

Abstract
Lamina-associated polypeptide 2α (LAP2α) is a nucleoplasmic protein that interacts with A-type lamins and the retinoblastoma protein (pRb) and affects pRb-mediated cell cycle regulation and chromatin organization. Mutations in lamin A/C and LAP2α cause late onset striated muscle diseases, but the molecular mechanisms are poorly understood. We have recently reported on the striated muscle phenotype of LAP2α-deficient mice, revealing new unexpected roles of LAP2α. Loss of LAP2α in skeletal muscle caused an upregulated stem cell-type gene expression in muscle satellite cell progeny and their delayed myogenic differentiation in vitro. In vivo, the myofiber-associated muscle stem cell pool was increased. In addition, absence of LAP2α promoted muscle remodeling towards fast myofiber types in the soleus muscle of old animals. In cardiac tissue, deletion of LAP2α caused systolic dysfunction in young mice with an increased susceptibility for fibrosis in old animals. The functional impairment in the heart was accompanied by a deregulation of major cardiac transcription factors, GATA4 and MEF2c and activation of compensatory pathways, including the downregulation of β-adrenergic receptor signaling.
Here we discuss potential functions of LAP2α in striated muscle at molecular level and how loss of these functions may cause the diverse muscle phenotypes. We propose that LAP2α serves as a transcriptional co-regulator, which controls muscle specific gene expression during muscle regeneration, muscle remodeling and stress response.
Key words: lamin, LAP2α, MyoD, MEF2c, HDAC, pRb, muscle differentiation, heart disease, fiber type specification
Introduction
Lamins are nuclear intermediate filaments in metazoans, which form a highly complex meshwork underneath the nuclear membrane bi-layer. For a long time they were considered merely as inert structural components of the nuclear backbone providing mechanical stability. However, in the past decade lamins have moved into the spot light of nuclear research and new functions ranging from nuclear shape maintenance and chromatin organization to DNA replication, transcription, signal transduction and cell cycle control were revealed.1 What is more, the importance of lamins in higher eukaryotes is increasingly emphasized by the discovery of a number of human diseases linked to alterations in lamin protein structure and function, termed laminopathies.2 Most laminopathy-linked mutations are attributed to LMNA, which represents one of the three mammalian lamin genes and encodes lamin proteins expressed predominantly in differentiated cells, named A-type lamins. The plethora of pathological changes caused by mutations in LMNA arise either in single tissues such as muscle, bone, fat, nerve and skin or multiple organ systems as in the case of Hutchinson-Gilford progeria syndrome. Intriguingly, ∼60% of LMNA mutations lead to defects in striated muscle tissue, such as Emey-Dreifuss Muscular Dystrophy (EDMD), Congenital Muscular Dystrophy and Dilated Cardiomyopathy (DCM).3
The search for laminopathy-linked mutations has been extended to genes encoding lamin-binding proteins and interaction partners, such as emerin and nesprin, whose alterations were shown to cause similar pathological effects in human and mice.3–6 Recently, we have demonstrated yet another link between a lamin-binding protein, called lamina-associated polypeptide 2α (LAP2α) and striated muscle disease.7,8 LAP2α is a nucleoplasmic chromatin-binding protein that arises as one of six splice isoforms of the mammalian LAP2 gene.9,10 While the other major LAP2 isoforms become integrated into the inner nuclear membrane, LAP2α localizes throughout the nuclear interior, because it lacks a transmembrane domain. Like all LAP proteins, LAP2α possesses a N-terminal structural motif called the LAP-Emerin-MAN1 (LEM) domain, which interacts with a conserved DNA-binding protein, Barrier-to-Autointegration Factor (BAF),11 and thus mediates its chromatin association. LAP2α is predominantly expressed in proliferating tissues and growing cultures where it has been proposed to function as a regulator of cell cycle progression and differentiation in association with lamin A/C and the retinoblastoma protein (pRb).9,12–14
A mutation in LAP2α (c. 2068C>T, p. R690C) which lowers its binding affinity to lamin A/C has previously been identified in a family suffering from DCM.15 Our newest studies in LAP2α-deficient mice confirm the link between LAP2α protein function and heart disease.7 What is more, the skeletal muscle defects found in Lap2α−/− mice make us confident that the discovery of a similar mutation-dependent condition in humans is just a matter of time.
By analyzing the Lap2α−/− phenotype in mice, we have discovered multiple and unexpected roles of LAP2α in the homeostasis and stress response of the striated muscle.7,8 However, the exact molecular mechanisms still remain elusive. Here we discuss the possible functions of LAP2α in striated muscle and propose several models of its potential action.
