

Abstract
Voltage sensors (VS) domains couple the activation of ion channels/enzymes to changes in membrane voltage. We used molecular dynamics simulations to examine interactions with lipids of several VS homologs. VSs in intact channels in the activated state are exposed to phospholipids, leading to a characteristic local distortion of the lipid bilayer which decreases its thickness by ∼10 Å. This effect is mediated by a conserved hydrophilic stretch in the S4–S5 segment linking the VS and the pore domains, and may favor gating charges crossing the membrane. In cationic lipid bilayers lacking phosphate groups, VSs form fewer contacts with lipid headgroups. The S3–S4 paddle motifs show persistent interactions of individual lipid molecules, influenced by the hairpin loop. In conclusion, our results suggest common interactions with phospholipids for various VS homologs, providing insights into the molecular basis of their stabilization in the membrane and how they are altered by lipid modification.
Introduction
Voltage-gated ion channels underlie various physiological processes, most notably the generation and propagation of action potentials (1). Having a basic topology of six transmembrane (TM) helices (S1–S6), they form a diverse superfamily which include voltage-activated potassium (Kv), sodium and calcium channels, in addition to more distant homologs such as hyperpolarization-activated cyclic-nucleotide-gated, cyclic nucleotide-gated, and transient receptor potential channels (2). They are either tetrameric or, in the case of sodium and calcium channels, monomers of four similar repeating subunits. Each subunit is equipped with a voltage sensor (VS) domain (S1–S4) which switches from a resting to an activated state in response to membrane depolarization, leading to the opening of a central pore domain (S5–S6) (Fig. 1 A). Recently VS domains have been found to regulate enzymes such as Ci-VSP phosphatase from Ciona intestinalis (3) or even to function as independent voltage-gated H+ channels such as Hv1 from human (4–6).
Figure 1.
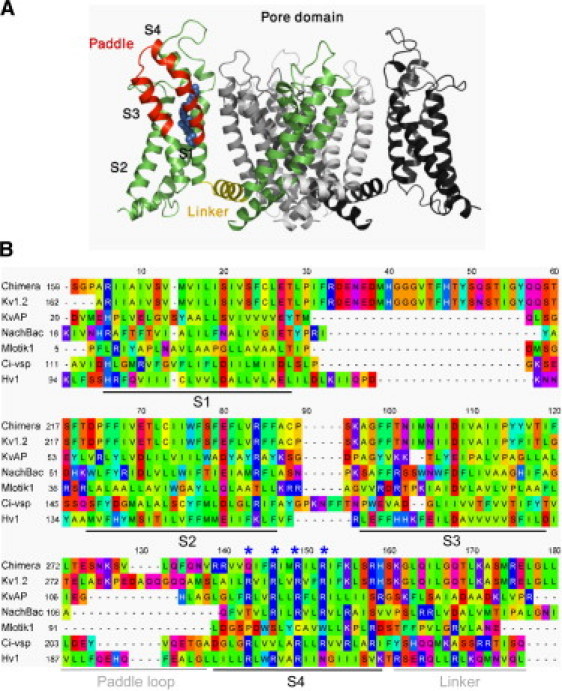
Voltage sensor (VS) domains share a common fold despite having highly divergent sequences. (A) Crystal structure of Kv paddle chimera (20) illustrating the architecture of the VS domain. One of the four subunits is colored, highlighting the paddle motif (red), the conserved positively charged residues (blue spheres), and the segment linking the VS to the pore (yellow). The other subunits are also shown (white, gray, and black). For clarity, the VS domain projecting toward the viewer is omitted. (B) Alignment of the VS domains homologs studied indicating the common topology. Residues are colored on the Taylor hydrophobicity scheme (91). Positions corresponding to R1, R2, R3, and R4 in Kv1.2 are highlighted (asterisks). Percentage sequence identity relative to Kv1.2 (Rattus norvegicus) are: Kv paddle chimera (Rattus norvegicus) 84.3%; KvAP (Aeropyrum pernix) 23.6%; Mlotik1 (Mesorhizobium loti) 12.6%; NachBac (Bacillus halodurans) 17.7%; Ci-VSP (Ciona intestinalis) 15.3%; and Hv1 (Homo sapiens) 19.9%.
The S4 helix of the VS domain contains several Arg or Lys residues which during the gating process move toward the external membrane surface carrying positive charges across the membrane electric field, making sequential interactions with acidic side chains at a central site capped by a highly conserved phenylalanine (Phe233 in Kv1.2) side chain on S2 (7–15). Each adjacent pair of these basic residues is separated by two hydrophobic residues, which seems to be crucial for shaping the electrical field around the VS domains (16).
Despite their high sequence diversity, VS domains share a common fold as revealed by the x-ray structures of several Kv and related channels, namely KvAP from Aeropyrum pernix (17), rat Kv1.2 (18,19), a chimeric channel consisting of the Kv2.1 paddle hairpin transplanted into Kv2.1 (Kv paddle chimera) (20), and a cyclic-nucleotide-gated channel from Mesorhizobium loti, Mlotik1 (21). That is also supported by spin-labeling and electron paramagnetic resonance data from the bacterial voltage-gated Na+ channel NachBac (22).
VS domains have been perceived as forming independent modules (17) because, for example, transplanting VS domains from Kv channels to KcsA confers voltage-sensitivity (23). However, only the central part of S3–S4 paddle motif can be transplanted among some Kv channels, Nav channels, and the VS proteins Ci-VSP and Hv1 (24,25), whereas other regions of the VS domain including the C-terminal part of the S4 helix cannot (24).
The membrane environment plays a key role in VS structure and function (26–30). For example, electron paramagnetic resonance studies of KvAP revealed exposure of the VS domains to the surrounding lipids (31). In addition, the Kv paddle chimera x-ray structure revealed several bound lipid molecules: at the interface between the VS and the pore domains, and between adjacent VS domains (20). Phospholipids such as DOPC (1,2-dioleoyl-glycero-3-phosphocholine) are necessary for KvAP function and can rescue nonfunctional channels which have been reconstituted into lipid bilayers lacking phosphate headgroups such as DOTAP (1,2-dioleoy-3-trimethylammonium propane) (26). Consistently, sphingomyelinase D (which removes positively-charged choline groups from the zwitterionic lipid sphingomyelin) leads to a stabilization of the activated VS domains of Kv2.1 (27), appearing to strengthen the interaction between exposed S4 Arg residues and lipid phosphate groups. In contrast, sphingomyelinase C (which removes the entire headgroup of the sphingolipid) immobilizes the VS in the resting state (28). Interestingly, in addition to influencing the VS function, lipid modifications also alter sensitivity to tarantula toxins which specifically target the paddle domain (32). However, lipid-modifying enzymes have been observed to affect certain types of Kv channels only, and sphingomyelin lipase C was also found to inhibit other non-VS channels such as KcsA and Kir1.1. Therefore, not all phospholipid interactions may be specific.
