

Abstract
Three HCM-causing tropomyosin (Tm) mutants (V95A, D175N, and E180G) were examined using the thin-filament extraction and reconstitution technique. The effects of Ca2+, ATP, phosphate, and ADP concentrations on cross-bridge kinetics in myocardium reconstituted with each of these mutants were studied at 25°C, and compared to wild-type (WT) Tm at physiological ionic strength (200 mM). All three mutants showed significantly higher (2–3.5 fold) low Ca2+ tension (TLC) and stiffness than WT at pCa 8.0. High Ca2+ tension (THC) was significantly higher for E180G than that for WT, whereas THC of V95A and D175N was similar to WT; high Ca2+ stiffness (YHC) had the same trend. The Ca2+ sensitivity of isometric force was significantly greater for V95A and E180G than for WT, whereas that of D175N remained the same as for WT; for all mutants, cooperativity was lower than for WT. Nine kinetic constants and the cross-bridge distribution were deduced using sinusoidal analysis. The number of force-generating cross bridges was similar among the D175N, E180G, and WT Tm forms, but it was significantly larger in the case of V95A than WT. We conclude that the increased number of actively cycling cross bridges at pCa 8 is the major cause of Tm mutation-related HCM pathogenesis, which may result in diastolic dysfunction. Decreased contractility (Tact) in V95A and D175N may further contribute to the severity of myocyte hypertrophy and related prognosis of the disease.
Introduction
Inherited forms of hypertrophic cardiomyopathy (HCM) are the most common genetic disorders of the heart caused by sarcomeric protein mutations (1,2). HCM affects ∼1 in 500 individuals; it is the leading cause of sudden death in young adults and a major cause of mortality in elderly people (3,4). The clinical and pathological manifestations of HCM are highly variable, and include myocyte hypertrophy and disarray, interstitial fibrosis, and thickening of the tunica media (smooth muscle layer) of coronary arteries. Affected individuals can remain asymptomatic, or experience consequences as serious as severe heart failure or sudden cardiac death (5,6).
Despite the interest these consequences have generated in the study of HCM, the molecular pathogenesis under the most common phenotypes of HCM, the maladaptive hypertrophy of the left ventricle, and the basis of phenotypic diversity, remain unknown (7). Several hypotheses have been proposed for possible molecular mechanisms leading to HCM pathogenesis, such as decreased myocardial contractility (8), disturbed myofibril formation (9,10), enhanced actin-activated ATPase activity and force generation (11), altered Ca2+ sensitivity (12), and impaired myocyte relaxation (13). Among these possibilities—which are not mutually exclusive—impaired myocyte relaxation has been extensively studied because HCM is considered to be a prototype for diastolic heart failure (14). Symptoms of impaired myocyte relaxation include decreased relaxation velocity (13), reduced left-ventricular end-diastolic volume (15), and enhanced left-ventricular end-diastolic pressure (14). The cause of impaired relaxation is not clear, although thickening of myocardium, structural changes related to cellular disorganization, enhanced interstitial fibrosis, and elevated Ca2+ sensitivity are all known to contribute to severe alterations in diastolic properties of the myocardium (12,16).
Although data from genotype-phenotype studies are limited, it is generally recognized that specific mutations do not always lead to a particular phenotype (17–19). Similar clinical manifestations can be found in patients with different mutations in sarcomeric proteins (20,21). It has therefore been suggested that the treatment of HCM patients can only be effective if it is different among HCM patients according to the specific sarcomere gene mutation that is involved (22). If this is the case, it is then of critical significance that HCM pathogenesis be addressed within the mutations of each individual gene.
So far, >400 mutations of sarcomeric proteins have been identified to cause HCM (23). Among these proteins are the cardiac thin filament proteins: actin (ACTC), troponin T (TNNT2), troponin I (TNNI3), and α-tropomyosin (Tm) (TPM1). In the case of α-Tm, 11 missense mutations have been associated with HCM. A helical coiled-coil Tm molecule has quasirepeating, loosely similar regions 1–7 (24), and regional deletion/replacement studies have suggested that each Tm region may contribute to a specific function (25–28). The mutation V95A is located in region 3 and mutations D175N and E180G are located in region 5. Although their pathophysiological mechanisms are not well understood, the mutant proteins D175N and E180G are thought to destabilize the coiled-coil structure of the Tm dimer, and to increase local flexibility (29,30) because they are at the g- and e-positions, respectively, of the heptad repeat. These positions are typically occupied by ionic amino-acid residues, which enable electrostatic attraction between the two α-helices of the Tm dimer. In addition, these mutations may affect the Tm-TnT interaction, because Tm residues 175–190 (region 5) are known to interact with TnT2 (26).
In Tm-associated HCM, it has been hypothesized that the disease state is related to impaired relaxation caused by increased Ca2+ sensitivity. For example, the D175N mutation is known to increase Ca2+ sensitivity of both biopsy tissue from patients (31) and the hearts of transgenic mice that expressed 65% D175N protein (32,33). In another study this mutation did not significantly affect Ca2+ sensitivity (34) after adenovirus-mediated gene transfer, which may be related to lower expression level (∼60%) obtained with this technique. E180G is known to increase Ca2+ sensitivity in hearts of transgenic mice (35) as well as in cardiac cells transfected using adenoviral vectors (34). E180G is also known to decrease actin affinity both in the presence and absence of troponin (Tn) (36). Both D175N and E180G have been shown to be defective in promoting the myosin-S1 activated switch of the thin filament from the closed to open state, presumably because of the impaired TnT2 interaction (36). Moreover, V95A, as well as A63V and K70T, are also known to cause an increase in Ca2+ sensitivity in vitro (37,38).
However, all of the previous studies lack the direct measurement of cross-bridge cycling kinetics, and concomitant force generation and stiffness increase within the myocardium system under diastolic conditions. In this report, the effect of three HCM-related Tm mutants (V95A, D175N, and E180G) on force generation, stiffness, and cross-bridge kinetics were studied using the thin filament removal/reconstitution technique combined with sinusoidal analysis (39–41). Our results demonstrate:
-
1.
Impaired relaxation of myocardium, shown by the significantly elevated cross-bridge cycling and increased isometric tension at pCa 8.0 with all three mutants.
-
2.
Decreased myocardial contractility, as indicated by the significant decrease in Ca2+-activatable tension with mutants V95A and D175N.
Material and Methods
Experimental material and thin-filament extraction and reconstitution technique
We used thin, slender bundles (length ∼2 mm, diameter 90–110 μm) from bovine cardiac muscle for thin-filament extraction and reconstitution and mechanical measurements. These bundles were dissected from trabeculae (see the Supporting Material) that were freely suspended in the right ventricle and stored in the skinning/storage solutions for at least seven days. Bundles were attached to the experimental apparatus by nail polish, and further skinned in 1% Triton X100 for 20 min in relaxing solution. They were then stretched to the sarcomere length of ∼2.1 μm, and thin-filament extraction and reconstitution was performed. This technique was originally developed in Ishiwata's laboratory (40), refined in Kawai's laboratory (41), and was performed as described (25,39–41) with minor modifications (see the Supporting Material).
