

Abstract
Thrombospondin-1 (TSP1) binding to calreticulin (CRT) on the cell surface signals focal adhesion disassembly leading to the intermediate adhesive phenotype, cell migration, anoikis resistance, and collagen stimulation. Residues Lys 24 and 32 in TSP1 and amino acids 24-26 and 32-34 in CRT have been shown through biochemical and cell-based approaches to be critical for TSP1-CRT binding and signaling. This study investigated the molecular and structural basis for these key TSP1 and CRT residues on TSP1-CRT binding. Based on a validated TSP1-CRT complex structure, we adopted steered molecular dynamics simulations to determine the effect of mutation of these key residues on TSP1-CRT binding and validated the simulation results with experimental observations. We further performed 30 ns molecular dynamics simulations for wild type TSP1, CRT, K24A & K32A mutant TSP1, and mutant CRT (residues 24-26 & 32-34 mutated to Ala) and studied the conformational and structural changes in TSP1 and CRT as the result of mutation of these critical residues. Results showed that mutation of residues 24 and 32 to Ala in TSP1 and of amino acids 24-26 and 32-34 to Ala in CRT result in a shortened β strand in the binding site, decreased hydrogen bond occupancy for β strand pairs that are located within or near the binding site, increased conformational flexibility of the binding site, a changed degree of dynamic correlated motion between the residues in the binding site and the other residues in protein, and a changed degree of overall correlated motions between the residues in protein. These changes could directly contribute to the loss or reduced binding between TSP1 and CRT and the resultant effects on TSP1-CRT binding induced cellular activities. Results from this study provide a molecular and structural insight for the role of these critical residues in TSP1 and CRT in TSP1-CRT binding.
Keywords: thrombospondin-1, calreticulin, critical binding residues, structural basis, molecular dynamics simulation
Cell adhesion between cells and the extracellular matrix is a multi-step process that is initiated by the binding of a ligand in the extracellular matrix to a cell surface receptor, triggering cell spreading and the formation of focal adhesions and stress fibers (1). Cell-matrix adhesion is also a reversible process in which a cell moves from a state of stronger adherence to a state of weaker adherence (1, 2). Intermediate adhesion is an adaptive condition and is characterized by a restructuring of focal adhesions and stress fibers while maintaining a spread cell shape, i.e. focal adhesion disassembly (1). Thrombospondin-1 (TSP1) is a protein in the extracellular matrix and in soluble form that interacts with the cell surface protein calreticulin (CRT) to signal intermediate adhesion and regulate cell migration, anoikis resistance, and collagen synthesis (3-9). TSP1 is a multifunctional matricellular protein that does not directly provide structure to the extracellular matrix, but interacts with cells and modifies cellular functions (10, 11). It is a large (420 kDa) disulfide-linked homotrimeric glycoprotein, with each monomer of TSP1 is composed of N and C-terminal globular domains connected by a rod-like segment (12). CRT, a ubiquitous calcium-binding protein, is composed of a globular β-sandwich N-domain, a proline-rich β-hairpin P-domain, and a calcium-binding C-domain (13). The conformation of CRT P-domain shows a spiral-like shape and indicates conformational flexibility of the CRT-P domain (14, 15). The N-terminal domain of TSP1 binds to the N-domain of CRT to enhance CRT binding to LDL receptor-related protein complex (LRP1), signaling intermediate adhesion, cell migration, anoikis resistance, and collagen synthesis in endothelial cells and fibroblasts (3-9). The CRT binding site in TSP1 has been localized to amino acids 17-35 (ELTGAARKGSGRRLVKGPD) and its functions can be mimicked by a peptide of this sequence, the hep I peptide (16). Two lysine residues in the hep I peptide (residues of 24 and 32 of TSP1 N-domain) are critical for hep I peptide binding to CRT (3, 16). The CRT binding site for TSP1 is an 18 residue sequence (CRT19.36: RWIESKHKSDFGKFVLSS) in the N-terminal domain of CRT (4). Two clusters of basic amino acids 24-26 and amino acids 32-34 in this sequence of CRT are critical for TSP1 binding and function (7). The crystal structure of the TSP1 N-domain has been solved (17). A validated three-dimensional structural model of CRT with N-domain, P-domain and partial C-domain has been constructed based on the crystal structure of calnexin and the NMR structure of the P-domain of CRT (14, 18, 19). A validated complex of TSP1 N-domain with CRT was constructed (20). The availability of the structures of TSP1 N-domain, CRT and the complex of TSP1 N-domain and CRT provide a basis to study the molecular and structural basis for the role of the key residues in TSP1 and CRT in TSP1-CRT binding.