The polar nature of VS domains have resulted in some debate on how they are stabilized in a lipid bilayer environment. While spectroscopic and functional studies suggest that the electric field is focused across the VS domains (33,34), it is not entirely clear whether this focusing is caused by a perturbation of the structure of the surrounding lipids or by the structure/chemistry of the protein. Neutron diffraction and solid-state NMR studies showed that KvAP VS domains cause some local reshaping of the surrounding bilayer while being largely hydrated (35).
Molecular dynamics (MD) simulations of VS domains of Kv channels, either as individual domains or as part of the intact channel, revealed how they might be stabilized in the membrane bilayer, showing close interactions between the Arg residues in the S4 helix and phosphate headgroups (36–43).
In this study, we use coarse-grained (CG) MD simulations (44–53) to study the nature of these VS-lipid interactions and the extent into which they are conserved among various distantly-related homologs of voltage-gated ion channels and other VS proteins. We compare how the interactions differ between conventional phospholipid bilayers (DOPC) and bilayers formed by cationic lipids (DOTAP). Our results point to a common mechanism by which phospholipid membranes adapt to VS domains by substantial distortion of the lower (intracellular) bilayer leaflet, reducing the bilayer thickness and ultimately lowering the barrier for gating charge movements.
Methods
Homology models
Homology models for the activated Hv1 VS monomer (UniProt Q96D96), Ci-VSP VS monomer (Q4W8A1) and NachBac channel tetramer (Q9KCR8) were built based on the x-ray structure of the KvAP VS domain (PDB ID 1ORS) (17), Kv paddle chimera VS domain (20), and Kv paddle chimera tetramer, respectively. Hv1 and Ci-VSP models also contained the S4–S5 linker segments. Homologous sequences were obtained for each of the target sequences and structures from UniRef100 (54) using noniterative BLAST (e value < 10). Each protein was aligned against its homologous sequences using MAFFT (55) based on the BLOSUM62 substitution matrix (56). Next, structural profiles (i.e., position-specific substitution matrices) were calculated for the structures and sequence profiles were calculated for the target sequences. The structural profiles were then aligned against the sequence profiles using FUGUE (57). The resulting structure-sequence alignments were manually adjusted to ensure conservation of key residues, then used as input for MODELER (58) generating 10 models per alignment. The best models was selected based on the energy and constraint violation values of MODELER and the sequence-structure compatibility scores of pG (59), PROSA2003 (60), and VERIFY3D (61). Any unreliable regions in the models were improved by altering the alignments manually using ViTO (62).
Coarse-grain molecular dynamics simulations
CG parameters are as described previously for proteins (48,49), lipids, and water (47). For Kv paddle chimera, Kv1.2, KvAP, Mlotik1, and KcsA the CG models were generated starting from the atomistic x-ray structures, having, respectively, the following PDB IDs: 2R9R (resolution 2.4 Å), 2A79 (2.9 Å), 1ORS (1.9 Å), 3BEH (3.1 Å), and 1K4C (2.0 Å). Missing side chains in Kv1.2 were modeled based on the Kv paddle chimera using MODELER (58). CG models for Hv1, Ci-VSP, and NachBac were generated from homology models as described above. In the CG representation adopted here and previously tested (47–49,63), the protein main chain was represented by a single particle whereas various types of amino-acid side chains were represented by 0–4 particles. Aromatic side chains of Phe, Trp, and Tyr were represented by planar rings of three particles (49). To maintain structure, an elastic network model was applied in which a harmonic restraint is between any two backbone particles which are within 7 Å, with force constant of 10 kJ mol−1 Å−2. Unless stated otherwise, water molecules, detergents, and other cofactors from the original PDB structures were not considered in the CG representations.
GROMACS was used for all simulations (64,65). Lenard-Jones and electrostatic interactions were shifted to 0 between 9 and 12 Å and between 0 and 12 Å, respectively. A relative dielectric constant of 20 was used. The list of nonbonded neighbors was updated every 10 steps. Simulations were run in constant temperature, pressure, and number of particles. The temperature of protein, lipids, and solvent was coupled separately at 323 K using the Berendsen algorithm (66), with a coupling constant τT = 40 ps. The system pressure was anisotropically coupled at 1 bar using the same algorithm with a coupling constant τP = 40 ps and compressibility of 10−5 bar.
In setting up the simulations, the protein was aligned along the z axis of the simulation box, adding randomly placed lipids and water molecules. Cl− or Na+ countercharge ions were then added if necessary to ensure there was no net charge for the overall system (see Table S1 in the Supporting Material). Next, the protein and lipids were energy-minimized to relax any steric clashes, using the steepest descent method up to a maximum of 100 steps. Production simulations (three repeats, each for 320 ns) were run. Molecular structure images were produced using PyMOL (DeLano Scientific, Palo Alto, CA).
Bilayer distortion analysis
For the analysis of bilayer distortion, the bilayer was centered on the protein center of mass and divided into 5 × 5 Å bins. For each bin, the distance between the phosphate atoms from the inner and outer leaflets (dPP) was calculated, in addition to the distance of phosphates from either leaflet relative to the central plane of the membrane (dZ). The physiological ion/out orientation was preserved for each protein. The calculations were made over the last 220 ns of each simulation and the mean values were taken together with 95% confidence intervals (1.96 × mean ± SE).
Results
Models and simulations
We examined a representative set of structures which comprise:
-
1.
X-ray structures of Kv1.2 (18), the Kv paddle chimera (20), the cyclic-nucleotide gated 6-TM channel Mlotik1 (21), and the VS domain from KvAP (17); and
-
2.
Homology models of the bacterial voltage-gated Na+ channel NachBac (67), of the VS domains of the voltage-sensitive phosphatase Ci-VSP (3) and of the voltage-activated proton channel Hv1 (4).