Proteins
G-actin was purified from rabbit fast-twitch skeletal muscles (43), and Tn was purified from bovine hearts in Kawai's Lab as described (44). Human wild-type (WT) and mutant α-Tms were expressed as recombinant proteins in Escherichia coli and purified at The Florida State University (Tallahassee, FL). Pure WT or mutant Tm was used in the reconstitution. These Tms have two extra amino acids, Gly-Ser, which remain at the N-terminus after removal of an affinity tag, and functionally substitute for acetylation (45). These residues remain at the N-terminus after removal of an affinity tag, and functionally substitute for acetylation (45). Previous studies showed that this N-terminal difference had little effect on Tm stability and function, either in vitro (46) or in fiber studies at physiological ionic strength (28).
pCa-tension and pCa-stiffness studies
pCa-tension and pCa-stiffness studies were performed as described in Kawai et al. (28) except that the pCa range used in this study was from 8.0 to 4.66. The tension and stiffness under pCa 8 were called low Ca2+ tension (TLC) and low Ca2+ stiffness (YLC). The tension and stiffness under pCa 4.66 were called high Ca2+ tension (THC) and high Ca2+ stiffness (YHC). Tension baseline was defined as that existing in the relaxing solution (see Table S2 in the Supporting Material), which contains 6 mM EGTA, no added Ca, and 40 mM BDM at 0°C. There is no significant tension in this condition. Subsequent tension measurements were performed as the increment from this baseline tension. The pCa-tension relationship of reconstituted fibers was studied with 12 different Ca2+ concentrations (pCa: 8.0, 7.5, 7.0, 6.4, 6.2, 6.0, 5.8, 5.6, 5.4, 5.2, 5.0, 4.66) with fixed MgATP (5 mM) and Pi (8 mM) concentrations. The pCa-tension data were fitted to the Hill equation,
| (1) |
where pCa = –log10[Ca2+], Tact is the Ca2+ activatable tension (Tact = THC – TLC), and Ca50 is the apparent Ca2+ dissociation constant, which represents the Ca2+ concentration at half-maximum tension. pCa50 (= –log10Ca50) represents Ca2+ sensitivity, and nH (Hill factor) represents the cooperativity. pCa-tension curves were individually fitted to Eq. 1, and the results were averaged. All tension values were normalized to unregulated maximum tension (Ta; see section S1.2 in the Supporting Material) of the actin-filament reconstituted myocardium without Tm or Tn, obtained under our standard activating condition with the 5S8P solution (5 mM MgATP, 8 mM Pi, pCa 4.66) at 25°C. Ta averaged to 13.9 ± 0.6 kPa (n = 51). Yact is defined as Yact = YHC–YLC.
Sinusoidal analysis
The elementary steps of the cross-bridge cycle based on six states (Scheme 1) were characterized by sinusoidal analysis performed as described in Kawai and Brandt (47) and Kawai et al. (48). Small amplitude (0.125% L0) sinusoidal length changes were applied to the reconstituted myocardium at 18 different frequencies (f) in the range 0.13–100 Hz. The resulting tension transients were analyzed and the complex modulus Y(f) was calculated. Y(f) is the ratio of the stress change to the strain change expressed in the frequency domain. Y(f) was then fitted to Eq. 2, which incorporates three exponential processes (47)—Process A, Process B, and Process C—as
| (2) |
where ; 2πa, 2πb, and 2πc are the apparent rate constants of exponential processes A, B, and C, respectively; A, B, and C are their respective magnitudes (sometimes called amplitudes); and H is a constant. A, B, C, and H have the same units as Y(f) or isometric tension, and thus are normalized to Ta. In cardiac muscle fibers, Process A was not observed (A = 0) at ≤ 25°C (41,48,49). The complex modulus thus measured includes the effect of series compliance, which may (50) or may not (51) affect the apparent rate constants depending on reports. However, the series compliance is not a concern for this report, because it is not changed between mutants and the WT control.
Scheme 1.
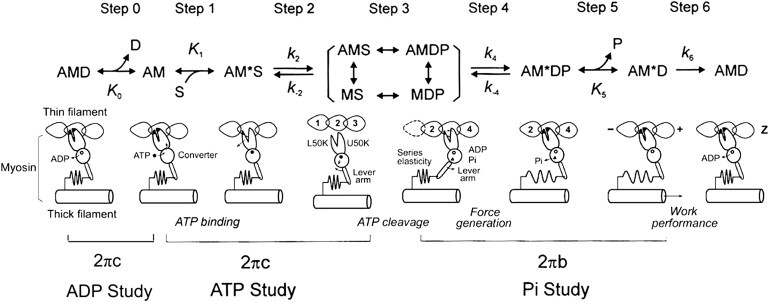
Elementary steps of the cross-bridge cycle. The uppercase letters K indicate the association or equilibrium constants, and the lowercase letters k indicate the rate constants of the elementary steps. Collectively these are called the kinetic constants, and they can be deduced experimentally. (A, actin; M, myosin; D, MgADP; S, MgATP; and P, phosphate (Pi).) Rearranged from Kawai and Candau (80).
The apparent rate constant 2πb and 2πc were studied as functions of the MgATP, Pi, and MgADP concentrations. The data were fitted to the following equations, which were derived from Scheme 1, assuming that steps 0, 1, and 5 are in fast equilibrium, and that step 6 is the slowest forward step of the cross-bridge cycle (52),
| (3) |
| (4) |
where
| (5) |
and S = [MgATP] and P = [Pi]. The value K1 is the ATP association constant, k2 is the forward rate constant of cross-bridge detachment step 2, and k−2 is its reversal step. The value k4 is the forward rate constant of the force generation step 4, and k−4 is its reversal step. K5 is the Pi association constant. The value σ in Eq. 5 was calculated from K1 and K2 obtained from the MgATP study and S = 5 mM. The reconstituted myocardium was studied with eight different MgATP concentrations (0.05, 0.1, 0.2, 0.5, 1, 2, 5, 10 mM) at a fixed Pi concentration (8 mM) and pCa 4.66 to deduce the rate and association constants of steps 1 and 2. The effect of phosphate (Pi) was studied at six different Pi concentrations (0, 2, 4, 8, 16, and 32 mM) in the presence of a fixed MgATP concentration (5 mM) and pCa 4.66 to deduce the rate and association constants of steps 4 and 5. The effect of MgADP was studied at four different MgADP concentrations (0, 1, 2, and 3 mM) at fixed Pi (8 mM) and MgATP (2 mM) concentrations and pCa 4.66 to deduce the association constant of step 0 (Scheme 1).