In our recent studies, we investigated the binding thermodynamics of the TSP1-CRT complex and the conformational changes in CRT induced by binding to TSP1 through combined binding free energy analysis, molecular dynamics simulation and anisotropic network model restrained molecular dynamics simulation (20). We constructed a validated TSP1-CRT complex as shown in Figure 1S. We performed binding free energy calculations for the wildtype TSP1-CRT complex, the TSP1 mutant-CRT complex with residues of Lys 24 and Lys 32 of TSP1 mutated to Ala (TSP1 K24A &K32 A mutant), and the TSP1-CRT mutant complex with residues 24-26 and 32-34 of CRT mutated to Ala, based on the 30 ns MD simulation trajectories. The calculated results of the binding free energy showed that either the K24A and K32A mutations in TSP1 or the mutation of residues 24-26 and 32-34 of CRT to Ala significantly decreased the TSP1-CRT complex binding, which was consistent with previous experimental observations (3, 7, 16). Analysis of the binding free energy components showed that mutations in TSP1 and CRT resulted in the significant changes in the electrostatic energy, van der Waals energy, polar solvation energy, and entropy of the TSP1-CRT complex, which results from the charge changes in the TSP1-CRT protein complex and the conformational changes in TSP1 and CRT induced by TSP1-CRT interactions. Conformational analyses showed that TSP1 binding to CRT resulted in a more “open” conformation and a significant rotational change for CRT N-domain with respect to CRT P-domain, which could expose the potential binding site(s) in CRT for binding to LRP1 to signal focal adhesion disassembly. Although we characterized the effect of the K24A and K32A mutations in TSP1 and the mutation of residues 24-26 and 32-34 of CRT to Ala on TSP1-CRT binding on binding thermodynamics, the effects of these binding site mutations on the structure of TSP1 and CRT and the impact of these structural alterations on TSP1-CRT interactions have not been investigated.
In this study, we simulated the effect of the key residues in TSP1 and CRT on TSP1-CRT binding with steered molecular dynamics simulations, which were consistent with previous biochemical data (3, 4, 7). We performed 30 ns molecular dynamics (MD) simulations for wild type TSP1, CRT, TSP1 mutant (residues 23 and 32 mutated to Ala), and mutant CRT (residues 24-26 & 32-34 mutated to Ala) and studied the conformational and structural changes in TSP1 and CRT as the result of mutation of these critical residues. Results from this study provide molecular and structural insight into the role of these critical residues of TSP1 and CRT in TSP1-CRT binding that signals focal adhesion disassembly.