Apart from the high sequence identity between Kv1.2 and the Kv paddle chimera (84.3%), these VS homologs share relatively low similarity, ∼20% on average (Fig. 1 B). Therefore, they form a suitable set for studying protein-lipid interactions in the context of divergence among VS sequences. The non-voltage-gated channel KcsA (68) was included as a control.
Starting from randomly positioned protein, lipids (DOPC), waters, and ions, we simulated the self-assembly of the membrane bilayer and the simultaneous insertion of the protein, which occurs typically within 30–40 ns of the start of the simulation (Fig. 2). As well as the intact native tetramers for Kv paddle chimera, Kv1.2, NachBac, and MlotiK1, individual VS domains from these channels were also simulated alone, to allow direct comparison to the VS proteins Ci-VSP and Hv1 (Table S1).
Figure 2.
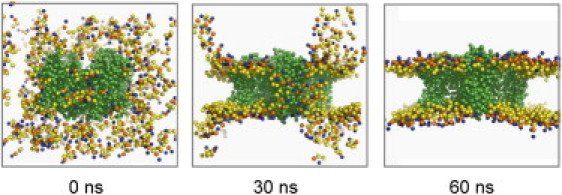
CG self-assembly MD simulations reveal protein-lipid interactions for various VS homologs. Snapshots of the self-assembly of a DOPC bilayer along with the associated insertion of the Kv paddle chimera channel. Lipid phosphate groups (orange), choline (blue), and glycerol (yellow). The lipid tails and water particles are omitted for clarity, and the protein is shown (green). Starting from protein, lipids, water, and ions placed in the simulation box (0 ns), the membrane bilayer starts to self-assembly around the protein (30 ns) and the process is typically complete in <60 ns.
Lipid bilayer distortion is a characteristic of voltage-gated channels
Structures of the VS domains examined here are considered to be in the channel-activated conformation because the x-ray structures were solved in the absence of an electrical field (i.e., equivalent to a depolarized membrane) (17,20). To see how the various activated VS domains are accommodated in phospholipid (DOPC) bilayer, we examined local distortions of the membrane surrounding the protein by measuring the distance of the phosphate groups in either the upper or lower leaflets relative to a central plane of the membrane and the distance between phosphate groups in the two leaflets (Figs. 3 and 4).
Figure 3.
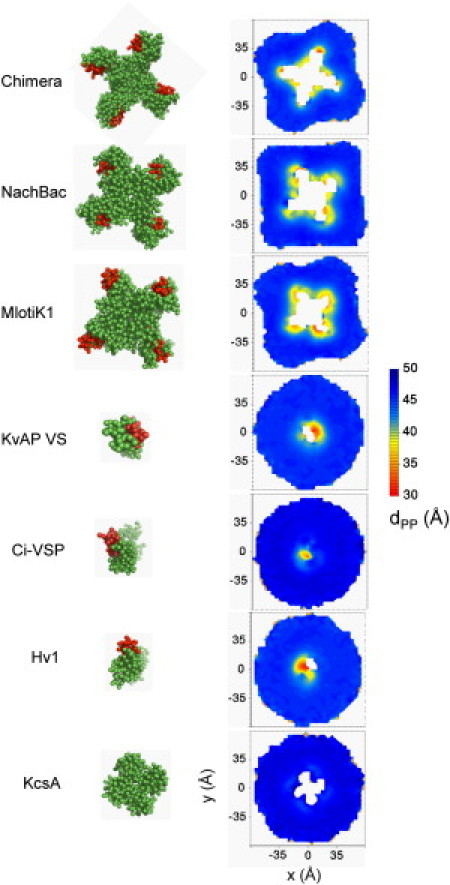
VS domains cause a major local distortion of the phospholipid bilayer. (Left) CG structures are shown with the S4 helix (red). (Right) Also viewed down the bilayer normal, contour plots show the phosphate-phosphate distance between the two membrane leaflets (dPP) as a function of position in the bilayer (xy) plane, averaged over the last 220 ns of the MD simulations. KcsA, a potassium channel lacking a voltage sensor domain, is shown for comparison. See also Fig. S1.
Figure 4.
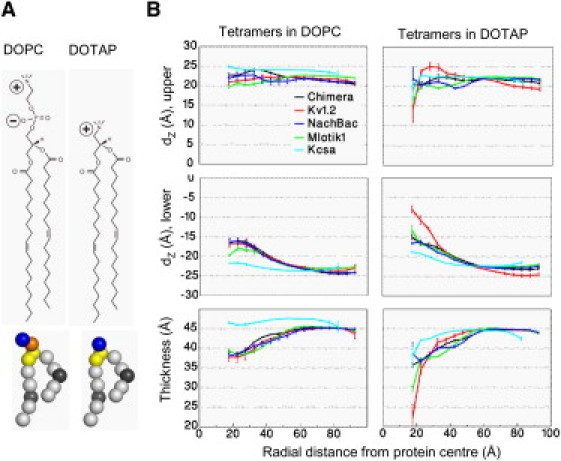
VS domains cause greater distortion to the lipid bilayer in the absence of lipid phosphate groups, as seen by comparing DOPC and DOTAP. (A) Covalent structures of the lipids DOPC and DOTAP lipids and their corresponding CG models (orange, phosphate; blue, choline; yellow, glycerol; gray, carbon; black, carbon particle with a double bond). (B) The average positions (dZ) of phosphate groups in the upper leaflet (top), the lower leaflet (middle) (both relative to the bilayer center at z = 0), and bilayer thickness (bottom), each as a function of the radial distance from the protein center of mass. The bilayer thickness is calculated as the phosphate-phosphate (dPP) and choline-choline (dCC) distance for DOPC and DOTAP, respectively. (Error bars) 95% confidence intervals. See also Fig. S2.