Results
pCa-tension and pCa-stiffness studies
To determine the effect of HCM-related Tm mutations on cooperative activation and Ca2+ sensitivity, we studied tension and stiffness of the thin filament-reconstituted myocardium as functions of [Ca2+]. pCa-tension plots comparing mutant and WT Tms are shown in Fig. 1. It is clear that all three mutants affected the pCa-tension relationship, with E180G having the greatest effect and D175 the smallest. The effects of the three mutations on several parameters are summarized in Fig. 2. All three mutants showed significantly increased (2–3.5×) TLC compared to WT (p < 0.0001), indicating that the relaxation of myocardium is partially impaired under diastolic condition.
Figure 1.
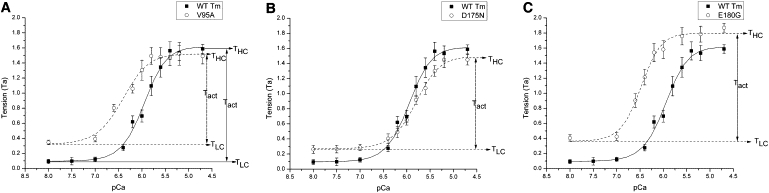
pCa-tension plots comparing the effects of three mutant Tms to those of WT-Tm. Experiments were performed at 25°C. Symbols represent the mean ± SE (solid line for WT, dashed line for mutants). Curves are the results of fitting the data to Eq. 1. Definitions of TLC, THC, and Tact are illustrated.
Figure 2.
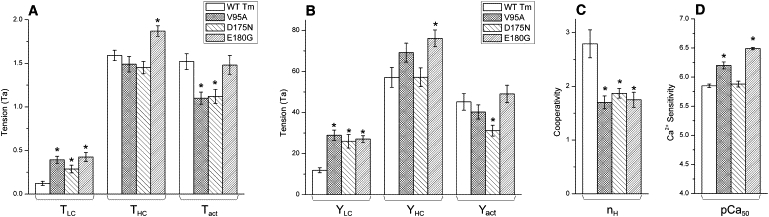
Tension, stiffness, cooperativity, and pCa50. (A) Result of the tension study. All three mutants showed significantly elevated TLC compared to WT. Only the E180G-Tm mutant showed significantly higher THC than WT. The Tact of V95A and D175N was significantly less than that of WT, but that of E180G remained similar to WT. (B) Result of the stiffness study. The trends are approximately the same as for the tension study in panel A, except that the active stiffness in the case of V95A was not significantly different from that of WT. (C) Results of the cooperativity study. In all three mutants the cooperativity was reduced compared to that for WT. (D) Results of the pCa50 (Ca2+ sensitivity) study. The V95A and E180G both exhibited pCa50 significantly greater than that of WT, but the pCa50 of D175N was similar to that of WT. Tension and stiffness are normalized to Ta. The asterisk (∗) is p < 0.05.
Cooperativity (nH) was calculated by averaging the nH of individual curves (Fig. 2 C). All three mutants showed decreased cooperativity compared to WT, as indicated by decreased nH (WT 2.79 ± 0.26, V95A 1.70 ± 0.12, D175N 1.87 ± 0.09, and E180G 1.75 ± 0.14; see Table S3). In the case of E180G-Tm, this may seem to contradict the fact that the slopes (∝nH) of the curve shown in Fig. 1 C appear to be similar to WT-Tm; however, this is the result of the averaging procedure. Here, the data points for the same pCa value were averaged, whereas averaged nH was obtained by averaging individually fitted nH. (For a further discussion on this subject, see the Supporting Material.) pCa50 of V95A (6.20 ± 0.06, N = 13) and E180G (6.49 ± 0.02, N = 11) was significantly larger than WT (5.85 ± 0.03, N = 7), indicating increased Ca2+ sensitivity of mutants, whereas pCa50 of D175N (5.88 ± 0.05 N = 15) remained similar to WT (Fig. 2 D).
Sinusoidal analysis and cross-bridge kinetics
Sinusoidal analysis was performed as described in Fujita et al. (41) and Kawai and Brandt (47), to characterize the elementary steps of the cross-bridge cycle of the reconstituted myocardium. Nyquist plots comparing mutant Tms (dashed line) and WT (solid line) are shown in Fig. 3; data in panels A–C were obtained at pCa 8, whereas panels D–F depict data obtained at pCa 4.66. The Nyquist plot of WT demonstrates that cross bridges actively cycled and performed energy transduction even at pCa 8 (Fig. 3 A), as reported earlier for the myocardium (53), because processes B and C (represented by two semicircles) are clearly present. The diameter of semicircles is proportional to the number of actively cycling cross bridges (52). Fig. 3, D–F, shows Nyquist plots at pCa 4.66 (25°C) together with those at the relaxing solution (containing 40 mM BDM; see Table S2) at 0°C.
Figure 3.
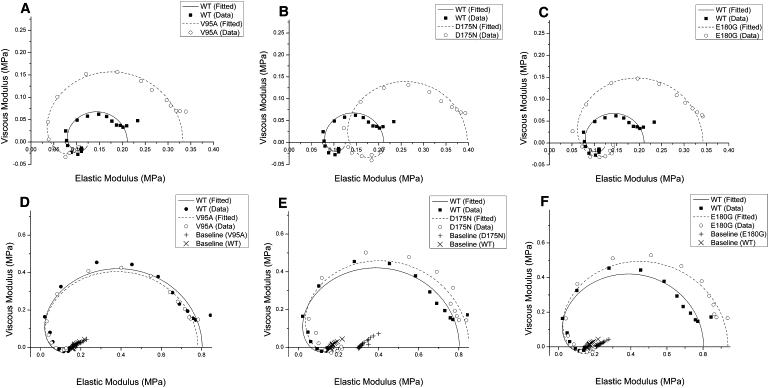
Nyquist plots of the mutants and WT. Nyquist plots of the mutants (dashed line) compared to WT (solid line) at 25°C. (A–C) Three mutants are compared to WT at pCa 8.0. (D–F) Three mutants are compared to WT at pCa 4.66. Also included in panels D–F is the baseline taken in the relaxing solution at 0°C (+ for the mutant, and × for WT). Range of frequency used is 0.13–100 Hz. The frequency increases in the clockwise direction, and the rightmost point corresponds to 100 Hz. Note the difference in scales for both axes in panels A–C versus panels D–F.