Materials and Methods
Steered molecular dynamics simulations
To evaluate the effects of mutations of residues 24 and 32 to Ala in TSP1 and of amino acids 24-26 and 32-34 to Ala in CRT on TSP1/CRT binding, steered molecular dynamics (SMD) simulations with constant velocity protocol was used to probe the unbinding procedure of TSP1-CRT complex, TSP1 K24A&K32A mutant-CRT complex and TSP1-CRT mutant complex. We used NAMD 2.6 MD package (21) for SMD simulations, and AMBER force field was used for the simulated systems. The validated TSP1-CRT complex obtained from the previous study (20) and its mutant were separately solvated with TIP3P water molecules (22) and a 150 mM NaCl physiological salt concentration in a periodic box, and 1 nm of solvent between the protein and the box boundaries was ensured to reduce potential artifacts arising from periodicity. Before SMD simulations, we performed energy minimization and equilibration of the water, and the warm up of the simulated system with a standard protocol similar to that of our previous MD simulation studies (23-27). For the system warmed up to 300K, we further implement the SMD simulations to probe the effect of mutations of residues 24 and 32 to Ala in TSP1 and of amino acids 24-26 and 32-34 in CRT to Ala on the unbinding of TSP1-CRT complex. Instead of applying the pulling force only on one point to mimic the atomic force microscopy (AFM) as in previous studies (28-30), which could break the secondary structure of the protein, we applied the pulling forces at a constant speed of 0.2 Å/ps, which was chosen as a balance of the complex unbinding procedure and proper simulation time scale, on the Ca atom of each residue on beta sheets of TSP1 except the residues in CRT binding site. The residues in the structurally stable N-domain and the partial C-domain of CRT, except the residues in the binding site for TSP1, were treated as rigid and were fixed, and leave the structurally flexible P-domain of CRT as unfixed and flexible region (Fig. 1). The pulling direction was along the center of the mass of the pulled residues and the fixed residues. Force experienced by the Ca atom of the pulled residues was calculated with Equation (4) (29):
FIGURE 1.
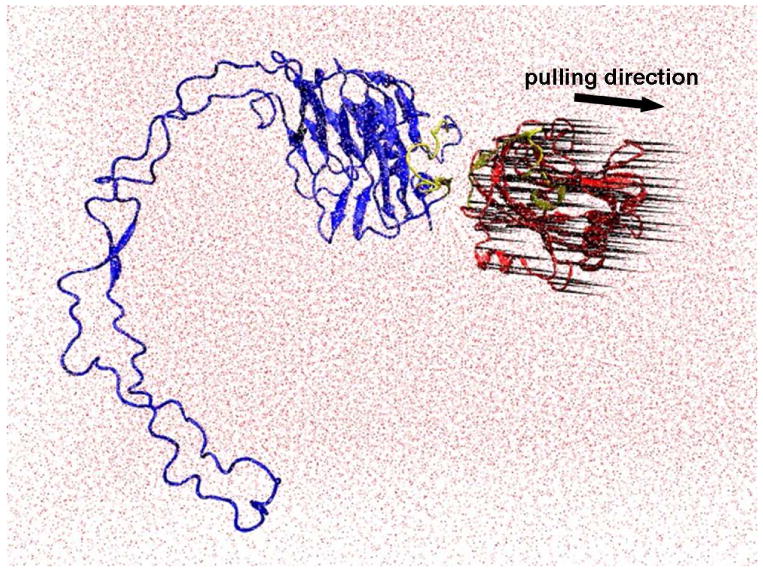
Setup of SMD simulation. TSP1-CRT complex was solvated in water. Pulling force was applied on the alpha carbons over beta sheets of TSP1 except the residues in the binding site for CRT. The residues in structurally stable N- domain and the partial C-domains of CRT, except the residues in the binding site for TSP1, were treated as rigid body and were fixed, and the structurally flexible P-domain of CRT was left as unfixed and flexible region. The pulling direction was along the center of the mass of the pulled residues and the fixed residues. Red: TSP1, blue: CRT, yellow: binding sites (TSP1 binding site for CRT is localized to amino acids 17-35: ELTGAARKGSGRRLVKGPD and the CRT binding site for TSP1 is an 18 residue sequence of CRT19.36: RWIESKHKSDFGKFVLSS), red dot: water. The black “porcupine needles” indicate the direction of pulling force applied on the alpha carbons over beta sheets of TSP1. The images were made with VMD software support.
| (1) |
Where K is the spring constant, set as 7kcal/mol/Ǻ2 (31), v is pulling velocity, t is time, and x is the movement of the pulled Ca atom from its original position. The force extension profiles over time experienced by TSP1-CRT complex and its mutant were compared to determine the effect of residues K24A and K32A mutations of TSP1 and the mutation of residues 24-26 and 32-34 to Ala in CRT on the unbinding ability of TSP1-CRT complex. The calculated result was compared with the experimental data for the validation.
