 1.5nM for all three templates (Figure 3B). These numbers represent an upper estimate of the Kd for the interaction between PSC and mononucleosomes since the template concentration used here (1nM) is too high to allow measurement of the true Kd. (Nucleosomes are not stable at concentrations below 1nM, so different methods will be required to measure the true Kd). Nevertheless, these data indicate that, under the conditions used for REA assays, binding to all of the templates (as determined by EMSA) is very similar.
Figure 3. PSC binds all mononucleosome templates at low concentrations that do not inhibit chromatin remodeling.
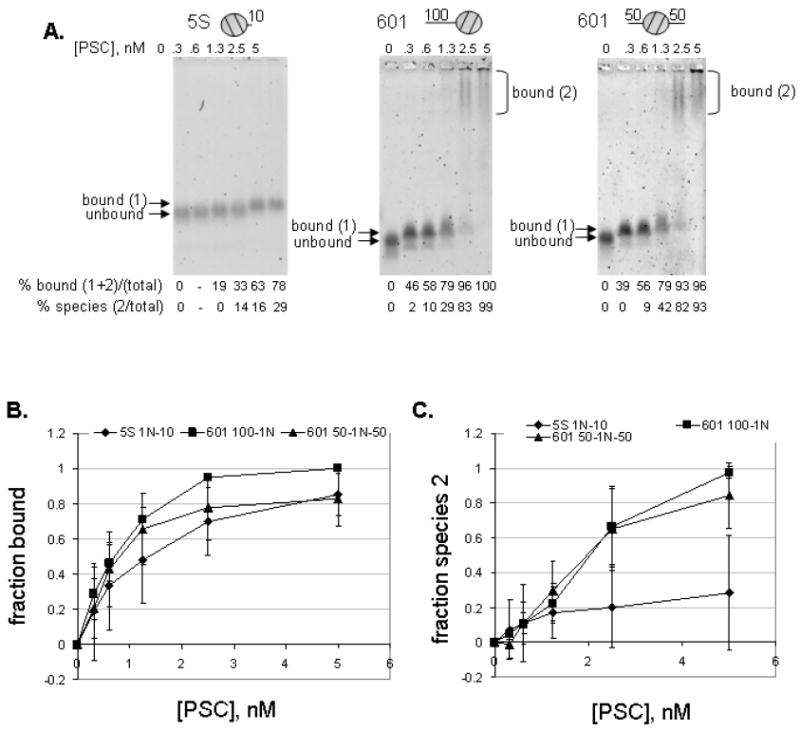
A. EMSAs with various mononucleosome substrates at 1nM. Details of quantification of EMSAs are presented in sFigure 5. At low concentrations, PSC binds mononucleosomes (bound (1)), while at higher concentrations it forms a second, slowly migrating species (bound (2)). B. Quantification of fraction bound (bound 1+2) for each template. C. Quantification of slowly migrating species (bound (2)) only. Error bars are SD.
For all of the nucleosomes, two bound species were observed. The first appeared at low concentrations of PSC. The second species migrated slowly in agarose gels and in some cases was retained in the well. Both species were included in the quantification of the fraction bound. However, when the second bound species is quantified separately, the 10-1N templates require higher concentrations of PSC to produce the slow mobility complex (bound species 2) (Figure 3C). 50% of the template formed the second bound species on the longer templates at about 2nM PSC, while less than 40% of the 10-1N template formed the second bound species at 5nM PSC. When nucleosomes were used at 5nM PSC, the 10-1N template formed the second bound species (sFigure 3F) with 10nM PSC. The concentration of PSC required to form the second bound species was higher when 5nM of the 50-1N-50 mononucleosomes were used as the template. This suggests that the ratio of PSC to nucleosomes might be important for forming this species, since at both 1 and 5nM template the second species forms at a ratio of 2 or more PSC per nucleosome.