The data in Fig. 3, A–C, demonstrates that, in the case of WT, ∼17% of cross bridges are active at pCa 8 compared to pCa 4.66. The diameter of the Nyquist plots doubled (2–2.3×) in all three mutants, demonstrating that 31–39% of cross bridges are actively cycling at pCa 8, which demonstrates impaired relaxation. We conclude, therefore, that the elevated TLC and YLC of mutants at pCa 8 are caused by actively cycling cross bridges. Tact of V95A (1.10 ± 0.07, N = 12) and D175N (1.12 ± 0.08, N = 8) were significantly reduced (∼28%) from WT (1.52 ± 0.09, N = 11), indicating decreased myocardial contractility in mutants, whereas E180G showed similar Tact (1.48 ± 0.11, N = 10) to WT. E180G showed significantly higher THC (∼18%) than WT, but THC of V95A and D175N remained similar to WT. This is mainly because E180G showed similar Tact. A similar trend can be seen in stiffness (Fig. 2 B).
Two apparent rate constants, 2πb and 2πc, were measured by sinusoidal analysis, as functions of [MgATP], [MgADP], and [Pi] (Fig. 4). It is clear that all mutants showed some changes in the apparent rate constants, but the changes were small. The rate and association constants of the elementary steps as defined in Scheme 1 are collectively referred to as kinetic constants. These were deduced by fitting the ligand concentration dependence of the apparent rate constants (52). The kinetic constants of the mutants and WT are compared in Fig. 5.
Figure 4.
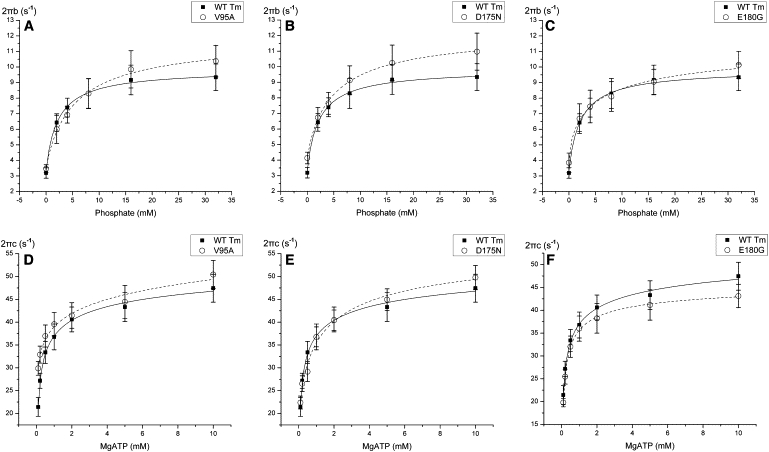
Apparent rate constants. The apparent rate constants for myocardium reconstituted with mutants (dashed line) and WT (solid line) Tm. Symbols represent the mean ± SE. Solid curves were generated by fitting the data to Eq. 3 (A–C) or Eq. 4 (D–F). (A–C) 2πc is plotted against [MgATP]. (D–F) 2πb is plotted against [Pi].
Figure 5.
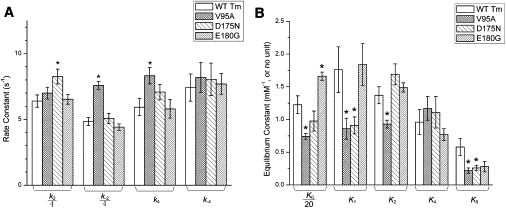
Kinetic constants. The kinetic constants of the reconstituted myocardium as shown in Scheme 1. These were deduced using sinusoidal analysis, as for Fig. 4. (A) Rate constants: V95A showed increased k−2 (30 ± 1 s−1) and k4 (8.3 ± 0.6 s−1) compared to WT (k−2 = 19 ± 1 s−1 and k4 = 5.9 ± 0.7 s−1); D175N showed increased k2 (33 ± 2 s−1) compared to WT (26 ± 2 s−1). (B) Equilibrium constants: V95A showed decreased K0 (15 ± 1 mM−1), K1 (0.9 ± 0.2 mM−1), K2 (0.9 ± 0.1), and K5 (0.22 ± 0.04 mM−1) compared to WT (K0 = 25 ± 3 mM−1; K1 = 1.8 ± 0.4 mM−1; K2 = 1.4 ± 0.1; and K5 = 0.58 ± 0.13 mM−1). D175N showed decreased K5 (0.26 ± 0.04 mM−1), and E180G showed increased K0 (33 ± 1 mM−1) compared to WT. None of the mutants showed any significant change in K4. The asterisk (∗) is p < 0.05.
All kinetic constants changed in mutants, but only the following changes were significant (p < 0.05): E180G exhibited ∼35% increase in K0 compared to WT. D175N exhibited ∼30% increase in k2 and ∼50% decrease in K5. V95A showed the most changes in the kinetic constants: it exhibited ∼40% decrease in K0, ∼50% decrease in K1, ∼30% decrease in K2, ∼60% decrease in K5, ∼56% increase in k−2, and ∼40% increase in k4. All three mutants showed reduced K5 (for E180G: p = 0.07), indicating that the Pi release step is accelerated in these HCM mutants.
Cross-bridge distribution and force per cross bridge
There are two main possibilities to explain the observed change in isometric tension (Fig. 2 A)—1), a change in either the number of force-generating cross bridges, or 2), the force generated by each cross bridge. To discriminate between these possibilities, we calculated the distribution of cross bridges in each state under the standard activating conditions (Table S2) as described in Zhao et al. (54), and the result is shown in Fig. 6. All three mutants showed a significant increase in the AM∗D state (∼2×, p < 0.05) compared to WT, which is consistent with the decrease in K5 (Fig. 4). D175N showed ∼30% decrease in AM∗S compared to WT. Att indicates the sum of all strongly attached (force-generating) cross bridges:
Figure 6.
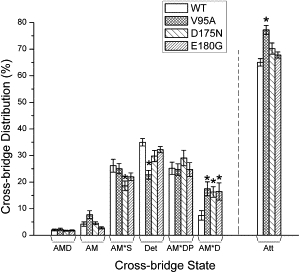
Cross-bridge distributions. The cross-bridge distribution over six states in three mutants in the standard activating solution (5S8P, see Table S2) was calculated from the equilibrium constants (Fig. 5B). The Att indicates the sum of all strongly attached (force generating) cross bridges. All three mutants showed significant increases (V95A, 18 ± 3%; D175N, 16 ± 2%; E180G, 17 ± 3%) in the AM∗D state compared to WT (8 ± 2%). D175N (19 ± 2%) showed a significant decrease in the AM∗S state compared to WT (26 ± 2%). V95A (77 ± 2%) showed an increase in the Att state compared to WT (65 ± 1%). The asterisk (∗) is p < 0.05.
Only V95A showed a significant increase in the Att state compared to WT (by 19 ± 4%), but Xatt of D175N or E180G is not significantly different from WT. Consequently, THC per cross bridge (= THC/Xatt) decreased from WT by 21 ± 6% in V95A (± with error propagation), remained similar in D175N, but increased by 13 ± 6% in E180G.