In addition to probing the force extension profile experienced by TSP1-CRT complex and its mutant with constant velocity SMD simulations, we also performed constant force SMD by applying constant force to the TSP1-CRT complex and its mutant to obtain the extension profile over time to determine the change of the extension profile of TSP1-CRT complex by the residues K24A and K32A mutations of TSP1 and the mutation of residues 24-26 and 32-34 to Ala in CRT. We compared the simulation results with the experimental results of biochemical studies (3, 16) for the further validation (available in Supporting Information).
Molecular dynamics simulations
We performed 30 ns MD simulations for TSP1 N-domain (TSP1) wildtype, TSP1 K24A & K32A mutant, CRT wildtype and CRT mutant with mutations of residues 24-26 and 32-34 to Ala (Table 1) to assess the conformational and structural changes of TSP1 N-domain and CRT resulted from the mutations of residues 24 and 32 to Ala in TSP1 and of amino acids 24-26 and 32-34 in CRT to Ala, thus providing molecular and structural insight for the role of the critical residues in TSP1 and CRT in TSP1-CRT binding. We used the AMBER 9 MD package (32) for the MD simulations. The MD simulations were performed in a periodic box (the size of the box depends on the simulated system). 1 nm of solvent between the protein and the box boundaries was ensured to reduce potential artifacts arising from periodicity. The periodic box was filled with TIP3P water molecules (22) and 150 mM NaCl (physiological salt concentration). Additional ions of Na+ or Cl- were added to the system to neutralize the charge of the protein complex. The AMBER force field was used for the simulated systems in combination with a standard MD simulation protocol similar to that of our previous studies (23-27). Briefly, the MD simulation protocol included: 1) steepest descent minimization for the solvent with the protein and ions restrained but with water mobile; 2) equilibration of water with mobile water molecules but with the protein and ions restrained at constant number-pressure-temperature (NpT) at 50K and 1 atm for 20 ps; 3) the warm up of the system via a series of 10 ps constant number-volume-temperature (NVT) MD simulations at 50, 100, 150, 200, 250, and 300K with SHAKE constraints and 2 fs time steps; 4) production simulation at NpT of 300K and 1 atm for the assigned time length of 30 ns in this study. In the production simulations, SHAKE constraints with relative tolerance of 1×10-5 were used on all hydrogen-heavy atom bonds to permit a dynamics time step of 2 fs. Electrostatic interactions were calculated by the particle-mesh Ewald method (PME) (33). The Lennard-Jones cutoffs were set at 1.0 nm. RMSD of each simulated system was calculated over the MD simulations to make sure that the system reached equilibration.
Table 1. Four systems for 30ns MD simulations.