Inhibition of nucleosome remodeling occurs at the PSC concentrations that produced the slow-moving species (compare Figure 3C with Figure 2E). However, formation of this species is not sufficient since 100% of 50-1N-50 or 100-1N templates form the second bound species at 5nM PSC, but remodeling is inhibited by about 40% (sFigure 3). This bound species may be necessary for inhibition of nucleosome remodeling, since the 10-1N template at 1 nM formed little of this species, and no inhibition of nucleosome remodeling was observed. In contrast, in experiments with 5nM template, the second species was formed and some inhibition of remodeling was observed (although at two-fold higher PSC concentrations than required to form the second bound species). Thus, PSC binds to mononucleosomes under conditions where it does not inhibit their remodeling, and inhibition of remodeling correlates with formation of higher order PSC-nucleosome complexes, although the precise nature of these complexes is not known.
PSC binds less stably to mononucleosomes than to dinucleosomes
EMSA suggests that binding of PSC to mononucleosomes occurs at concentrations where remodeling of polynucleosomes is inhibited but does not result in inhibition of mononucleosome remodeling. To determine whether PSC preferentially binds polynucleosome templates, binding to mononucleosomes was compared to binding to 2N templates in the presence of competitor nucleosomes. PSC was incubated with a mixture of Cy3 or Cy5-labeled 1N (50-1N-50) or 2N templates (probe) and increasing amounts of each unlabeled competitor, and binding was analyzed by EMSA (Figure 4A). In these experiments, PSC was used at 1nM, a concentration that does not form the slowly migrating species. Under these conditions, binding to the 50-1N-50 template is prevented by excess 50-1N-50, or 2N. In contrast, binding to a 2N template is prevented by 2N competitor, but not a 10-fold excess of 50-1N-50.
Figure 4. Competition binding assays demonstrate that PSC binds 2N templates preferentially over 1N templates but that binding to both templates is stable.
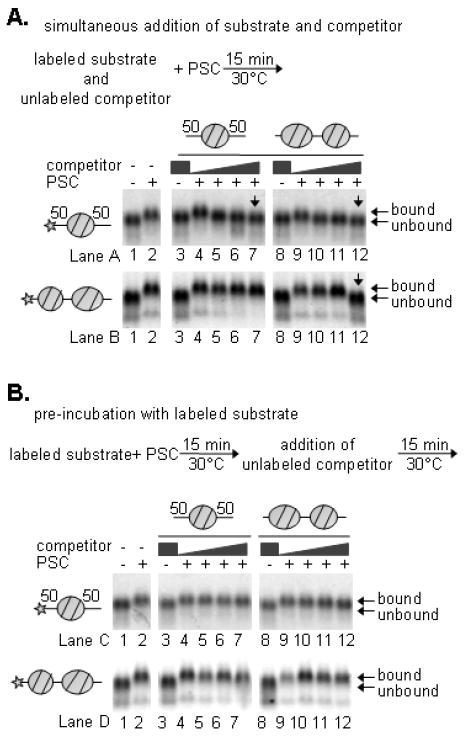
A. Competition assays in which competitor was added simultaneously with labeled substrates, as indicated. B. Competition assays in which competitor was added after a 15-minute pre-incubation with labeled substrate. PSC was used at 1nM and labeled nucleosomes at 0.5nM. Competitor was used up to 10nM.
The experiments above address partitioning of PSC among different templates when binding to all of them is initiated simultaneously. To probe the stability of binding of PSC to the two templates, PSC was pre-bound to either template, and binding was challenged by the addition of excess unlabelled competitor. In this case, after 15 minutes of incubation with the competitor, PSC remained bound to both the 50-1N-50 and the 2N templates in the presence of an excess of either competitor (Figure 4B). Thus, PSC binds more tightly to 2N than 50-1N-50 templates, which might explain how it is able to inhibit remodeling of 2N templates more effectively than 1N ones. Nevertheless, once PSC is bound to the 50-1N-50, the complex is stable for at least 15 minutes.