Discussion
The clinical manifestations of HCM are highly diverse and appear to be linked to both genetic and nongenetic factors (55). However, there are emerging lines of evidence supporting the concept that sarcomeres should be considered as a key part in cardiac contractile function and its regulation, rather than a simple assembly of force generators (56–59). The biochemical properties of cardiac sarcomeres have been shown to be directly responsible for many, if not all, of the mechanical properties of the heart (60–63). As the key component of the thin-filament regulatory mechanism and the excitation-contraction coupling process, Tm's normal function is vital for maintaining the efficient contractility of the heart. In this report, we investigated early consequences of the gene alterations of Tm, which eventually leads into HCM.
Previous studies have shown that the mutant protein expression level may be an important factor, because human HMC patients are heterozygotes, and 100% expression level cannot be achieved in transgenic animals (64). In our studies, we started from the other extreme (100% mutant proteins) where the effects are expected to be the largest. Some of our results (increased pCa50 in V95A-Tm and E180G-Tm) are consistent with previous studies (34,35,37). In other studies, D175N also showed increased pCa50 (31,33) at a lower expression level, which is at variance from our result. This difference may indicate that the interaction between normal and mutant protein might contribute to the sarcomeric dysfunction.
Tm mutations D175N and E180G are located in a putative TnT2 binding region (65) and both cause a charge change from −1 to 0, which is thought to increase the local flexibility of Tm and affect its interaction with the Tn complex. V95A is located at the d-position of the heptad repeat, which is critical for stabilization of the local coiled-coil structure of Tm. This mutation introduces an Ala residue into region 2 of Tm. Consequently, it is possible that these three mutants cause a shift in equilibrium between the closed state and the open state in the three-state model (66) and subsequently result in elevated tension under diastolic conditions (as shown by elevated TLC). This also indicates that there exist at least two different mechanisms affecting three-state equilibrium, because these three mutants are located in two different regions. D175N and E180G altered cross-bridge kinetics to a lesser extent than V95A, which implies that residue 95 of Tm is not only involved in Ca2+-activation but also in the cross-bridge cycle during the open state. Evidence from in vitro studies has indicated that the substitution of Gly for Glu in the e-position of the coiled-coil structure destabilizes α-helix formation by 3×, whereas the effect is less dramatic with the mutation D175N (67,68). E180G, despite its close proximity to D175N and the similar role it plays in heptad repeat, showed a higher TLC (P = 0.067), Tact (P < 0.05), THC (P < 0.0005), and pCa50 (P < 0.001) than D175N. From these results, we infer that E180G can induce greater flexibility of Tm into the adjacent region, causing a greater shift in equilibrium between the closed state and the open state, and possibly enhancing the hydrophobic interaction that takes place in force generation.
Left ventricular hypertrophy (LVH) is the most obvious and important clinical feature of HCM. From the functional perspective, LVH is generally considered to be a compensatory mechanism for the decreased contractility of the HCM heart (5). The sarcomeric dysfunction deduced from this study, maximum left ventricular wall thickness (LVWT), and cumulative survival rate of the three Tm mutants are summarized in Table 1. Results from clinical studies indicate that patients with HCM caused by D175N exhibit more serious LVH than E180G and higher cumulative survival rate than V95A (69); E180G has been associated with milder LVH than D175N, but its cumulative survival rate is not reported (70,71). In a transgenic mouse study, E180G exhibited severe LVH and significantly lower cumulative survival rate than D175N at an expression level of ∼60% (33,35), which is at variance with the patient studies. Such differences are also seen in some patient studies and likely reflect different genetic backgrounds and environmental factors.
Table 1.
Comparison of HCM symptoms and possible molecular pathogenesis
| Sarcomeric dysfunction | Maximum LVWT (Ref.) | Cumulative survival rate (Ref.) | |
|---|---|---|---|
| V95A | nH ↓ TLC↑ Tact ↓ pCa50 ↑ K0 ↓ K1 ↓ K2 ↓ K5 ↓ k−2 ↑ k4 ↑ | 16 ± 6 mm (37) | Low, similar to that of MHC7 mutation R403Q (37) |
| D175N | nH ↓ TLC ↑ Tact ↓ K5 ↓ k2 ↑ | 20.3 ± 3.1 mm, significantly greater than for E180G (70) | Significantly higher than for MHC7 mutation R403Q (69) |
| E180G | nH ↓ TLC ↑ pCa50↑ K0 ↓ | 12.5 ± 4.7 mm (70) | Not studied |
After comparing sarcomeric dysfunction, maximum LVWT, and cumulative survival rate for the three Tm mutants, we note the following:
a) HCM pathogenesis may be related to impaired relaxation of the myocardium
All three α-Tm mutants showed a significantly increase in the number of cycling cross bridges (from 17% to 31–39%) at pCa 8, as evidenced by increased magnitudes B and C (Fig. 3) (47,52). This results in significantly elevated tension (TLC: 2–3.5×) and stiffness (YLC: 2×) (Fig. 2). We conclude, therefore, that the increase in cycling cross bridges at pCa 8 is the direct cause of the diastolic failure manifested in Tm-related HCM. This mechanism has been previously proposed based on indirect evidence (34,37), but we now demonstrate this directly. Elevated number of cycling cross bridges at pCa 8 will inevitably contribute to diastolic dysfunction: pCa∼7 was reported as the diastolic Ca2+ concentration in rat (74). This intrinsic and permanent diastolic pressure overload is capable of stimulating cardiac growth, which leads to an increased release of angiotension II and aldosterone (75), and results in phenotypic alterations at the myocyte level (76)—hypertrophy (77) and abnormal extracellular matrix formation (75).
Thus, elevated tension at pCa 8 can lead to impaired LV filling causing diastolic dysfunction. The thickened myocardium, structural changes including cellular disorganization, and enhanced interstitial fibrosis (16) should thus be considered as either compensatory mechanisms or symptoms caused by HCM. Elevated diastolic tension and stiffness can also help explain the enhanced LV end-diastolic pressure and decreased LV end-diastolic volume. In addition, reduced cooperativity for all three mutants indicates impaired coupling among Tn, Tm, actin, and myosin, which also occurs in dilated cardiomyopathy-related α-Tm mutations (12).
b) LVH and elevated Ca2+-sensitivity may compensate for decreased contractility
Marian (8) has proposed that decreased contractility would result in increased stimulus for cardiomyocytes to produce trophic factors, resulting in hypertrophy and interstitial fibrosis. This proposal is supported by the comparison between E180G and D175N mutations. Tact was similar to WT in E180G-Tm containing myocardium, whereas Tact was ∼28% less than WT with D175N-Tm myocardium (Fig. 2); LVWT of E180G-Tm is significantly less than that with D175N-Tm (Table 1) (70,71). V95A myocardium, however, exhibited an ∼28% reduction in Tact as with D175N (Fig. 2), but LVWT was similar to E180G (Table 1), which may explain the lower cumulative survival rate for patients with V95A mutation than D175N mutation due to inadequate compensation from LVH.