The Lap2α−/− Phenotype
The absence of LAP2α in mice causes sustained left ventricular systolic dysfunction in males present already at the age of 10 weeks, which is followed by an elevated incidence of cardiac fibrosis in old animals (10–12 months).7 At the molecular level, the defect in heart function is accompanied by deregulated expression of major cardiac transcription factors MEF2c and GATA4 and some of their target genes at different ages. In addition, LAP2α-deficient hearts show a weaker response to chronic stress induced by the administration of the β-adrenergic agent isoproterenol. The impairment of the cardiac stress response coincides with the downregulation of β2-adrenergic receptors and is presumably a manifestation of heart pre-sensitization processes due to the persisting systolic dysfunction in the absence of LAP2α.
Our second study revealed an increase of the skeletal muscle stem cell pool in young Lap2α−/− mice, characterized by an excess of CD45−Sca1−Mac1−CXCR4+β1-integrin+ (CSM4B) satellite cells.8 In vitro cultured progeny of Lap2α−/− satellite cells showed delayed differentiation and deregulation of the major MyoD-driven myogenic transcription pathway. Old LAP2α-deficient skeletal muscle presented qualitative changes in muscle fiber composition, with a reduction in the number of slow-twitch oxidative fibers in favor of fast-twitch glycolytic myofibers.
In addition, the absence of LAP2α in mice causes defects in other tissues. Its deficiency in skin induces enhanced proliferation of progenitor cells in the basal layer, leading to hyperplasia of the paw epidermis. Similarly, the haematopoietic system of Lap2α−/− mice exhibits increased proliferation of erythroid progenitors, accompanied with their delayed differentiation. As a result, the regeneration process in the spleen after acute stress proceeds faster.13 Despite the observed changes in multiple tissues, the behavioral profile and physical fitness as well as the life expectancy of LAP2α-deficient mice remain largely unaffected.8
Pathways of LAP2α Action—The Molecular Point of View
The hyperproliferation of erythropoietic and skin progenitors in Lap2α−/− mice was linked to an impairment of pRb protein function, which regulates the expression of E2F-target genes and thereby controls cell cycle progression.13 However, besides its role in cell cycle control, pRb has been found essential for the differentiation of various tissues including skeletal muscle.16,17 There, in addition to promoting cell cycle exit of proliferating myoblasts, the hypophosphorylated form of pRb supports the subsequent progression towards terminal myogenic differentiation by regulating the transcriptional activity of MyoD (Fig. 1).16,18 In proliferating myoblasts MyoD is rendered inactive by association with histone deacetylases. At the onset of differentiation pRb is thought to sequester the inhibitory class I histone deacethylase (HDAC1) away from the protein, thereby allowing the transcription of MyoD-specific target genes.19 Additionally, pRb has been found to promote functional cooperation between MyoD and myocyte enhancer factor 2c (MEF2c) and thus enable the execution of the muscle-specific transcriptional program.20
Figure 1.
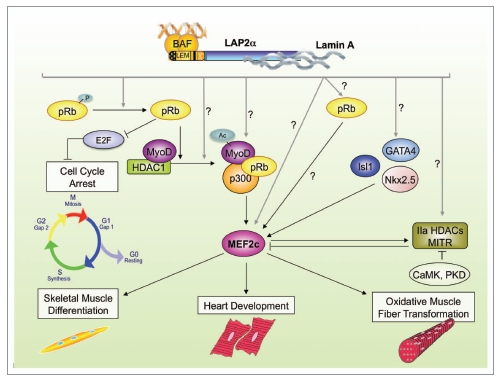
Potential gene regulatory functions of LAP2α in striated muscle. Many of the observed changes in gene expression in LAP2α-deficient striated muscle tissue converge on an impaired activity of MEF2c. This impairment could contribute to the phenotypes of LAP2α-deficient mice, including impaired differentiation of myoblasts, decrease in slow, oxidative fiber types in soleus muscle and defects in heart development and stress response. For details see the text.