PSC self-interacts
The correlation between inhibition of chromatin remodeling and formation of the slowly migrating species that might represent interaction between PSC-bound nucleosomes, and the requirement for 2 nucleosomes for an efficiently used substrate suggests that PSC may bring nucleosomes together to inhibit their remodeling. This could occur if PSC has two binding sites or if the protein interacts to form dimers or multimers. Several strategies were used to determine whether PSC can interact with itself. First, PSC was epitope tagged with either FLAG or HA and expressed in Sf9 cells. When both versions of PSC were co-expressed, they could be co-immunoprecipitated with anti-FLAG beads (Figure 5A). Second, treatment of PSC with two different chemical cross-linking reagents results in formation of high molecular weight complexes (Figure 5B). In the cross-linking experiments, an initial high molecular weight complex is observed, which could be a dimer, but with increasing cross-linking, additional higher molecular weight species are formed. Third, PSC was analyzed by glycerol gradient sedimentation. The peak of PSC migrated further in the gradient than Immunoglobulin (150kDa) but not as far as Ferritin (440kDa) (Figure 5C). PSC also migrates further into the gradient than PCC (composed of PSC, Pc, and dRING), which has a predicted molecular weight of 261kDa. Previous scanning transmission electron microscopy (STEM) measurements suggest that PCC is a monomeric complex [9]. A subcomplex composed of PSC+dRING migrates similarly to PSC. PSC, PCC, and PSC+dRING migrate as discrete peaks at KCl concentrations from 300-900mM, although at lower KCl concentrations, variable levels of aggregates were observed in the pellet (data not shown). These results are consistent with PSC forming salt-stable dimers that are prevented by assembly into PCC. Efforts to confirm that PSC is indeed the size of a dimer using size exclusion chromatography were not successful. Under all conditions tested, PSC migrates in the void volume and as an extended peak in the earliest eluting fractions from a Superose 6 column (date not shown). Thus, we conclude that PSC can self-interact but confirming the stoichiometry of the interaction will require additional methods. The behavior of PSC could be explained by its predicted unstructured C-terminal region which may adopt an extended, flexible shape.
Figure 5. PSC interacts with itself to likely form dimers.
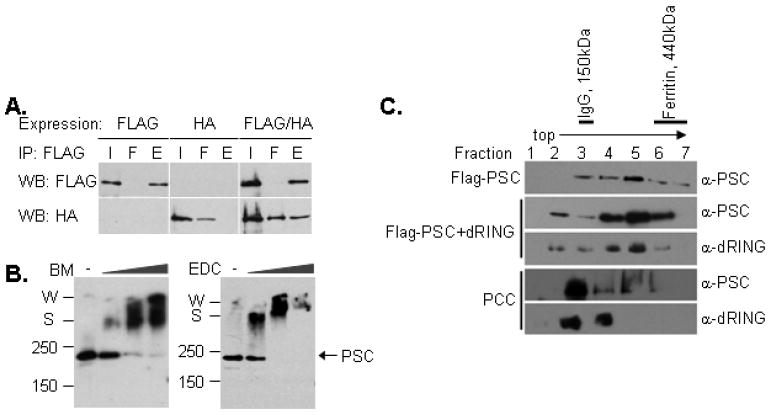
A. FLAG- and HA-tagged PSC were expressed individually or together, and FLAG-PSC immunoprecipitated. When FLAG-PSC and HA-PSC were co-infected (third panel), HA-PSC co-purifies with FLAG-PSC. B. Crosslinking with BM (left) or EDC (right) followed by Western blot analysis. PSC cross-links into high molecular weight species when treated with either cross-linker. C. Glycerol gradient sedimentation of PSC, PSC-dRING, or PCC (PSC+dRING+Pc) followed by Western blot analysis. The peak of FLAG-PSC is in fraction 5, as is the peak for PSC-dRING, while the peak of PCC is in fraction 3. The molecular weight of FLAG-PSC is 171 kDa. Gradients shown were run in 900mM KCl, although similar results were obtained at 300 and 600mM KCl.
Inhibition of chromatin remodeling requires PSC sequences that are not essential for nucleosome binding
Previous data indicate that the N-terminal region of PSC (aa1-572) can assemble into PCC but does not inhibit chromatin remodeling or compact chromatin [9, 18]. In contrast, the C-terminal half of the protein (aa456-1603) has both activities. To narrow the region of PSC required for inhibition of chromatin remodeling, a series of PSC constructs truncated from either or both the N and C termini (Figure 6A,B) was tested for inhibition of binding to and chromatin remodeling of a mononucleosome substrate with two linkers (Figure 6C). These experiments were carried out with 5S 50-1N-50 mononucleosomes at 5nM. PSC1-572 did not bind mononucleosomes or inhibit their remodeling, but both PSC1-910 and PSC456-1603 bind to mononucleosomes, induce formation of the slowly migrating species, and inhibits nucleosome remodeling. PSC456-909, which encompasses the overlap between the two active truncations, binds the mononucleosomes but does not induce formation of the slowly migrating species or inhibit nucleosome remodeling. Thus, this truncation separates nucleosome binding activity of PSC from inhibition of chromatin remodeling. The activity of PSC456-909 was also tested on 601 50-1N-50 mononucleosome templates at 1nM; despite binding to these templates, it does not inhibit their remodeling (not shown).