The tetrameric channels Kv paddle chimera, Kv1.2, NachBac, and Mlotik1 all show a common pattern of large (∼10 Å) deformation of the inner membrane leaflet close to the protein. This is attributed to a common cluster of polar and charged residues in the S4–S5 linker, connecting the VS domains to the pore region, which form close contacts with lipid headgroups from the inner leaflet pulling them upwards. In addition, there are prominent sites of deformations of the outer leaflets near the outer parts of the VS domains (Fig. S1). A lesser degree (∼3 Å) of membrane distortion was also observed for the VS monomers of Ci-VSP and Hv1 and the isolated VS domains from the channel tetramers (all of which included the S4–S5 linker; Fig. S2). Here, the unrestricted S4–S5 linker has the freedom to adjust itself in the membrane-lipid interface/solvent, allowing the VS domains to adapt more favorably to the membrane. On the other hand, the non-voltage-gated channel KcsA does not locally distort the bilayer. This suggests that membrane distortion is a distinctive property of (activated) VS domains, imposing specific pattern of interactions with the surrounding lipids. This may facilitate the movement of voltage-sensing charges across the membrane.
Environment and modularity of VS domains
To measure the extent to which each VS homolog is accessible to the surrounding lipids during the simulations, we calculated the average number of contacts made between individual amino acids and lipid head- or tail groups sampled over the last 220 ns of the simulations. By mapping these values on each structure, the various homologs can be seen to have a common spatial distribution of residues spanning the membrane interface and hydrocarbon regions (Fig. 5 A). The contacts made by VS domains to either the lipid headgroups or tails are broadly similar among the homologs, although slightly fewer interactions can be seen for NachBac and Mlotik1 (12% and 15% less than Kv paddle chimera, respectively) (Fig. 5 B). Compared to the VS domains, the pore domains in the tetrameric channels are less accessible to lipid headgroups yet make similar levels of contact with lipid tails (Fig. S3 A).
Figure 5.
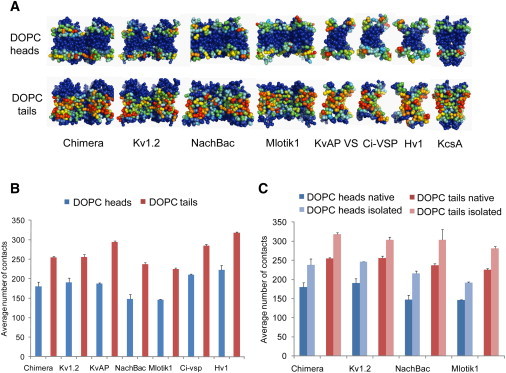
Patterns of protein lipid contacts are conserved among VS homologs. (A) Average number of contacts over the MD simulations between amino-acid side chains and DOPC head/tail groups mapped onto the channel and VS CG structures. Densities range from low contact number (blue) to high contact number (red). A cutoff distance of 6 Å was used to score a contact (as is standard for CG models). (B) Average number of contacts between the VS domains of various homologs and DOPC heads or tail groups. (C) Average number of contacts between the VS domains and DOPC heads or tail groups, comparing the VS domain simulated in isolation versus being part of the intact native channel tetramer. See also Fig. S3B.
To examine how loosely the VS domains are packed against the pore regions in the channels, we compared VS domains simulated in their intact native tetramers and on their own in lipid bilayers (Fig. 5 C). In all of the intact channels, the VS domains are largely exposed to the lipid bilayer, making ∼80% of the total lipid interactions they make in isolation. This also indicates that similar areas of contact exist in various tetrameric channels between the VS domains and the pore regions, which ensure functional coupling between voltage sensing and ion conduction. The S3–S4 paddle regions are a little less exposed to the lipid bilayer in the tetramers relative to the isolated VS domains, suggesting that they form part of this functional interface (Fig. S3 B).
Interaction with lipid phosphate groups
To understand how the absence of phosphate groups affects the channels, we performed simulations similar to those in DOPC (above) but instead using the cationic lipid DOTAP, which lacks a phosphate group (Fig. 4 A). DOTAP has been shown to form stable bilayers, to form mixed PC/DOTAP bilayers (69), and to lead to loss of function of KvAP (26). Thus, the absence of the anionic phosphate group in DOTAP was anticipated to alter VS/bilayer interactions.
In all channel tetramers, there is a clear increase in the degree of local bilayer distortion of both the inner and outer leaflets in DOTAP relative to DOPC, drastically reducing the local thickness of the bilayer adjacent to the protein by up ∼20 Å (Fig. 4 B). Therefore, activated channels are still accommodated in such cationic bilayers but with extensive rearrangement of the surrounding lipids that is likely to destabilize the VS domain in the active state where the gating charges need to have access to the outer membrane leaflet interface. This agrees with KvAP being nonfunctional when in DOTAP bilayers (26), and with recent simulation studies (70) indicating that the free energy of transfer of an arginine side chain from bulk water to the interface of a DOTAP membrane is +4 kcal/mol compared to −3 kcal/mol for a DOPC membrane.
VS domains interact less with membrane in absence of phosphate groups while pore domains are unaffected
To identify which parts of the channels are most affected by the absence of the phosphate groups in the membrane, we plot the average number of contacts made between the VS or pore domains and either DOPC or DOTAP (Fig. 6 A, Fig. S3 A). The VS domains show a systematic decrease in interaction with the lipid headgroups in DOTAP compared to DOPC (22% lower) while interactions with lipid tail groups are unaltered (3% lower). This effect is uniform over the four VS domains in each channel (Fig. S4 B). On the other hand, lipid interactions for the pore domains are shown to be similar between DOPC and DOTAP (Fig. S3 A).
Figure 6.
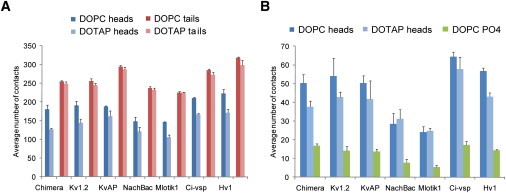
VS domains interact less strongly with lipid heads in the absence of phosphate groups. (A) Average number of contacts between VS domains and lipid heads or tails in MD simulations in which the VS is part of the intact native tetramer and either in a DOPC or DOTAP bilayer. (B) Average number of contacts between the paddle motif (S3–S4) and the lipid headgroups in simulations of the intact native channel tetramers in either DOPC (dark blue) or DOTAP (light blue). Contacts made to the phosphate groups of DOPC (plotted in green). (Error bars) 95% confidence intervals. See also Fig. S3A.