A direct interaction between MyoD and pRb has not been proven yet. However, as pRb is a known lamin A/C and LAP2α binding partner21,22 and MyoD co-exists in complexes containing lamin A/C,4 one could envisage lamin A/C-LAP2α complexes as mediators of the interaction between the two proteins. The evidence for this hypothesis comes from studies performed on lamin A/C- and emerin-deficient mice, as well as lamin-linked EDMD patient samples, which all exhibit alterations in pRb and MyoD expression and/or activity.4,5,23,24 Furthermore, the expression of pRb, lamin A/C and LAP2 proteins increases during initial phases of muscle differentiation coinciding with the peak of MyoD activity.4 In line with these data, we found reduced MyoD protein levels and deregulated expression of its target genes during differentiation of Lap2α−/− mouse primary myoblasts. Our observation that MyoD mRNA was even slightly increased in Lap2α−/− myoblasts—likely due to a compensatory response—suggests a potential defect in MyoD protein stability and transcriptional activity in the LAP2α-deficient background. MyoD is a short-lived protein with a half-life of ∼30 min which is degraded by the ubiquitin pathway in the absence of DNA-association.25,26 Since the absence of lamin A/C causes downregulation of MyoD, similar to the loss of LAP2α, and overexpression of MyoD in Lmna−/− myoblasts is sufficient to rescue their impaired in vitro differentiation,23 it is tempting to hypothesize that LAP2α and lamin A/C might serve to protect MyoD from proteasomal degradation, just as they have been shown to stabilize pRb protein.22 In addition, as DNA- and chromatin-binding proteins,10 LAP2α or LAP2α-lamin A/C complexes might recruit or influence the recruitment of chromatin modifying factors to MyoD-target promoters, which have been shown to influence the MyoD-pRb pathway, including HDACs, histone acetyl transferases and histone methyltransferases.27 In line with this hypothesis, artificial tethering of lamin A to promoters was found to be sufficient for induction of transcriptional repression.28
A potential deregulation of the MyoD-pRb pathway might also account for the observed shift in the muscle fiber type content in old Lap2α−/− animals.8 Namely, concomitant with MyoD activity, pRb levels rise during muscle differentiation29,30 and, even though pRb is not required for maintenance of terminal muscle differentiation,18 a change in its activity in the absence of LAP2α might affect fiber type specification driven by differential regulation of MyoD and its targets MEF2C and myogenin. Therefore, it would be interesting to see whether the loss of pRb during later stages of differentiation in Rbfl/fl:Mck-Cre mice18 causes a similar change in muscle fiber type distribution (despite unaltered fiber size) as shown in LAP2α-deficient animals.8 Whether the absence of LAP2α in adult muscle fibers affects pRb activity and if so, whether this has any consequence on the function of MyoD and its target-genes myogenin and MEF2c in coordinating the fast and slow fiber transcription programs remains to be determined. Also, the mechanistic details of LAP2α—pRb interaction are still not fully understood. Our data, showing that pRb levels are unaltered in LAP2α-deficient myoblasts and skeletal muscle tissue (unpublished data), suggest that loss of LAP2α does not affect pRb protein stability as reported in Lmna−/− mice,24 but rather may affect upstream pRb regulating pathways, such as its phosphorylation/dephosphorylation.
Besides the potential involvement of LAP2α in MyoD-dependent processes in skeletal muscle, the functional defect observed in LAP2α-deficient hearts suggests yet another pathway potentially regulated by LAP2α,7 as MyoD is normally not expressed in cardiac muscle.31 pRb expression in the heart transiently decreases between the blastocyst stage and adult age in mice.32 Its absence in embryonic stem cells causes a delay in cardiac differentiation, but pRb seems to be dispensable in differentiated cardiomyocytes just as LAP2α.7,32 The systolic dysfunction in Lap2α−/− mice, which apparently originates during fetal heart development,7 might therefore be a result of an impaired LAP2α—pRb pathway in cardiac progenitor cells.
The Missing Link
One common factor in molecular pathways driving heart development and stress response, skeletal muscle differentiation and oxidative fiber type specification, among others, is MEF2c,33 whose expression was reduced in both LAP2α-deficient skeletal and cardiac muscle.7,8 MEF2c belongs to an evolutionary ancient MADS (MCM1, agamous, deficiens, serum response factor) family of transcription factors that regulate the differentiation of various tissues such as muscle, nerve, bone, fat and skin.33 The expression of MEF2c in the heart is under the control of many signaling pathways such as the Transforming Growth Factor β, Notch and Sonic Hedgehog pathways. It is a direct transcriptional target and a co-factor of major cardiac transcription factors Nkx2.5, GATA4, Isl1 and FOXH1.34,35 The downregulation of MEF2c in the heart tissue of newborn Lap2α−/− mice implicates LAP2α in the regulation of cardiac-specific transcription at the level of MEF2c expression regulation (Fig. 1).7 Normal levels of Nkx2.5, GATA4 and Isl1 mRNA in the heart tissue of Lap2α−/− mice during late embryonic stages and early postnatal life (unpublished data and reviewed in ref. 7), point to a role of LAP2α in the regulation of the activity rather than the stability of these transcription factors, either by direct interaction or through modification and recruitment of co-factors. In addition, as MEF2c has been shown to autoregulate its own promoter during differentiation,36 LAP2α may also directly or indirectly affect MEF2c activity.