| System | TSP1 N-domain | CRT |
|---|---|---|
| 1 | TSP1 wild type | |
| 2 | TSP1 K24A & K32A mutant | |
| 3 | CRT wild type | |
| 4 | CRT mutant with mutations of residues 24-26 and 32-34 to Ala |
Conformational and structural analyses
With the MD simulation trajectories after equilibration, we performed conformational analysis, secondary structure and hydrogen bond analysis to understand the molecular and structural basis for the critical role of TSP1 residues 24 and 32 and CRT residues 24-26 and 32-34 in TSP1-CRT binding. We calculated the root mean square fluctuations (RMSFs) of TSP1 N-domain (TSP1) wildtype, TSP1 K24A & K32A mutant, CRT wildtype and CRT mutant with mutations of residues 24-26 and 32-34 to Ala. In addition to RMSF analyses, we also analyzed the dynamical cross-correlation map between the residues of TSP1, CRT and their mutants. Through these analyses, we investigated the changes of the conformational flexibility and the degree of correlated motion between residues in the protein by the mutation of the key residues. We calculated the changes of the hydrogen bond formation in the β sheet region and the changes of the residues of β strand occupancy for TSP1 N-domain and CRT to evaluate structural changes of the protein by the key residues' mutation. A hydrogen bond was assigned when the distance of hydrogen and acceptor was less than 4 Å, and the angle of donor – hydrogen – acceptor is less than 30°. OH and NH groups were treated as donors, oxygen or nitrogen was defined as acceptor. The occupancy of each hydrogen bond was calculated based on the percentage of time that the hydrogen bond existed over the entire simulation time. The hydrogen bond occupancy of each β- strand pair in TSP1 N-domain and CRT was considered to be the average occupancy of the hydrogen bonds of each β-strand pair. A β strand is typically 3 to 10 amino acids long with backbone in an almost fully extended conformation; however, β strands are rarely perfectly extended. If the dihedral angles (φ, ψ) of an amino acid with its adjacent residues were near −135° and 135°, the amino acid was assigned to be contained in the β strand. The occupancy of each residue in β strand was determined based on the percentage of time that the residue existed in β strand over the simulation.
Results and Discussion
Effect of the mutations of the key residues in TSP1 and CRT on TSP1-CRT binding from SMD simulation
We first validated the SMD simulation results about the effect of the mutations of the key residues in TSP1 and CRT on TSP1-CRT binding with the experimental observation (3, 4, 7). Under constant velocity SMD simulations, it was shown that around 60 ps, the buried protein surface area (BPSA) for the TSP1-CRT complex was decreased from 2142 Å2 to 1482 Å2, the BPSA of TSP1 K24A & K32A mutant-CRT complex was decreased from 2182 Å2 to 1279Å2 and the BPSA of the TSP1-CRT mutant complex with residues 24-26 and 32-34 of CRT mutated to Ala was decreased from 2160Å2 to 1087 Å2 (Fig. 2 A). The significantly decreased BPSA indicated the reduced binding of TSP1 and CRT. The force required to break the binding of the wildtype TSP1-CRT complex (around 4000 pN) was much larger than that of the TSP1 mutant-CRT complex and of the TSP1-CRT mutant complex (around 3000 pN) (Fig. 2B). These calculated results showed that mutation of residues 24 and 32 to Ala in TSP1 and of amino acids 24-26 and 32-34 to Ala in CRT resulted in decreased binding between TSP1 and CRT. TSP1 K24A & K32A mutations and mutations of amino acids 24-26 and 32-34 to Ala in CRT directly affect the electrostatic interactions between TSP1 and CRT as observed in our previous thermodynamics study of the TSP1-CRT complex (20). In addition to the effect on electrostatic interactions between TSP1 and CRT, TSP1 K24A & K32A mutations and mutations of amino acids 24-26 and 32-34 to Ala in CRT caused significant changes in van der Waals energy, polar solvation energy, and entropy of the TSP1-CRT complex, and thereby, significantly decreased the binding free energy of the TSP1-CRT complex (20). Experimental studies also observed that a peptide with mutation of residues 24 and 32 to Ala does not signal focal adhesion disassembly or stimulate collagen (9). Simple electrostatic interactions are not the sole determinant of binding since a scrambled CRT binding sequence peptide (scrambled hep I) does not signal focal adhesion disassembly nor does it block TSP1 stimulation of collagen (9, 16). Our previous binding thermodynamic results (20) help to interpret the results from the SMD simulations that TSP1 K24A & K32A mutations and mutations of amino acids 24-26 and 32-34 to Ala in CRT decreased the resistance to force-induced dissociation of the TSP1-CRT complex. Biochemical binding studies (immunoprecipitation) have been used to show that purified TSP1 and CRT bind to each other and that complex formation can be disrupted by the hep I peptide, but not by the hep I peptide mutated in at residues 24 and 32 to Ala (3). Furthermore, CRT null cells engineered to express CRT lacking the TSP1 binding site were refractory to TSP1-induced signaling of resistance to anoikis and a peptide of the TSP binding sequence of CRT, but not the mutated binding sequence, blocks TSP1 stimulation of collagen expression (4, 7, 9). The calculated results obtained in these current studies are thus consistent with these previous biochemical and cell-based studies on the role of these sequences in mediating TSP1-CRT binding and signaling (3, 4, 7). Moreover, the CRT mutations could also affect CRT binding to LRP1, thereby, disrupting the formation of the TSP1-CRT-LRP1 ternary complex for signaling the cellular activities (6).