Loop affects lipid-head interactions of the S3-S4 paddle motif
The S3–S4 paddle motif of the VS domain has been suggested to undergo substantial movement during VS activation (71). Its transferability between Kv, Nav channels and VS proteins (24,25) and its targeting by tarantula toxins known to partition into the membrane (72) suggests it moves largely within a lipid environment (24). When the paddle motifs from the various homologs are compared, they show a similar decrease in the number of contacts to lipid headgroups between DOPC and DOTAP except for NachBac and Mlotik1 (Fig. 6 B), both of which are lower than for the other VS domains regardless of lipid species. It can be noted the loop regions connecting S3 to S4 in these two homologs are shorter and contain a smaller number of charged/polar residues compared to the other VS homologs (Fig. 1 B). Therefore, the loop is likely to be an important component of the paddle motif and may affect the way it moves in the membrane.
Where do lipids bind?
Lipid interactions with specific residues of the paddle region (in S3 facing the membrane and on S4 facing S5) have been inferred based on sphingomyelinase D perturbation of sensitivity to tarantula toxins (32). To examine which residues of the VS sequence interact with phospholipids, we calculated the average number of interactions made for each residue with either lipid head- or tail groups (Fig. S5). The VS domains in all homologs show common trends in which the cores of TM segments interact extensively with lipid tails whereas the ends of helices and loops interact with lipid headgroups. The S3–S4 paddle in particular makes comparable interactions to other segments, but binds persistently to individual lipids (binds to at least one lipid for >50 ns). For the pore regions, the S5 helices seem to be as much exposed to the lipid tail groups as other helices in the VS whereas only the N-terminal half of the S6 helix seem to be exposed. For the VS proteins Ci-VSP and Hv1, similar patterns are observed as for the channel tetramers although the number of contacts is generally higher, presumably reflecting the difference in the oligomerization states.
Discussion
We used extended CG self-assembly MD simulations to study protein-lipid interactions in a number of homologs of voltage-gated ion channels. The activated-state structures of all of the tetrameric channels cause a characteristic distortion of the DOPC phospholipid bilayer, leading to substantial thinning of the membrane (Fig. 4). Although the exposed positive side chains on S4 helices are involved, most of this distortion is caused by a common cluster of hydrophilic side chains in the S4–S5 segments. These segments, which mechanistically link the VS and the pore domains, and are thought to move relatively toward the external side upon channel activation (71), may have the extra role of reshaping the local membrane environment to favor gating charge movement across the membrane. These segments are also present in VS proteins which lack conventional pore domains, suggesting they could play an equivalent role.
It is useful to compare our results with a recent study of the biophysical properties of an isolated VS domain in a lipid bilayer (35). In the latter study, neutron scattering was used to provide an average profile across a membrane into which the KvAP VS domain had been reconstituted. The presence of the VS domain resulted in a decrease in the bilayer thickness of Δd = −3 Å at a high protein/lipid ratio (P/L = 1:100). This may be compared to the local decrease in bilayer thickness close to the protein in a number of simulation studies. Thus, for the KvAP VS domain, coarse-grained simulations (73) with P/L = 1:250 yielded Δd = −3 Å at a distance of 10 Å from the center of the protein domain, whereas all-atom simulations (39) with P/L = 1:125 yielded Δd = −4 Å at the same distance from the center of the domain (albeit with poorer statistics due to shorter simulations). Comparable more-recent all-atom simulations (35) gave Δd = −5 Å for lipids in the first and second solvation shell of the protein. Thus, experiment and simulations (both CG and all-atom) are in good agreement for the isolated VS domain. In this study, at a P/L 1:350, Δd = −7 Å at 15 Å from the center of the protein. This is for an intact Kv channel, not just the isolated VS domain and as discussed below, this results in a somewhat more pronounced local distortion.
The smaller level of membrane distortion observed around isolated VS domains (∼3 Å, Fig. S2) is remarkably consistent with the neutron diffraction and solid-state NMR experiments performed on isolated KvAP VS domains (35). Our results show that a greater level of distortion is caused by the native quaternary structures, which is both an accumulative effect from multiple VS domains and importantly due to the conformational restrictions on the S4–S5 linker. The view that the conformation of active state VS causes effective membrane thickness to decrease reconciles between the ranges of motion of S4 during activation inferred from accessibility and toxin-binding studies suggesting a movement of ∼15 Å perpendicular to the membrane plane (14,74), and fluorescence distance measurements pointing to somewhat smaller motions (up to ∼5 Å (75–77)).
VS domains from various homologs make similar number of contacts overall with phospholipid head- or tail groups (Fig. 5, A and B). The VS of the channels are effectively symmetrical, as expected from the homotetrameric structure (Fig. S4 A). They are all substantially exposed to the membrane, which supports experimental data (31), and uniformly show that 20% of the total interface is hidden from the membrane (Fig. 5 C). Therefore, although VS domains are largely embedded in lipids, they form a persistent interface with the pore domains, which might explain the limited portability of full-length VS domains, and highlights the potential use of this specialized interface as a target for small molecules.
In a DOTAP bilayer which lacks phosphate groups, VS homologs are accommodated but with a considerably higher distortion of the membrane. When comparing interaction with DOPC versus DOTAP, pore domains contacts are shown to be largely unaffected, whereas the VS domains make significantly fewer contacts with DOTAP headgroups but show similar interaction to lipid tail groups (Fig. 6 A). This distorted lipid interaction is likely to destabilize of activated VS, because the enzymatic removal of phosphate headgroups from sphingomyelin bilayer containing Kv channels abolishes VS function. Our observation that the central pore domains are highly accessible to the lipid tails suggests that they might be more sensitive to the modification of the lipid hydrocarbon chain rather than the headgroup.
The paddle motifs of the VS homologs show various levels of contacts with DOPC headgroups, especially in their interaction with phosphate groups. This is associated with differences in the composition and length of the loop regions between S3 and S4. In particular, NachBac and Mlotik1, which have short loops, make relatively fewer contacts with phosphate groups and do not show a significant difference between DOPC and DOTAP (Fig. 6 B). Based on this observation, these two channels may well be unaffected by the absence of phosphate groups. By interacting with lipid headgroups, the S3–S4 loop could form a conformational restraint on paddle movement during VS activation. Therefore, the nature of the paddle loop and presumably, the number of positive charges on S4, might ultimately determine the level of sensitivity of the VS function to the removal of phospholipid headgroups from the membrane. In the case of a smaller number of charges needing to be exposed during activation, water and/or polar groups of the lipid head molecules would be sufficient to stabilize them. This would explain why not all voltage-gated channels are sensitive to sphingomyelinase C.