Figure 6. Inhibition of chromatin remodeling requires sequences in PSC not required for stable nucleosome binding.
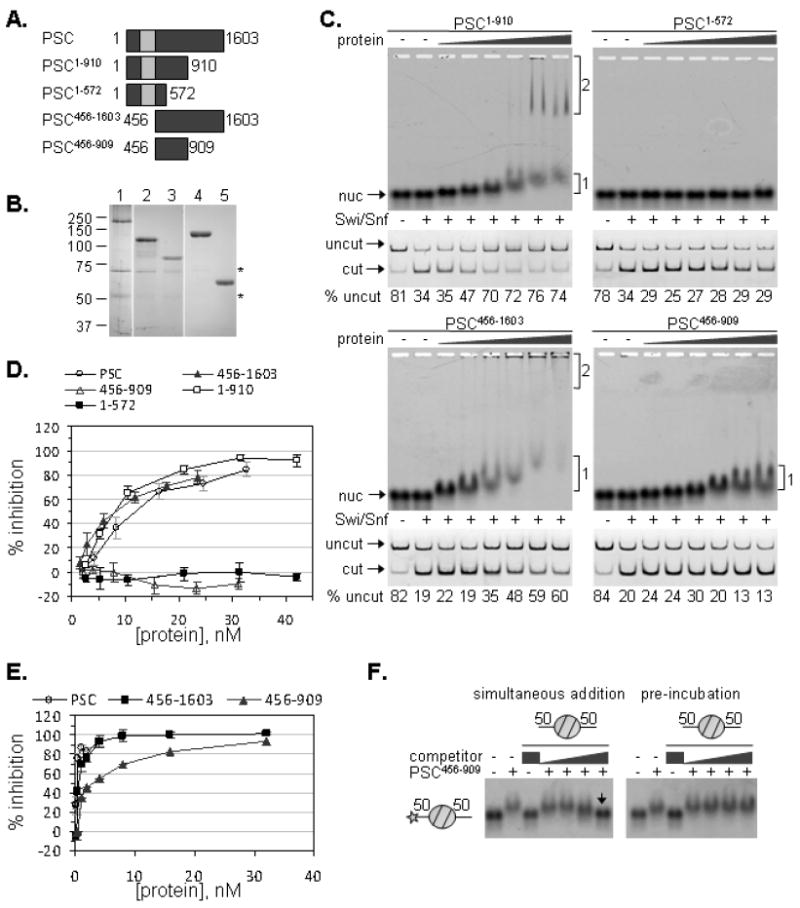
A. Schematic diagram of PSC truncations. Note that some of these truncations have been previously characterized [9, 18]. B. Colloidal Blue stained gel of representative preparations of PSC and PSC truncations. Asterisks (*) indicate contaminants that are likely Hsc70 (~70 kDa) and β-tubulin (~55 kDa). C. EMSA and REA of each truncation with a 50-1N-50 mononucleosome (2 linkers). The two shifted species are indicated next to the gels in cases where they form. D. Summary of REAs on two-linker 1N substrate with PSC and the various truncations. E. PSC456-909 was tested for inhibition of remodeling of a 12N array. Full length PSC, and PSC456-1603 were included for comparison. F. PSC456-909 was tested for stable binding to the two-linker 1N substrate, using competition assays as in Figure 4.
To further investigate the activity of PSC456-909, its ability to inhibit remodeling of 12N arrays was compared with PSC and PSC456-1603 (Figure 6E). PSC456-909 was able to inhibit remodeling of a 12N array but required at least 10-fold higher concentrations than PSC or PSC456-1603. PSC456-909 was also tested for stable binding to the 50-1N-50 5S mononucleosome in the two competition assays used in Figure 4 (Figure 6F). PSC456-909 behaved like PSC in this assay: its binding is stable to competitor added after pre-incubation. Thus, inhibition of chromatin remodeling and formation of the slow-migrating mononucleosome-PSC species require additional sequences in PSC that are not necessary for stable nucleosome binding.