FIGURE 2.


(A) Buried protein surface area (BPSA) for the TSP1-CRT complex, the TSP1 K24A & K32A mutant-CRT complex and the TSP1-CRT mutant over the course of constant velocity SMD simulation. (B) Force experienced in the TSP1-CRT complex, the TSP1 K24A & K32A mutant-CRT complex and the TSP1-CRT mutant over the course of constant velocity SMD simulation.
Under the constant force SMD simulations, the extension profile for the TSP1-CRT complex, the TSP1 K24A & K32A mutant-CRT complex and the TSP1-CRT mutant complex showed that under the same external load pulling, it took a longer time to significantly increase the distance between the centers of the mass of the binding sites of the TSP1-CRT complex for the wildtype TSP1-CRT complex than its mutants (Fig. 2S). The structure of TSP1-CRT complex with the binding sites of the TSP1-CRT complex highlighted is shown in Figure 1S. During the calculated disruption of the TSP1-CRT complex(s) under external load pulling, the relative rotational motion between TSP1 and CRT could increases, and therefore, it is not surprising that the distance between the centers of the mass of the binding sites of TSP1 K24A & K32A mutant-CRT complex decreased before unbinding as observed in Fig. 2S. The results showed that TSP1 K24A & K32A mutations and mutations of amino acids 24-26 and 32-34 to Ala in CRT significantly decreased the binding strength between TSP1 and CRT, consistent with the biochemical and cell biological experimental results (3, 4, 7).
Conformational and structural changes in TSP1 caused by the mutations of residues 24 and 32 to Ala
Conformational changes in TSP1
The RMSD of TSP1 and the K24A & K32A mutant over the 30 ns MD simulations exhibited initial equilibration after 10 ns of MD simulations (Fig. 3S). The simulation trajectories from 10 ns to 30 ns were used for conformational and structural analyses. The crystal structure of the TSP1 N-domain shows that the TSP1 binding site for CRT spans the α1 helix to the β2 strand, which is a flexible and exposed loop region with a short β strand as shown in Figure 1S (17). For the wild type TSP1 N-domain and the TSP1 K24A and K32A mutant, the root mean square fluctuation (RMSF), calculated by averaging MD trajectories from 10 ns to 30 ns, showed that TSP1 K24A and K32A mutations resulted in the changes in TSP1 conformational flexibility, including an increased conformational fluctuation of the TSP1 binding site for CRT and the decreased fluctuation for residues 94-95, 108, 120, 161-163 (Fig. 3 A). The increased fluctuation of the TSP1 binding site for CRT and the changed fluctuation of other residues could directly affect the electrostatic interactions and van der Waals interactions between TSP1 and CRT as observed in our previous thermodynamics study (20), including the prevention of the specific side chain interactions at the binding sites from being formed. These changes could contribute to the loss of binding between TSP1 and CRT as observed in biochemical experiments (3, 16). The changed conformational fluctuation of TSP1 in the regions other than binding site of CRT in TSP1 could also affect binding to other ligands for the TSP1 N domain such as integrins. The dynamical cross-correlation maps for TSP1 and its mutant are shown in Fig. 3 B, in which wild type TSP1 is represented in the top left half and the TSP1 K24A and K32A mutant in the bottom right half. Dynamical cross-correlation mapping showed that K24A and K32A mutations in TSP1 resulted in an overall decreased degree of correlated motion between the residues in TSP1, evidenced by less red patches (correlated motions) and blue patches (anti-correlated motions) compared to those for wildtype TSP1. Results also showed that the degree of correlated motion between the residues in the TSP1 binding site for CRT and the other residues in CRT was reduced by the K24A and K32A mutations in TSP1 (circled in Fig. 3 B). Our previous study of the binding thermodynamics of the TSP1-CRT complex showed that the TSP1 K24A and K32A mutations significantly decreased the binding free energy of TSP1-CRT complex, including the significant changes in the electrostatic energy, van der Waals energy, polar solvation energy, and entropy of the TSP1-CRT complex (20). These changes in the degree of the correlated motion for the residues in TSP1 and in the conformational flexibility of TSP1 by TSP1 K24A and K32A mutations could directly cause the changes in the electrostatic energy, van der Waals energy, polar solvation energy, and entropy for TSP1 interactions with CRT, and thereby, contribute to loss of TSP1 and CRT binding.