One of the direct MEF2c transcriptional targets is the MEF2c-interacting transcription repressor (MITR), a major splice isoform of the class II histone deacetylase HDAC9.37 In a feedback mechanism, class II HDACs are thought to impair the activity of MEF2c by facilitating its sumoylation38 and by recruiting inhibitory class I HDACs to the protein complex.39 The MEF2c-HDAC II pathway plays an important role in transcriptional regulation specific for oxidative muscle types, such as slow skeletal and heart muscle.40 The phosphorylation of HDAC II, as a consequence of calcium-dependent signaling, results in its translocation outside the nucleus and active transcription of MEF2c target genes.40 The observed decrease in the expression of both MEF2c and MITR in the absence of LAP2α might therefore explain the reduction in the number of slow fibers in the soleus muscle of Lap2α−/− mice.8
LAP2α—A Potential HDAC-Mediator?
Most of the aforementioned pathways affected by the loss of LAP2α directly or indirectly rely on the activity of HDACs. In addition, the ablation of various HDACs in mice results in similar muscle phenotypes as in the absence of LAP2α.41 Therefore, it is tempting to speculate about a role of LAP2α in the recruitment/exclusion of HDACs at sites of tissue-specific transcription. Such a function has already been demonstrated for another LAP2 isoform. LAP2β has been shown to specifically bind HDAC3 and thus mediates gene repression.42 Although, the HDAC3-interacting domain of LAP2β resides within the β-specific region of the protein,42 the α-specific tail domain may have different binding sites for HDACs. It would therefore be interesting to test whether loss of LAP2α has any effect on the activity of class I HDACs, the subcellular localization of class II HDACs and/or general acetylation status of chromatin. Intriguingly, faulty chromatin acetylation has been shown in muscle tissue of lamin A/C-deficient mice and attributed to the deregulated activity of MITR,43 again pointing to a close relation of lamin A/C and their interacting proteins to HDAC-mediated gene regulation.
Conclusion
Altogether, the cascade of events triggered by the ablation of Lap2α in mouse striated muscle points to a role of the protein in tissue-specific transcriptional regulation. LAP2α may function as a co-factor directly influencing the activity of tissue-specific transcription factors and/or as a mediator between transcription factors and chromatin remodeling complexes (Fig. 1). Targeting of LAP2α to promoters may involve both specific interaction with transcription factors (conferring binding specificity to specific promoters) and general non-sequence specific binding to DNA via the association of BAF with LAP2α's LEM motif. The details of the role of LAP2α and lamins in transcriptional regulation are just beginning to emerge and we can expect new intriguing mechanistic details to be identified in the coming years.
Acknowledgements
We gratefully acknowledge grant support from the Austrian Science Research Fund (FWF P17871 and P22043) and the EURO-Laminopathies research project of the European Commission (Contract LSHM-CT-2005-018690) to R.F. I.G. was a fellow in the International Ph.D. Program at the Vienna Biocenter supported by grant WK001 from the Austrian Science Research Fund (FWF).