DISCUSSION
The protein and substrate requirements for nucleosome binding and inhibition of chromatin remodeling by PSC were investigated. The key conclusions are: 1) PSC binds mononucleosomes under conditions where it does not inhibit their remodeling; 2) mononucleosomes with two 50 bp linkers or a single 100 bp linker are better substrates for inhibition of remodeling than mononucleosomes with a single 10 base pair linker; 3) a region of PSC sufficient for chromatin binding does not inhibit chromatin remodeling; 4) full-length PSC interacts with itself. The analysis of substrate and PSC sequence requirements thus reveals that inhibition of chromatin remodeling requires additional steps beyond nucleosome binding.
The relationship between nucleosome binding by PSC and inhibition of remodeling
Our data indicate that while two nucleosomes are required for full inhibition of chromatin remodeling at low concentrations of PSC, remodeling of mononucleosomes with DNA linkers can be inhibited more than 50%, albeit at higher PSC concentrations. We consider two models for how this can occur. The first is based solely on differences in PSC affinity for different substrates, while the second suggests that inhibition of nucleosome remodeling requires that nucleosomes are brought together to form a structure that is more difficult to remodel.
Our data indicate that PSC binds nucleosomes and interacts with itself, suggesting a simple explanation for higher affinity binding of PSC to 2N than 1N templates: two PSC molecules could bind 2N templates (one per nucleosome), and interact with each other. Similar interactions may occur when PSC-bound mononucleosomes are brought together, as seems to occur in the slowly migrating species observed by EMSA. The data in Figure 4B, as well as unpublished results with multinucleosome templates, suggest that once PSC is bound, little exchange is observed for at least 15 minutes with either 1N or 2N templates. In our REA assays, PSC is pre-bound to the template prior to the addition of hSwi/Snf. The amount of inhibition may therefore reflect the fraction of the templates that are bound by PSC in the initial binding step, which remain stably bound and inhibited over the entire reaction. Although our stability experiment does not provide evidence for this, it is possible that in the one hour mononucleosome remodeling experiments, PSC binds and falls off nucleosomes during the reaction, so that the level of inhibition observed reflects an ongoing competition between PSC and hSwi/Snf for substrate binding. Another possibility is that hSwi/Snf itself is able to displace PSC from 1N templates more effectively than from polynucleosome templates.
A second model for inhibition of chromatin remodeling by PSC suggests an additional step beyond nucleosome binding in which nucleosomes are brought together to form a structure that is refractory to binding or remodeling by hSwi/Snf. This interaction is by definition intermolecular for 1N templates but intramolecular (and therefore more efficient) for 2N ones. The finding that inhibition of remodeling of mononucleosomes occurs at lower ratios of PSC to nucleosomes when nucleosomes are at 5nM vs. 1nM is consistent with the idea that the concentration of PSC bound mononucleosomes is important for inhibition of chromatin remodeling. These data also suggest that inhibition does not involve non-specific aggregation of PSC, since this would occur at the same PSC concentration when mononucleosomes are used at either 1 or 5nM, but inhibition is more effective with 5nM template. The finding that inhibition of chromatin remodeling is correlated with the formation of the slow-migrating PSC-1N species by EMSA also suggests that PSC bound nucleosomes can interact, likely through PSC-PSC interactions. These interactions could be important for inhibition of remodeling, and could be analogous to the interactions that occur on polynucleosome templates and lead to chromatin compaction [9], although we cannot entirely rule out that the slowly migrating species are non-specific aggregates whose formation is facilitated by the extra DNA present on mononucleosome templates with linkers. Bringing nucleosomes together could inhibit chromatin remodeling either by decreasing the affinity of hSwi/Snf for its substrate, as was suggested by the exclusion of hSwi/Snf from PSC bound multinucleosome arrays (see Figure 6 of [14]), or allowing binding but not remodeling. It is difficult to distinguish these two possibilities at present, in part because we do not yet know if hSwi/Snf and PSC can simultaneously bind mononucleosomes (as would need to occur if PSC binding alone does not inhibit remodeling).