Mlotik1 is unique in that it lacks most of the positive residues on S4 found in other Kv channels, which has led to the suggestion it belongs to the non-voltage-gated family of cyclic-nucleotide-gated channels (21). However, based on its high K+ selectivity and its substantial basal activity in the absence of cAMP, it is also possible that it is a member of hyperpolarization-activated cyclic nucleotide-gated channels (78). Although this Mlotki1 lacks many of the characteristic positive residues from the N-terminal part of S4, it has Arg and Lys at the C-terminus which could, in principle, sense hyperpolarizing potential and drive the movement of S4 (79–81). If Mlotik1 is indeed voltage-sensitive, in the current x-ray structure the VS would be in the up-state and the main pore in the closed state. For Motik1 we observe that its lipid bilayer surrounding is similarly distorted that around the other VS homologs. This is consistent with recent studies showing that voltage-sensitivity in hyperpolarization-activated cyclic nucleotide-gated channels can be reversed to that of Kv channels by changing the coupling between the S4–S5 liner and S6 (82). This suggests that reducing the effective membrane thickness is a common mechanism for facilitating movement of S4 in both the inward and outward directions. However, additional functional data are needed for this channel before a more detailed interpretation can be made.
Examination of the lipid interaction of individual amino-acid side chains show that the VS interact canonically with the membrane. On the other hand, the paddle region in particular binds intimately to specific lipids, which highlights the current view on the special role of lipids in VS function. Furthermore, the large accessibility of the S5 helix of the pore to the membrane also raises the possibility that mutations in S5 affecting VS activation (83) may be acting indirectly through the membrane, highlighting again the complexity of interplay between membrane and voltage-gated channel function.
It is important to consider the possible limitations of the CG model used, derived from the widely used MARTINI (84) CG force field for lipids and peptides. As has been noted (85), CG models such as MARTINI do not fully reproduce the thermodynamics of Arg side chain insertion into a lipid bilayer. For example., a detailed comparison using our CG model (63) suggested an approximately twofold underestimation of the free energy barrier for burying a single Arg side chain in the bilayer core. However, comparisons of CG (73) and atomistic (36) simulations of the S4 helix from Kv channels (with its multiple Arg side chains) suggest that CG simulations are capable of (qualitatively) reproducing the local bilayer distortions caused by Arg insertion into a bilayer. Furthermore, CG simulations of model peptide helices containing an arginine side chain at different positions within the hydrophobic host sequence yielded good agreement with solid-state NMR experiments (86) suggesting that CG simulations are capable of correctly capturing the structure and interactions of Arg side chains with lipid molecules, despite approximations to the energetics of such interactions.
Our observations concerning the membrane lipid interactions with various VS domain homologs fit well with the accumulating evidence for the crucial role played by lipid molecules in the function of membrane proteins (87). Examples of such interactions include not only Kv channels, but also a number of receptors and transporters, as exemplified, respectively, by the interaction of cholesterol with the nicotinic acetylcholine receptor (88), and interactions with cardiolipin with the bacterial translocon (89). Therefore, the membrane protein function could be modulated by changes in bilayer properties, exploiting compositional and other differences between various subcellular membrane domains (90).
Acknowledgments
We are grateful to Peter Bond and Kathryn Scott who wrote the original script for membrane structure and contacts analysis and to Kia Balali-Mood, Chze-ling Wee, Ranjit Vijayan, and Kaishu Tai for help at various stages of this work.
Funding was provided by the Biotechnology and Biological Sciences Research Council and the Wellcome Trust.
Supporting Material
References
- 1.Hille B. Sinauer; Sunderland, MA: 2001. Ionic Channels of Excitable Membranes. [Google Scholar]
- 2.Ashcroft F.M. From molecule to malady. Nature. 2006;440:440–447. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- 3.Murata Y., Iwasaki H., Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature. 2005;435:1239–1243. doi: 10.1038/nature03650. [DOI] [PubMed] [Google Scholar]
- 4.Ramsey I.S., Moran M.M., Clapham D.E. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sasaki M., Takagi M., Okamura Y. A voltage sensor-domain protein is a voltage-gated proton channel. Science. 2006;312:589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- 6.Ramsey I.S., Mokrab Y., Clapham D.E. An aqueous H+ permeation pathway in the voltage-gated proton channel Hv1. Nat. Struct. Mol. Biol. 2010;17:869–875. doi: 10.1038/nsmb.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schoppa N.E., McCormack K., Sigworth F.J. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 1992;255:1712–1715. doi: 10.1126/science.1553560. [DOI] [PubMed] [Google Scholar]
- 8.Aggarwal S.K., MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 9.Noceti F., Baldelli P., Stefani E. Effective gating charges per channel in voltage-dependent K+ and Ca2+ channels. J. Gen. Physiol. 1996;108:143–155. doi: 10.1085/jgp.108.3.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsson H.P., Baker O.S., Isacoff E.Y. Transmembrane movement of the Shaker K+ channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 11.Yusaf S.P., Wray D., Sivaprasadarao A. Measurement of the movement of the S4 segment during the activation of a voltage-gated potassium channel. Pflugers Arch. 1996;433:91–97. doi: 10.1007/s004240050253. [DOI] [PubMed] [Google Scholar]