Abbreviations
- BAF
barrier-to-autointegration factor
- CSM4B
CD45−Sca1−Mac1−CXCR4+β1-integrin+
- DCM
Dilated Cardiomyopathy
- EDMD
Emey-Dreifuss Muscular Dystrophy
- HDAC
histone deacethylase
- LAP2α
lamina-associated polypeptide 2α
- LEM
LAP2-emerin-MAN1
- MADS
MCM1, agamous, deficiens, serum response factor
- MEF2c
myocyte enhancer factor 2c
- MITR
MEF2c-interacting transcription repressor
- pRb
retinoblastoma protein
and
Footnotes
Previously published online: www.landesbioscience.com/journals/nucleus/article/12394
References
- 1.Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7:940–952. doi: 10.1038/nrg1906. [DOI] [PubMed] [Google Scholar]
- 3.Worman HJ, Bonne G. “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res. 2007;313:2121–2133. doi: 10.1016/j.yexcr.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakay M, Wang Z, Melcon G, Schiltz L, Xuan J, Zhao P, et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain. 2006;129:996–1013. doi: 10.1093/brain/awl023. [DOI] [PubMed] [Google Scholar]
- 5.Melcon G, Kozlov S, Cutler DA, Sullivan T, Hernandez L, Zhao P, et al. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet. 2006;15:637–651. doi: 10.1093/hmg/ddi479. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16:2816–2833. doi: 10.1093/hmg/ddm238. [DOI] [PubMed] [Google Scholar]
- 7.Gotic I, Leschnik M, Kolm U, Markovic M, Haubner BJ, Biadasiewicz K, et al. Lamina-associated polypeptide 2alpha loss impairs heart function and stress response in mice. Circ Res. 106:346–353. doi: 10.1161/CIRCRESAHA.109.205724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gotic I, Schmidt WM, Biadasiewicz K, Leschnik M, Spilka R, Braun J, et al. Loss of LAP2alpha delays satellite cell differentiation and affects postnatal fiber-type determination. Stem Cells. 28:480–488. doi: 10.1002/stem.292. [DOI] [PubMed] [Google Scholar]
- 9.Berger R, Theodor L, Shoham J, Gokkel E, Brok-Simoni F, Avraham KB, et al. The characterization and localization of the mouse thymopoietin/lamina-associated polypeptide 2 gene and its alternatively spliced products. Genome Res. 1996;6:361–370. doi: 10.1101/gr.6.5.361. [DOI] [PubMed] [Google Scholar]
- 10.Vlcek S, Korbei B, Foisner R. Distinct functions of the unique C terminus of LAP2alpha in cell proliferation and nuclear assembly. J Biol Chem. 2002;277:18898–18907. doi: 10.1074/jbc.M200048200. [DOI] [PubMed] [Google Scholar]
- 11.Margalit A, Brachner A, Gotzmann J, Foisner R, Gruenbaum Y. Barrier-to-autointegration factor—a BAFfling little protein. Trends Cell Biol. 2007;17:202–208. doi: 10.1016/j.tcb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Ishijima Y, Toda T, Matsushita H, Yoshida M, Kimura N. Expression of thymopoietin beta/lamina-associated polypeptide 2 (TPbeta/LAP2) and its family proteins as revealed by specific antibody induced against recombinant human thymopoietin. Biochem Biophys Res Commun. 1996;226:431–438. doi: 10.1006/bbrc.1996.1373. [DOI] [PubMed] [Google Scholar]
- 13.Naetar NKB, Kozlov S, Kerenyi MA, Dorner D, Kral I, Gotic I, et al. Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat Cell Biol. 2008;10:1341–1348. doi: 10.1038/ncb1793. [DOI] [PubMed] [Google Scholar]
- 14.Parise P, Finocchiaro G, Masciadri B, Quarto M, Francois S, Mancuso F, et al. Lap2alpha expression is controlled by E2F and deregulated in various human tumors. Cell Cycle. 2006;5:1331–1341. doi: 10.4161/cc.5.12.2833. [DOI] [PubMed] [Google Scholar]
- 15.Taylor MR, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR, et al. Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat. 2005;26:566–574. doi: 10.1002/humu.20250. [DOI] [PubMed] [Google Scholar]
- 16.De Falco G, Comes F, Simone C. pRb: master of differentiation. Coupling irreversible cell cycle withdrawal with induction of muscle-specific transcription. Oncogene. 2006;25:5244–5249. doi: 10.1038/sj.onc.1209623. [DOI] [PubMed] [Google Scholar]
- 17.Vidal A, Carneiro C, Zalvide JB. Of mice without pockets: mouse models to study the function of Rb family proteins. Front Biosci. 2007;12:4483–4496. doi: 10.2741/2403. [DOI] [PubMed] [Google Scholar]
- 18.Huh MS, Parker MH, Scime A, Parks R, Rudnicki MA. Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J Cell Biol. 2004;166:865–876. doi: 10.1083/jcb.200403004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puri PL, Iezzi S, Stiegler P, Chen TT, Schiltz RL, Muscat GE, et al. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol Cell. 2001;8:885–897. doi: 10.1016/s1097-2765(01)00373-2. [DOI] [PubMed] [Google Scholar]