Mapping the minimal sequence requirements for inhibition of chromatin remodeling
The data with PSC truncations (Figure 6) further supports the idea that nucleosome binding is not sufficient for inhibition of chromatin remodeling since PSC456-909 binds stably to 1N templates but does not inhibit their remodeling (Figure 6). PSC456-909 also does not promote formation of the slow-migrating species in EMSA, suggesting that additional regions of PSC mediate this activity. PSC 456-1097 was also tested for inhibition of remodeling of nucleosomal arrays (data not shown) and behaves similarly to PSC456-909. Thus, sequences required for formation of the slow-migrating species in EMSA and inhibition of remodeling may reside between 1097 and 1603. Because PSC1-910, which lacks these sequences, can form the slow-migrating species and inhibit chromatin remodeling, sequences between aa1 and 456 may functionally replace the C-terminal sequences in this setting. Psc alleles truncated at aa910, 1075, or 1098 have milder phenotypes than those truncated at aa521, but nevertheless have phenotypes, indicating that the truncated region of the protein (aa 1098-1603) is important for PSC function in vivo [21]. The proteins encoded by these alleles do not have defects in vitro [18], and it seems that either an intact N-terminal or C-terminal domain, in conjunction with the sequences from 456-909 are sufficient for chromatin compaction and inhibition of chromatin remodeling in vitro.
PSC self-interacts
Our data indicate that PSC can interact with itself; the idea of self-interaction is consistent with previously observed intragenic complementation of Psc alleles in Drosophila (20). In mammalian systems, Mel-18 (a homolog- of PSC) was demonstrated to form homodimers [37], even though most other analyses of PSC homologs have not detected homodimers. Computation and structural analysis suggest that both PSC and dRING have a RING domain and a predicted ubiquitin fold domain. The ubiquitin fold in the C-terminal region of Ring1b (mammalian homologue of dRING) can form homodimers [38, 39] although Ring1b preferentially forms heterodimers with Bmi-1. The Ring domains of Bmi-1 and Ring1b form heterodimers [25, 26], while full-length Bmi-1 and Ring1b form heterotetramers. Our analysis of a PSC-dRING subcomplex (Figure. 5C), indicates that it migrates similarly to a likely PSC dimer on a glycerol gradient; this complex may be a heterotetramer whose migration is dominated by the behavior of PSC. Our finding that PSC may form stable dimers suggests the Drosophila proteins differ from their mammalian homologs in their interactions.
Our glycerol gradient data also suggest that PSC assembled into PCC does not form salt-stable multimers. This is consistent with our previous STEM analysis of PCC, which measured the mass of PCC as that expected for a monomer [9]. It is also consistent with structural analysis of Ring1b, which indicates that dimerization is blocked by interaction with the C-terminal region of a Pc homologue [40]. Thus, addition of Pc may be the key to inhibiting PSC and dRING dimerization. It is interesting in this regard that Pc is missing from the dRAF complex that mediates histone ubiquitylation [24]. Perhaps PSC dimerization is important in this complex. Since PSC can inhibit chromatin remodeling on its own or in PCC, it seems that dimerization is not central to inhibition of chromatin remodeling or chromatin compaction. Although our glycerol gradient data are consistent with PSC forming dimers, it should be noted that this stoichiometry has not been rigorously confirmed. It will be interesting to determine the stoichiometry of PSC-PSC interactions in the presence and absence of mononucleosome substrates.
In summary, we have dissected the protein and substrate requirements for the Polycomb protein PSC to inhibit chromatin remodeling. Inhibition of chromatin remodeling and chromatin compaction may reflect the same activity since all PSC truncations that are active in chromatin remodeling can compact chromatin. Taken together, our results suggest inhibition of chromatin remodeling, and likely chromatin compaction, by PSC involve at least two distinct molecular events: nucleosome binding, and interactions among PSC bound nucleosomes.
Supplementary Material
Acknowledgments
The authors thank Dr. Nancy Kleckner for useful suggestions on the manuscript.
Abbreviations
- PcG
Polycomb Group
- PSC
Posterior Sex Combs
- PRC1
Polycomb Repressive Complex 1
- dRAF
dRING Associated Factors
- Ph
Polyhomeotic
- Pc
Polycomb
- SCM
Sex Comb on Midleg
- PCC
PRC1 Core Complex
- N
nucleosome
- EMSA
electrophoretic mobility shift assay
- REA
restriction enzyme accessibility assay
- STEM
scanning transmission electron microscopy
- aa
amino acid
Footnotes
This study was supported by NIH grant GM078456-01 to NJF.