It has been reported that elevated Ca2+ sensitivity is associated with HCM (12,38), consistent with our observations on myocardium containing V95A- and E180G-Tm (Figs. 1 and 2). Previous studies also reported decreased Ca2+ sensitivity in dilated cardiomyopathy α-Tm mutants (12,37). HCM myosin mutant R403Q has been shown to cause a similar decrease in Tact (∼30%) (78) and a significantly higher level of LVH than V95A (37), but their cumulative survival rate is similar. This may be because, similar to our results with D175N-Tm (Figs. 1 and 2), the myosin mutation R403Q has little effect on the Ca2+ sensitivity of isometric force production (79).
Conclusions
Based on the results we present here, we conclude that the impaired relaxation and elevated Ca2+ sensitivity are the major cause of Tm mutation-related HCM pathogenesis. A decrease in active tension may contribute to the hypertrophy of cardiomyocytes, as well as to the severity of symptoms and disease prognosis.
Acknowledgments
We thank Dr. Fang Wang for cloning and generating the α-Tm mutants.
This work was supported by National Institutes of Health grants No. HL70041 and No. AHA0850184Z to M.K., and grants No. HL63974 and No. GM079592 to P.B.C. Note that the work reported here and the conclusions drawn are solely the responsibility of the authors, and do not necessarily represent the official view of the awarding organizations.
Supporting Material
References
- 1.Elliott P., McKenna W.J. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–1891. doi: 10.1016/S0140-6736(04)16358-7. [DOI] [PubMed] [Google Scholar]
- 2.Ho C.Y., Seidman C.E. A contemporary approach to hypertrophic cardiomyopathy. Circulation. 2006;113:e858–e862. doi: 10.1161/CIRCULATIONAHA.105.591982. [DOI] [PubMed] [Google Scholar]
- 3.Maron B.J., McKenna W.J., Wigle E.D. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J. Am. Coll. Cardiol. 2003;42:1687–1713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 4.Ommen S.R., Gersh B.J. Sudden cardiac death risk in hypertrophic cardiomyopathy. Eur. Heart J. 2009;30:2558–2559. doi: 10.1093/eurheartj/ehp307. [DOI] [PubMed] [Google Scholar]
- 5.Marian A.J., Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2001;33:655–670. doi: 10.1006/jmcc.2001.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barron J.T. Hypertrophic cardiomyopathy. Curr. Treat. Options Cardiovasc. Med. 1999;1:277–282. doi: 10.1007/s11936-999-0044-2. [DOI] [PubMed] [Google Scholar]
- 7.Elliott P., McKenna W. Hypertrophic cardiomyopathy: a 50th anniversary. Preface. Heart. 2008;94:1247–1248. doi: 10.1136/hrt.2008.154344. [DOI] [PubMed] [Google Scholar]
- 8.Marian A.J. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet. 2000;355:58–60. doi: 10.1016/s0140-6736(99)06187-5. [DOI] [PubMed] [Google Scholar]
- 9.Yang Q., Sanbe A., Robbins J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J. Clin. Invest. 1998;102:1292–1300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marian A.J., Yu Q.T., Roberts R. Expression of a mutation causing hypertrophic cardiomyopathy disrupts sarcomere assembly in adult feline cardiac myocytes. Circ. Res. 1995;77:98–106. doi: 10.1161/01.res.77.1.98. [DOI] [PubMed] [Google Scholar]
- 11.Tyska M.J., Hayes E., Warshaw D.M. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 2000;86:737–744. doi: 10.1161/01.res.86.7.737. [DOI] [PubMed] [Google Scholar]
- 12.Chang A.N., Harada K., Potter J.D. Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in α-tropomyosin. J. Biol. Chem. 2005;280:34343–34349. doi: 10.1074/jbc.M505014200. [DOI] [PubMed] [Google Scholar]
- 13.Kim S.J., Iizuka K., Vatner S.F. An α-cardiac myosin heavy chain gene mutation impairs contraction and relaxation function of cardiac myocytes. Am. J. Physiol. 1999;276:H1780–H1787. doi: 10.1152/ajpheart.1999.276.5.H1780. [DOI] [PubMed] [Google Scholar]
- 14.Mandinov L., Eberli F.R., Hess O.M. Diastolic heart failure. Cardiovasc. Res. 2000;45:813–825. doi: 10.1016/s0008-6363(99)00399-5. [DOI] [PubMed] [Google Scholar]
- 15.Du J., Liu J., Huang X.P. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H2604–H2613. doi: 10.1152/ajpheart.91506.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zieman S.J., Fortuin N.J. Hypertrophic and restrictive cardiomyopathies in the elderly. Cardiol. Clin. 1999;17 doi: 10.1016/s0733-8651(05)70062-x. 159–172., ix. [DOI] [PubMed] [Google Scholar]
- 17.Marian A.J. On predictors of sudden cardiac death in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2003;41:994–996. doi: 10.1016/s0735-1097(02)03003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fananapazir L., Epstein N.D. Genotype-phenotype correlations in hypertrophic cardiomyopathy. Insights provided by comparisons of kindreds with distinct and identical β-myosin heavy chain gene mutations. Circulation. 1994;89:22–32. doi: 10.1161/01.cir.89.1.22. [DOI] [PubMed] [Google Scholar]
- 19.Bos J.M., Poley R.N., Ackerman M.J. Genotype-phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol. Genet. Metab. 2006;88:78–85. doi: 10.1016/j.ymgme.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niimura H., Bachinski L.L., Seidman C.E. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N. Engl. J. Med. 1998;338:1248–1257. doi: 10.1056/NEJM199804303381802. [DOI] [PubMed] [Google Scholar]