- 20.Novitch BG, Spicer DB, Kim PS, Cheung WL, Lassar AB. pRb is required for MEF2-dependent gene expression as well as cell cycle arrest during skeletal muscle differentiation. Curr Biol. 1999;9:449–459. doi: 10.1016/s0960-9822(99)80210-3. [DOI] [PubMed] [Google Scholar]
- 21.Dorner D, Gotzmann J, Foisner R. Nucleoplasmic lamins and their interaction partners, LAP2alpha, Rb and BAF, in transcriptional regulation. Febs J. 2007;274:1362–1373. doi: 10.1111/j.1742-4658.2007.05695.x. [DOI] [PubMed] [Google Scholar]
- 22.Markiewicz E, Dechat T, Foisner R, Quinlan RA, Hutchison CJ. Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol Biol Cell. 2002;13:4401–4413. doi: 10.1091/mbc.E02-07-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frock RL, Kudlow BA, Evans AM, Jameson SA, Hauschka SD, Kennedy BK. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006;20:486–500. doi: 10.1101/gad.1364906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, et al. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci USA. 2004;101:9677–9682. doi: 10.1073/pnas.0403250101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abu Hatoum O, Gross-Mesilaty S, Breitschopf K, Hoffman A, Gonen H, Ciechanover A, et al. Degradation of myogenic transcription factor MyoD by the ubiquitin pathway in vivo and in vitro: regulation by specific DNA binding. Mol Cell Biol. 1998;18:5670–5677. doi: 10.1128/mcb.18.10.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thayer MJ, Tapscott SJ, Davis RL, Wright WE, Lassar AB, Weintraub H. Positive autoregulation of the myogenic determination gene MyoD1. Cell. 1989;58:241–248. doi: 10.1016/0092-8674(89)90838-6. [DOI] [PubMed] [Google Scholar]
- 27.Sartorelli V, Caretti G. Mechanisms underlying the transcriptional regulation of skeletal myogenesis. Curr Opin Genet Dev. 2005;15:528–535. doi: 10.1016/j.gde.2005.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee DC, Welton KL, Smith ED, Kennedy BK. A-type nuclear lamins act as transcriptional repressors when targeted to promoters. Exp Cell Res. 2009;315:996–1007. doi: 10.1016/j.yexcr.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coppola JA, Lewis BA, Cole MD. Increased retinoblastoma gene expression is associated with late stages of differentiation in many different cell types. Oncogene. 1990;5:1731–1733. [PubMed] [Google Scholar]
- 30.Martelli F, Cenciarelli C, Santarelli G, Polikar B, Felsani A, Caruso M. MyoD induces retinoblastoma gene expression during myogenic differentiation. Oncogene. 1994;9:3579–90. [PubMed] [Google Scholar]
- 31.Miner JH, Miller JB, Wold BJ. Skeletal muscle phenotypes initiated by ectopic MyoD in transgenic mouse heart. Development. 1992;114:853–860. doi: 10.1242/dev.114.4.853. [DOI] [PubMed] [Google Scholar]
- 32.MacLellan WR, Garcia A, Oh H, Frenkel P, Jordan MC, Roos KP, et al. Overlapping roles of pocket proteins in the myocardium are unmasked by germ line deletion of p130 plus heart-specific deletion of Rb. Mol Cell Biol. 2005;25:2486–2497. doi: 10.1128/MCB.25.6.2486-2497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 34.Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–1407. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava D. Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–1048. doi: 10.1016/j.cell.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Wang DZ, Valdez MR, McAnally J, Richardson J, Olson EN. The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development. 2001;128:4623–4633. doi: 10.1242/dev.128.22.4623. [DOI] [PubMed] [Google Scholar]
- 37.Haberland M, Arnold MA, McAnally J, Phan D, Kim Y, Olson EN. Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol Cell Biol. 2007;27:518–525. doi: 10.1128/MCB.01415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gregoire S, Yang XJ. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol Cell Biol. 2005;25:2273–2287. doi: 10.1128/MCB.25.6.2273-2287.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sparrow DB, Miska EA, Langley E, Reynaud-Deonauth S, Kotecha S, Towers N, et al. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999;18:5085–5098. doi: 10.1093/emboj/18.18.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37. doi: 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- 41.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Somech R, Shaklai S, Geller O, Amariglio N, Simon AJ, Rechavi G, et al. The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery and induces histone H4 deacetylation. J Cell Sci. 2005;118:4017–4025. doi: 10.1242/jcs.02521. [DOI] [PubMed] [Google Scholar]
- 43.Mejat A, Decostre V, Li J, Renou L, Kesari A, Hantai D, et al. Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy. J Cell Biol. 2009;184:31–44. doi: 10.1083/jcb.200811035. [DOI] [PMC free article] [PubMed] [Google Scholar]