- 12.Starace D.M., Stefani E., Bezanilla F. Voltage-dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron. 1997;19:1319–1327. doi: 10.1016/s0896-6273(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 13.Starace D.M., Bezanilla F. Histidine scanning mutagenesis of basic residues of the S4 segment of the Shaker K+ channel. J. Gen. Physiol. 2001;117:469–490. doi: 10.1085/jgp.117.5.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruta V., Chen J., MacKinnon R. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell. 2005;123:463–475. doi: 10.1016/j.cell.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 15.Tao X., Lee A., MacKinnon R. A gating charge transfer center in voltage sensors. Science. 2010;328:67–73. doi: 10.1126/science.1185954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.González-Pérez V., Stack K., Naranjo D. Reduced voltage sensitivity in a K+-channel voltage sensor by electric field remodeling. Proc. Natl. Acad. Sci. USA. 2010;107:5178–5183. doi: 10.1073/pnas.1000963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Y., Lee A., MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 18.Long S.B., Campbell E.B., Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 19.Chen X., Wang Q., Ma J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based x-ray crystallographic refinement. Proc. Natl. Acad. Sci. USA. 2010;107:11352–11357. doi: 10.1073/pnas.1000142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long S.B., Tao X., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 21.Clayton G.M., Altieri S., Morais-Cabral J.H. Structure of the transmembrane regions of a bacterial cyclic nucleotide-regulated channel. Proc. Natl. Acad. Sci. USA. 2008;105:1511–1515. doi: 10.1073/pnas.0711533105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakrapani S., Sompornpisut P., Perozo E. The activated state of a sodium channel voltage sensor in a membrane environment. Proc. Natl. Acad. Sci. USA. 2010;107:5435–5440. doi: 10.1073/pnas.0914109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu Z., Klem A.M., Ramu Y. Ion conduction pore is conserved among potassium channels. Nature. 2001;413:809–813. doi: 10.1038/35101535. [DOI] [PubMed] [Google Scholar]
- 24.Alabi A.A., Bahamonde M.I., Swartz K.J. Portability of paddle motif function and pharmacology in voltage sensors. Nature. 2007;450:370–375. doi: 10.1038/nature06266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosmans F., Martin-Eauclaire M.F., Swartz K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature. 2008;456:202–208. doi: 10.1038/nature07473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt D., Jiang Q.X., MacKinnon R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 2006;444:775–779. doi: 10.1038/nature05416. [DOI] [PubMed] [Google Scholar]
- 27.Ramu Y., Xu Y., Lu Z. Enzymatic activation of voltage-gated potassium channels. Nature. 2006;442:696–699. doi: 10.1038/nature04880. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y., Ramu Y., Lu Z. Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature. 2008;451:826–829. doi: 10.1038/nature06618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milescu M., Swartz K.J. Influence of membrane alterations on the inhibition of K-v channels by voltage sensor toxins. Biophys. J. 2007;1(Suppl Pt):295A–296A. [Google Scholar]
- 30.Biverståhl H., Lind J., Mäler L. Biophysical studies of the membrane location of the voltage-gated sensors in the HsapBK and KvAP K+ channels. Biochim. Biophys. Acta. 2009;1788:1976–1986. doi: 10.1016/j.bbamem.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Cuello L.G., Cortes D.M., Perozo E. Molecular architecture of the KvAP voltage-dependent K+ channel in a lipid bilayer. Science. 2004;306:491–495. doi: 10.1126/science.1101373. [DOI] [PubMed] [Google Scholar]
- 32.Milescu M., Bosmans F., Swartz K.J. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 2009;16:1080–1085. doi: 10.1038/nsmb.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asamoah O.K., Wuskell J.P., Bezanilla F. A fluorometric approach to local electric field measurements in a voltage-gated ion channel. Neuron. 2003;37:85–97. doi: 10.1016/s0896-6273(02)01126-1. [DOI] [PubMed] [Google Scholar]
- 34.Ahern C.A., Horn R. Focused electric field across the voltage sensor of potassium channels. Neuron. 2005;48:25–29. doi: 10.1016/j.neuron.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 35.Krepkiy D., Mihailescu M., Swartz K.J. Structure and hydration of membranes embedded with voltage-sensing domains. Nature. 2009;462:473–479. doi: 10.1038/nature08542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freites J.A., Tobias D.J., White S.H. Interface connections of a transmembrane voltage sensor. Proc. Natl. Acad. Sci. USA. 2005;102:15059–15064. doi: 10.1073/pnas.0507618102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Treptow W., Tarek M. Environment of the gating charges in the Kv1.2 Shaker potassium channel. Biophys. J. 2006;90:L64–L66. doi: 10.1529/biophysj.106.080754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jogini V., Roux B. Dynamics of the Kv1.2 voltage-gated K+ channel in a membrane environment. Biophys. J. 2007;93:3070–3082. doi: 10.1529/biophysj.107.112540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sands Z.A., Sansom M.S.P. How does a voltage sensor interact with a lipid bilayer? Simulations of a potassium channel domain. Structure. 2007;15:235–244. doi: 10.1016/j.str.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishizawa M., Nishizawa K. Molecular dynamics simulation of Kv channel voltage sensor helix in a lipid membrane with applied electric field. Biophys. J. 2008;95:1729–1744. doi: 10.1529/biophysj.108.130658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Treptow W., Tarek M., Klein M.L. Initial response of the potassium channel voltage sensor to a transmembrane potential. J. Am. Chem. Soc. 2009;131:2107–2109. doi: 10.1021/ja807330g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bjelkmar P., Niemela P.S., Lindahl E. Conformational changes and slow dynamics through microsecond polarized atomistic molecular simulation of an integral Kv1.2 ion channel. PLoS Comp. Biol. 2009;5:e1000289. doi: 10.1371/journal.pcbi.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schow E.V., Freites J.A., Tobias D.J. Down-state model of the voltage-sensing domain of a potassium channel. Biophys. J. 2010;98:2857–2866. doi: 10.1016/j.bpj.2010.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shelley J.C., Shelley M.Y., Klein M.L. A coarse grain model for phospholipid simulations. J. Phys. Chem. B. 2001;105:4464–4470. [Google Scholar]
- 45.Nielsen S.O., Lopez C.F., Klein M.L. Coarse grain models and the computer simulation of soft materials. J. Phys. Condens. Matter. 2004;16:R481–R512. [Google Scholar]
- 46.Lopez C.F., Nielsen S.O., Klein M.L. Structure and dynamics of model pore insertion into a membrane. Biophys. J. 2005;88:3083–3094. doi: 10.1529/biophysj.104.053769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marrink S.J., de Vries A.H., Mark A.E. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B. 2004;108:750–760. [Google Scholar]
- 48.Bond P.J., Sansom M.S.P. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 2006;128:2697–2704. doi: 10.1021/ja0569104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bond P.J., Holyoake J., Sansom M.S. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol. 2007;157:593–605. doi: 10.1016/j.jsb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 50.Marrink S.J., Risselada H.J., de Vries A.H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 51.Scott K.A., Bond P.J., Sansom M.S.P. Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure. 2008;16:621–630. doi: 10.1016/j.str.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 52.Treptow W., Marrink S.-J., Tarek M. Gating motions in voltage-gated potassium channels revealed by coarse-grained molecular dynamics simulations. J. Phys. Chem. B. 2008;112:3277–3282. doi: 10.1021/jp709675e. [DOI] [PubMed] [Google Scholar]