SUPPORTING INFORMATION: Supporting information for this manuscript consists of five supporting figures. sFigure 1 is an analysis of nucleosome positioning of the mononucleosome templates used in this study. sFigure 2 is an anlysis of the kinetics of remodeling of the mononucleosome templates used in this study. sFigure 3 shows binding and inhibition of chromatin remodeling of mononucleosomes by PSC using 5nM substrate rather than 1nM. sFigure 4 shows inhibition of nucleosome remodeling by PSC at a lower MgCl2 concentration (2mM free Mg++ instead of 4mM used in the main figures). sFigure 5 demonstrates how EMSA experiments were quantified. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jurgens G. A group of genes controlling the spatial expression of the bithorax complex in Drosophila. Nature. 1985;316:153–155. [Google Scholar]
- 2.Lewis EB. A gene complex controlling segmentation in Drosophila. Nature. 1978;276(5688):565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- 3.Schuettengruber B, et al. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128(4):735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. 2004;38:413–443. doi: 10.1146/annurev.genet.38.072902.091907. [DOI] [PubMed] [Google Scholar]
- 5.Heard E, Disteche CM. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 2006;20(14):1848–1867. doi: 10.1101/gad.1422906. [DOI] [PubMed] [Google Scholar]
- 6.Delaval K, Feil R. Epigenetic regulation of mammalian genomic imprinting. Curr Opin Genet Dev. 2004;14(2):188–195. doi: 10.1016/j.gde.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet. 2007;8(1):9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- 8.Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10(10):697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 9.Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. 2004;306(5701):1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- 10.King IF, Francis NJ, Kingston RE. Native and recombinant polycomb group complexes establish a selective block to template accessibility to repress transcription in vitro. Mol Cell Biol. 2002;22(22):7919–7928. doi: 10.1128/MCB.22.22.7919-7928.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shao Z, et al. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell. 1999;98(1):37–46. doi: 10.1016/S0092-8674(00)80604-2. [DOI] [PubMed] [Google Scholar]
- 12.Margueron R, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32(4):503–518. doi: 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohd-Sarip A, et al. Architecture of a polycomb nucleoprotein complex. Mol Cell. 2006;24(1):91–100. doi: 10.1016/j.molcel.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Francis NJ, et al. Reconstitution of a functional core polycomb repressive complex. Mol Cell. 2001;8(3):545–556. doi: 10.1016/s1097-2765(01)00316-1. [DOI] [PubMed] [Google Scholar]
- 15.Saurin AJ, et al. A Drosophila Polycomb group complex includes Zeste and dTAFII proteins. Nature. 2001;412(6847):655–660. doi: 10.1038/35088096. [DOI] [PubMed] [Google Scholar]
- 16.Levine SS, et al. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol Cell Biol. 2002;22(17):6070–6078. doi: 10.1128/MCB.22.17.6070-6078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavigne M, et al. Propagation of silencing; recruitment and repression of naive chromatin in trans by polycomb repressed chromatin. Mol Cell. 2004;13(3):415–425. doi: 10.1016/s1097-2765(04)00006-1. [DOI] [PubMed] [Google Scholar]
- 18.King IF, et al. Analysis of a polycomb group protein defines regions that link repressive activity on nucleosomal templates to in vivo function. Mol Cell Biol. 2005;25(15):6578–6591. doi: 10.1128/MCB.25.15.6578-6591.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emmons RB, et al. Molecular genetic analysis of Suppressor 2 of zeste identifies key functional domains. Genetics. 2009;182(4):999–1013. doi: 10.1534/genetics.108.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beuchle D, Struhl G, Muller J. Polycomb group proteins and heritable silencing of Drosophila Hox genes. Development. 2001;128(6):993–1004. doi: 10.1242/dev.128.6.993. [DOI] [PubMed] [Google Scholar]