- 21.Charron P., Dubourg O., Komajda M. Genotype-phenotype correlations in familial hypertrophic cardiomyopathy. A comparison between mutations in the cardiac protein-C and the β-myosin heavy chain genes. Eur. Heart J. 1998;19:139–145. doi: 10.1053/euhj.1997.0575. [DOI] [PubMed] [Google Scholar]
- 22.Keren A., Syrris P., McKenna W.J. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat. Clin. Pract. Cardiovasc. Med. 2008;5:158–168. doi: 10.1038/ncpcardio1110. [DOI] [PubMed] [Google Scholar]
- 23.Morita H., Rehm H.L., Seidman C.E. Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 2008;358:1899–1908. doi: 10.1056/NEJMoa075463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLachlan A.D., Stewart M. Tropomyosin coiled-coil interactions: evidence for an unstaggered structure. J. Mol. Biol. 1975;98:293–304. doi: 10.1016/s0022-2836(75)80119-7. [DOI] [PubMed] [Google Scholar]
- 25.Lu X., Tobacman L.S., Kawai M. Effects of tropomyosin internal deletion Δ23Tm on isometric tension and the cross-bridge kinetics in bovine myocardium. J. Physiol. 2003;553:457–471. doi: 10.1113/jphysiol.2003.053694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hitchcock-DeGregori S.E., Song Y., Greenfield N.J. Functions of tropomyosin's periodic repeats. Biochemistry. 2002;41:15036–15044. doi: 10.1021/bi026519k. [DOI] [PubMed] [Google Scholar]
- 27.Lu X., Tobacman L.S., Kawai M. Temperature-dependence of isometric tension and cross-bridge kinetics of cardiac muscle fibers reconstituted with a tropomyosin internal deletion mutant. Biophys. J. 2006;91:4230–4240. doi: 10.1529/biophysj.106.084608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawai M., Lu X., Wandling M.W. Tropomyosin period 3 is essential for enhancement of isometric tension in thin filament-reconstituted bovine myocardium. J. Biophys. (Japan) 2009 doi: 10.1155/2009/380967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh A., Hitchcock-DeGregori S.E. Dual requirement for flexibility and specificity for binding of the coiled-coil tropomyosin to its target, actin. Structure. 2006;14:43–50. doi: 10.1016/j.str.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 30.Singh A., Hitchcock-DeGregori S.E. Local destabilization of the tropomyosin coiled coil gives the molecular flexibility required for actin binding. Biochemistry. 2003;42:14114–14121. doi: 10.1021/bi0348462. [DOI] [PubMed] [Google Scholar]
- 31.Bottinelli R., Coviello D.A., Reggiani C. A mutant tropomyosin that causes hypertrophic cardiomyopathy is expressed in vivo and associated with an increased calcium sensitivity. Circ. Res. 1998;82:106–115. doi: 10.1161/01.res.82.1.106. [DOI] [PubMed] [Google Scholar]
- 32.Evans C.C., Pena J.R., Wolska B.M. Altered hemodynamics in transgenic mice harboring mutant tropomyosin linked to hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H2414–H2423. doi: 10.1152/ajpheart.2000.279.5.H2414. [DOI] [PubMed] [Google Scholar]
- 33.Muthuchamy M., Pieples K., Wieczorek D.F. Mouse model of a familial hypertrophic cardiomyopathy mutation in α-tropomyosin manifests cardiac dysfunction. Circ. Res. 1999;85:47–56. doi: 10.1161/01.res.85.1.47. [DOI] [PubMed] [Google Scholar]
- 34.Michele D.E., Albayya F.P., Metzger J.M. Direct, convergent hypersensitivity of calcium-activated force generation produced by hypertrophic cardiomyopathy mutant α-tropomyosins in adult cardiac myocytes. Nat. Med. 1999;5:1413–1417. doi: 10.1038/70990. [DOI] [PubMed] [Google Scholar]
- 35.Prabhakar R., Boivin G.P., Wieczorek D.F. A familial hypertrophic cardiomyopathy α-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J. Mol. Cell. Cardiol. 2001;33:1815–1828. doi: 10.1006/jmcc.2001.1445. [DOI] [PubMed] [Google Scholar]
- 36.Golitsina N., An Y., Hitchcock-DeGregori S.E. Effects of two familial hypertrophic cardiomyopathy-causing mutations on R-tropomyosin structure and function. Biochemistry. 1999;36:4637–4642. doi: 10.1021/bi962970y. [DOI] [PubMed] [Google Scholar]
- 37.Karibe A., Tobacman L.S., Fananapazir L. Hypertrophic cardiomyopathy caused by a novel α-tropomyosin mutation (V95A) is associated with mild cardiac phenotype, abnormal calcium binding to troponin, abnormal myosin cycling, and poor prognosis. Circulation. 2001;103:65–71. doi: 10.1161/01.cir.103.1.65. [DOI] [PubMed] [Google Scholar]
- 38.Heller M.J., Nili M., Tobacman L.S. Cardiomyopathic tropomyosin mutations that increase thin filament Ca2+ sensitivity and tropomyosin N-domain flexibility. J. Biol. Chem. 2003;278:41742–41748. doi: 10.1074/jbc.M303408200. [DOI] [PubMed] [Google Scholar]
- 39.Kawai M., Ishiwata S. Use of thin filament reconstituted muscle fibers to probe the mechanism of force generation. J. Muscle Res. Cell Motil. 2006;27:455–468. doi: 10.1007/s10974-006-9075-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujita H., Yasuda K., Ishiwata S. Structural and functional reconstitution of thin filaments in the contractile apparatus of cardiac muscle. Biophys. J. 1996;71:2307–2318. doi: 10.1016/S0006-3495(96)79465-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita H., Sasaki D., Kawai M. Elementary steps of the cross-bridge cycle in bovine myocardium with and without regulatory proteins. Biophys. J. 2002;82:915–928. doi: 10.1016/S0006-3495(02)75453-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reference deleted in proof.