- 53.Ayton G.S., Voth G.A. Systematic multiscale simulation of membrane protein systems. Curr. Opin. Struct. Biol. 2009;19:138–144. doi: 10.1016/j.sbi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bairoch A., Apweiler R., Yeh L.S. The universal protein resource (UniProt) Nucleic Acids Res. 2005;33(Database issue):D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katoh K., Misawa K., Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi J., Blundell T.L., Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- 58.Sali A., Blundell T.L. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 59.Sánchez R., Sali A. Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. USA. 1998;95:13597–13602. doi: 10.1073/pnas.95.23.13597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sippl M.J. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 61.Lüthy R., Bowie J.U., Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 62.Catherinot V., Labesse G. ViTO: tool for refinement of protein sequence-structure alignments. Bioinformatics. 2004;20:3694–3696. doi: 10.1093/bioinformatics/bth429. [DOI] [PubMed] [Google Scholar]
- 63.Bond P.J., Wee C.L., Sansom M.S.P. Coarse-grained molecular dynamics simulations of the energetics of helix insertion into a lipid bilayer. Biochemistry. 2008;47:11321–11331. doi: 10.1021/bi800642m. [DOI] [PubMed] [Google Scholar]
- 64.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 65.Van Der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 66.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 67.Ren D.J., Navarro B., Clapham D.E. A prokaryotic voltage-gated sodium channel. Science. 2001;294:2372–2375. doi: 10.1126/science.1065635. [DOI] [PubMed] [Google Scholar]
- 68.Zhou Y., Morais-Cabral J.H., MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 69.Cinelli S., Onori G., Diociaiuti M. Properties of mixed DOTAP-DPPC bilayer membranes as reported by differential scanning calorimetry and dynamic light scattering measurements. J. Phys. Chem. B. 2007;111:10032–10039. doi: 10.1021/jp071722g. [DOI] [PubMed] [Google Scholar]
- 70.Johansson A.C.V., Lindahl E. Protein contents in biological membranes can explain abnormal solvation of charged and polar residues. Proc. Natl. Acad. Sci. USA. 2009;106:15684–15689. doi: 10.1073/pnas.0905394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pathak M.M., Yarov-Yarovoy V., Isacoff E.Y. Closing in on the resting state of the Shaker K+ channel. Neuron. 2007;56:124–140. doi: 10.1016/j.neuron.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 72.Milescu M., Vobecky J., Swartz K.J. Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 2007;130:497–511. doi: 10.1085/jgp.200709869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bond P.J., Sansom M.S.P. Bilayer deformation by the Kv channel voltage sensor domain revealed by self-assembly simulations. Proc. Natl. Acad. Sci. USA. 2007;104:2631–2636. doi: 10.1073/pnas.0606822104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang Y., Ruta V., MacKinnon R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 2003;423:42–48. doi: 10.1038/nature01581. [DOI] [PubMed] [Google Scholar]
- 75.Glauner K.S., Mannuzzu L.M., Isacoff E.Y. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 1999;402:813–817. doi: 10.1038/45561. [DOI] [PubMed] [Google Scholar]
- 76.Cha A., Snyder G.E., Bezanilla F. Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy. Nature. 1999;402:809–813. doi: 10.1038/45552. [DOI] [PubMed] [Google Scholar]
- 77.Posson D.J., Selvin P.R. Extent of voltage sensor movement during gating of Shaker K+ channels. Neuron. 2008;59:98–109. doi: 10.1016/j.neuron.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nimigean C.M., Shane T., Miller C. A cyclic nucleotide modulated prokaryotic K+ channel. J. Gen. Physiol. 2004;124:203–210. doi: 10.1085/jgp.200409133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Männikkö R., Elinder F., Larsson H.P. Voltage-sensing mechanism is conserved among ion channels gated by opposite voltages. Nature. 2002;419:837–841. doi: 10.1038/nature01038. [DOI] [PubMed] [Google Scholar]
- 80.Prole D.L., Yellen G. Reversal of HCN channel voltage dependence via bridging of the S4-S5 linker and Post-S6. J. Gen. Physiol. 2006;128:273–282. doi: 10.1085/jgp.200609590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rothberg B.S., Shin K.S., Yellen G. Voltage-controlled gating at the intracellular entrance to a hyperpolarization-activated cation channel. J. Gen. Physiol. 2002;119:83–91. doi: 10.1085/jgp.119.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bell D.C., Yao H., Siegelbaum S.A. Changes in local S4 environment provide a voltage-sensing mechanism for mammalian hyperpolarization-activated HCN channels. J. Gen. Physiol. 2004;123:5–19. doi: 10.1085/jgp.200308918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Soler-Llavina G.J., Chang T.H., Swartz K.J. Functional interactions at the interface between voltage-sensing and pore domains in the Shaker Kv channel. Neuron. 2006;52:623–634. doi: 10.1016/j.neuron.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 84.Monticelli L., Kandasamy S.K., Marrink S.J. The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 85.Vorobyov I., Li L., Allen T.W. Assessing atomistic and coarse-grained force fields for protein-lipid interactions: the formidable challenge of an ionizable side chain in a membrane. J. Phys. Chem. B. 2008;112:9588–9602. doi: 10.1021/jp711492h. [DOI] [PubMed] [Google Scholar]
- 86.Vostrikov V.V., Hall B.A., Sansom M.S. Changes in transmembrane helix alignment by arginine residues revealed by solid-state NMR experiments and coarse-grained MD simulations. J. Am. Chem. Soc. 2010;132:5803–5811. doi: 10.1021/ja100598e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 88.Brannigan G., Hénin J., Klein M.L. Embedded cholesterol in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 2008;105:14418–14423. doi: 10.1073/pnas.0803029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gold V.A.M., Robson A., Collinson I. The action of cardiolipin on the bacterial translocon. Proc. Natl. Acad. Sci. USA. 2010;107:10044–10049. doi: 10.1073/pnas.0914680107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sharpe H.J., Stevens T.J., Munro S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell. 2010;142:158–169. doi: 10.1016/j.cell.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taylor W.R. Identification of protein sequence homology by consensus template alignment. J. Mol. Biol. 1986;188:233–258. doi: 10.1016/0022-2836(86)90308-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.