- 21.Wu CT, Howe M. A genetic analysis of the Suppressor 2 of zeste complex of Drosophila melanogaster. Genetics. 1995;140(1):139–181. doi: 10.1093/genetics/140.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo SM, Ahuja NK, Francis NJ. Polycomb group protein Suppressor 2 of zeste is a functional homolog of Posterior Sex Combs. Mol Cell Biol. 2009;29(2):515–525. doi: 10.1128/MCB.01044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brunk BP, Martin EC, Adler PN. Drosophila genes Posterior Sex Combs and Suppressor two of zeste encode proteins with homology to the murine bmi-1 oncogene. Nature. 1991;353(6342):351–353. doi: 10.1038/353351a0. [DOI] [PubMed] [Google Scholar]
- 24.Lagarou A, et al. dKDM2 couples histone H2A ubiquitylation to histone H3 demethylation during Polycomb group silencing. Genes Dev. 2008;22(20):2799–2810. doi: 10.1101/gad.484208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z, et al. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J Biol Chem. 2006;281(29):20643–20649. doi: 10.1074/jbc.M602461200. [DOI] [PubMed] [Google Scholar]
- 26.Buchwald G, et al. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. Embo J. 2006;25(11):2465–2474. doi: 10.1038/sj.emboj.7601144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ben-Saadon R, et al. The polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol Cell. 2006;24(5):701–711. doi: 10.1016/j.molcel.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 28.Sif S, et al. Mitotic inactivation of a human SWI/SNF chromatin remodeling complex. Genes Dev. 1998;12(18):2842–2851. doi: 10.1101/gad.12.18.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simpson RT, Stafford DW. Structural features of a phased nucleosome core particle. Proc Natl Acad Sci U S A. 1983;80(1):51–55. doi: 10.1073/pnas.80.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276(1):19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 31.Lee KM, Narlikar G. Assembly of nucleosomal templates by salt dialysis. Curr Protoc Mol Biol. 2001;Chapter 21:Unit 21 6. doi: 10.1002/0471142727.mb2106s54. [DOI] [PubMed] [Google Scholar]
- 32.Sif S, et al. Purification and characterization of mSin3A-containing Brg1 and hBrm chromatin remodeling complexes. Genes Dev. 2001;15(5):603–618. doi: 10.1101/gad.872801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carruthers LM, et al. Assembly of defined nucleosomal and chromatin arrays from pure components. Methods Enzymol. 1999;304:19–35. doi: 10.1016/s0076-6879(99)04004-5. [DOI] [PubMed] [Google Scholar]
- 34.Abmayr SM, et al. Preparation of nuclear and cytoplasmic extracts from mammalian cells. Curr Protoc Mol Biol. 2006;Chapter 12:Unit 12–1. doi: 10.1002/0471142727.mb1201s75. [DOI] [PubMed] [Google Scholar]
- 35.Narlikar GJ, Phelan ML, Kingston RE. Generation and interconversion of multiple distinct nucleosomal states as a mechanism for catalyzing chromatin fluidity. Mol Cell. 2001;8(6):1219–1230. doi: 10.1016/s1097-2765(01)00412-9. [DOI] [PubMed] [Google Scholar]
- 36.Bondarenko VA, et al. Nucleosomes can form a polar barrier to transcript elongation by RNA polymerase II. Mol Cell. 2006;24(3):469–479. doi: 10.1016/j.molcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 37.Fujisaki S, et al. Dimerization of the Polycomb-group protein Mel-18 is regulated by PKC phosphorylation. Biochem Biophys Res Commun. 2003;300(1):135–140. doi: 10.1016/s0006-291x(02)02791-2. [DOI] [PubMed] [Google Scholar]
- 38.Bezsonova I, et al. Ring1B contains a ubiquitin-like docking module for interaction with Cbx proteins. Biochemistry. 2009;48(44):10542–10548. doi: 10.1021/bi901131u. [DOI] [PubMed] [Google Scholar]
- 39.Czypionka A, et al. The isolated C-terminal domain of Ring1B is a dimer made of stable, well-structured monomers. Biochemistry. 2007;46(44):12764–12776. doi: 10.1021/bi701343q. [DOI] [PubMed] [Google Scholar]
- 40.Wang R, et al. Structural transitions of the RING1B C-terminal region upon binding the polycomb cbox domain. Biochemistry. 2008;47(31):8007–8015. doi: 10.1021/bi800857f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.