- 43.Spudich J.A., Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 44.Holroyde M.J., Robertson S.P., Potter J.D. The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J. Biol. Chem. 1980;255:11688–11693. [PubMed] [Google Scholar]
- 45.Schoffstall B., Brunet N.M., Chase P.B. Ca2+ sensitivity of regulated cardiac thin filament sliding does not depend on myosin isoform. J. Physiol. 2006;577:935–944. doi: 10.1113/jphysiol.2006.120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urbancikova M., Hitchcock-DeGregori S.E. Requirement of amino-terminal modification for striated muscle α-tropomyosin function. J. Biol. Chem. 1994;269:24310–24315. [PubMed] [Google Scholar]
- 47.Kawai M., Brandt P.W. Sinusoidal analysis: a high resolution method for correlating biochemical reactions with physiological processes in activated skeletal muscles of rabbit, frog and crayfish. J. Muscle Res. Cell Motil. 1980;1:279–303. doi: 10.1007/BF00711932. [DOI] [PubMed] [Google Scholar]
- 48.Kawai M., Saeki Y., Zhao Y. Crossbridge scheme and the kinetic constants of elementary steps deduced from chemically skinned papillary and trabecular muscles of the ferret. Circ. Res. 1993;73:35–50. doi: 10.1161/01.res.73.1.35. [DOI] [PubMed] [Google Scholar]
- 49.Wannenburg T., Heijne G.H., De Tombe P.P. Cross-bridge kinetics in rat myocardium: effect of sarcomere length and calcium activation. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H779–H790. doi: 10.1152/ajpheart.2000.279.2.H779. [DOI] [PubMed] [Google Scholar]
- 50.Martyn D.A., Chase P.B., Gordon A.M. A simple model with myofilament compliance predicts activation-dependent crossbridge kinetics in skinned skeletal fibers. Biophys. J. 2002;83:3425–3434. doi: 10.1016/S0006-3495(02)75342-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang G.W., Ding W., Kawai M. Does thin filament compliance diminish the cross-bridge kinetics? A study in rabbit psoas fibers. Biophys. J. 1999;76:978–984. doi: 10.1016/S0006-3495(99)77261-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawai M., Halvorson H.R. Two step mechanism of phosphate release and the mechanism of force generation in chemically skinned fibers of rabbit psoas muscle. Biophys. J. 1991;59:329–342. doi: 10.1016/S0006-3495(91)82227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saeki Y., Kawai M., Zhao Y. Comparison of crossbridge dynamics between intact and skinned myocardium from ferret right ventricles. Circ. Res. 1991;68:772–781. doi: 10.1161/01.res.68.3.772. [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y., Swamy P.M., Kawai M. The effect of partial extraction of troponin C on the elementary steps of the cross-bridge cycle in rabbit psoas muscle fibers. Biophys. J. 1996;71:2759–2773. doi: 10.1016/S0006-3495(96)79469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spirito P., Seidman C.E., Maron B.J. The management of hypertrophic cardiomyopathy. N. Engl. J. Med. 1997;336:775–785. doi: 10.1056/NEJM199703133361107. [DOI] [PubMed] [Google Scholar]
- 56.Sagawa K., Maughan L., Sunagawa K. Oxford University Press; Oxford, UK: 1988. Cardiac Contraction and the Pressure-Volume Relationship. [Google Scholar]
- 57.Fozzard H.A., Haber E., Katz A.M. 2nd Ed. Raven Press; New York: 1992. The Heart and Cardiovascular System: Scientific Foundations. [Google Scholar]
- 58.Solaro R.J. Integration of myofilament response to Ca2+ with cardiac pump regulation and pump dynamics. Am. J. Physiol. 1999;277:S155–S163. doi: 10.1152/advances.1999.277.6.S155. [DOI] [PubMed] [Google Scholar]
- 59.Solaro R.J., de Tombe P.P. Review focus series: sarcomeric proteins as key elements in integrated control of cardiac function. Cardiovasc. Res. 2008;77:616–618. doi: 10.1093/cvr/cvn004. [DOI] [PubMed] [Google Scholar]
- 60.Solaro R.J., Wolska B.M., Tombe P.P.d. Modulation of thin filament activity in long and short term regulation of cardiac function. In: Solaro M.R., editor. Molecular Control Mechanisms in Striated Muscle Contraction. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2002. pp. 291–327. [Google Scholar]
- 61.Davis J.P., Tikunova S.B. Ca2+ exchange with troponin C and cardiac muscle dynamics. Cardiovasc. Res. 2008;77:619–626. doi: 10.1093/cvr/cvm098. [DOI] [PubMed] [Google Scholar]
- 62.Hanft L.M., Korte F.S., McDonald K.S. Cardiac function and modulation of sarcomeric function by length. Cardiovasc. Res. 2008;77:627–636. doi: 10.1093/cvr/cvm099. [DOI] [PubMed] [Google Scholar]
- 63.Hamdani N., Kooij V., van der Velden J. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2008;77:649–658. doi: 10.1093/cvr/cvm079. [DOI] [PubMed] [Google Scholar]
- 64.Michele D.E., Gomez C.A., Metzger J.M. Cardiac dysfunction in hypertrophic cardiomyopathy mutant tropomyosin mice is transgene-dependent, hypertrophy-independent, and improved by β-blockade. Circ. Res. 2002;91:255–262. doi: 10.1161/01.res.0000027530.58419.82. [DOI] [PubMed] [Google Scholar]
- 65.White S.P., Cohen C., Phillips G.N., Jr. Structure of co-crystals of tropomyosin and troponin. Nature. 1987;325:826–828. doi: 10.1038/325826a0. [DOI] [PubMed] [Google Scholar]
- 66.McKillop D.F.A., Geeves M.A. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chou P.Y., Fasman G.D. Conformational parameters for amino acids in helical, β-sheet, and random coil regions calculated from proteins. Biochemistry. 1974;13:211–222. doi: 10.1021/bi00699a001. [DOI] [PubMed] [Google Scholar]
- 68.Michele D.E., Metzger J.M. Physiological consequences of tropomyosin mutations associated with cardiac and skeletal myopathies. J. Mol. Med. 2000;78:543–553. doi: 10.1007/s001090000161. [DOI] [PubMed] [Google Scholar]
- 69.Coviello D.A., Maron B.J., Seidman C.E. Clinical features of hypertrophic cardiomyopathy caused by mutation of a “hot spot” in the α-tropomyosin gene. J. Am. Coll. Cardiol. 1997;29:635–640. doi: 10.1016/s0735-1097(96)00538-4. [DOI] [PubMed] [Google Scholar]
- 70.Thierfelder L., MacRae C., Seidman C. A familial hypertrophic cardiomyopathy locus maps to chromosome 15q2. Proc. Natl. Acad. Sci. USA. 1993;90:6270–6274. doi: 10.1073/pnas.90.13.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thierfelder L., Watkins H., Seidman C.E. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 72.Reference deleted in proof.
- 73.Reference deleted in proof.
- 74.Berlin J.R., Bassani J.W., Bers D.M. Intrinsic cytosolic calcium buffering properties of single rat cardiac myocytes. Biophys. J. 1994;67:1775–1787. doi: 10.1016/S0006-3495(94)80652-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sadoshima J., Xu Y., Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- 76.Obert P., Stecken F., Guenon P. Effect of long-term intensive endurance training on left ventricular structure and diastolic function in prepubertal children. Int. J. Sports Med. 1998;19:149–154. doi: 10.1055/s-2007-971897. [DOI] [PubMed] [Google Scholar]
- 77.Koide M., Carabello B.A., Zile M.R. Hypertrophic response to hemodynamic overload: role of load vs. renin-angiotensin system activation. Am. J. Physiol. 1999;276:H350–H358. doi: 10.1152/ajpheart.1999.276.2.h350. [DOI] [PubMed] [Google Scholar]
- 78.Belus A., Piroddi N., Poggesi C. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J. Physiol. 2008;586:3639–3644. doi: 10.1113/jphysiol.2008.155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Palmer B.M., Fishbaugher D.E., Maughan D.W. Differential cross-bridge kinetics of FHC myosin mutations R403Q and R453C in heterozygous mouse myocardium. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H91–H99. doi: 10.1152/ajpheart.01015.2003. [DOI] [PubMed] [Google Scholar]
- 80.Kawai M., Candau R. Muscle contraction and supplying ATP to muscle cells. In: Connes P., Hue O., Perrey S., editors. Handbook of Exercise Physiology—From a Cellular to an Integrative Approach. IOS Press; Amsterdam, The Netherlands: 2010. pp. 1–23. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.