FIGURE 3.
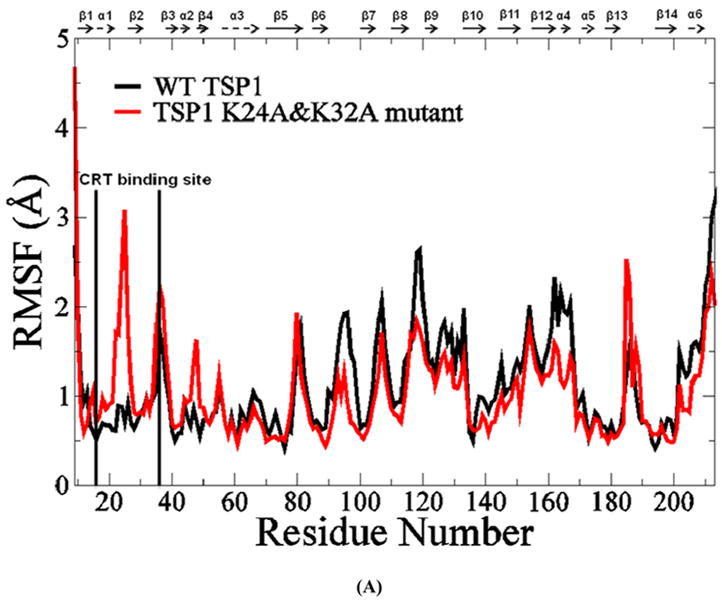
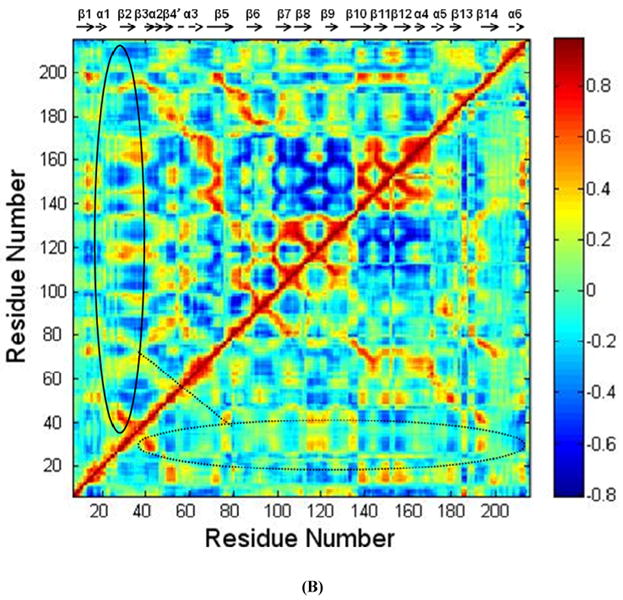
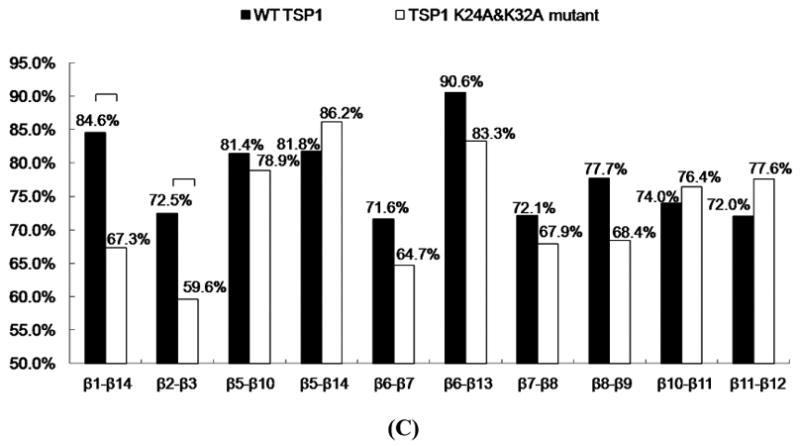
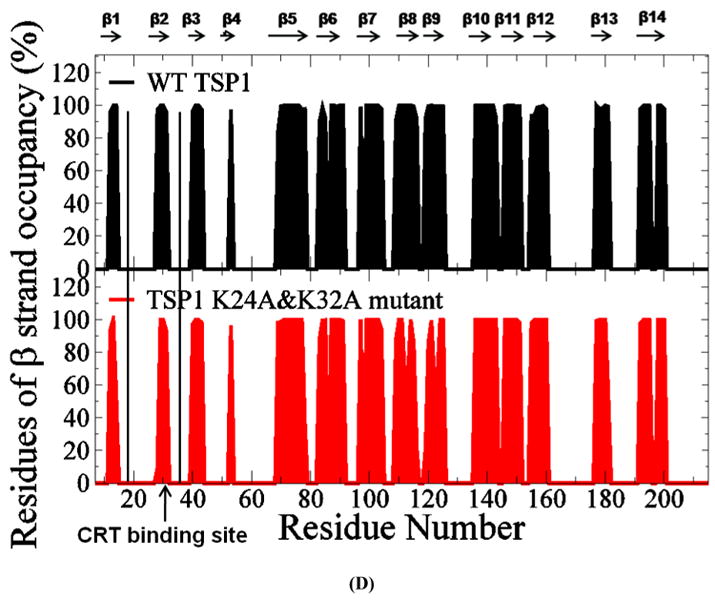
(A) Root mean squared fluctuation (RMSF) of TSP1 N-domain. β strands and α helices corresponding to the residues in TSP1 were marked on the top of RMSF figure. (B) Dynamical cross-correlation map for the degree of correlated motion of the residues in TSP1 (red: correlated motion between residues; blue: anti-correlated motion between residues; circled region showed the correlated motion between the residues in the binding site of TSP1 for CRT and the other residues of TSP1). Wild type TSP1 (top left) compared to TSP1 K24A & K32A mutant (right bottom). β strands and α helices corresponding to the residues in TSP1 were marked on the top of the dynamical cross-correlation map. (C) Hydrogen bond occupancy of each β strand pair in TSP1. (D) Residues of β strand occupancy in TSP1.
Changes of hydrogen bond and β strand formations in TSP1
The globular TSP1 N-domain is primarily composed of 13 antiparallel β strands, one irregular strand-like segment, and six α helices that are located either between β strands or at the C-terminal area (17). The hydrogen bonds formed between β strands play an important role in protein structural stability. Analysis of hydrogen bond formation between β strands and their occupancy over the last 20 ns of MD simulation trajectories showed that the TSP1 K24A and K32A mutations resulted in a more than 10% decrease in hydrogen bond occupancy between the β1 and β14 strands and the β2 and β3 strands when compared to the wildtype TSP1 N-domain (Fig. 3 C). Structures of wild-type TSP1 N-domain and TSP1 N-domain K24A & K32A mutant to compare β1-14 and β2-3 strands of the wild-type and mutant TSP1 were shown as Figure 5S and Figure 6S. The β2 strand is located within the region of residues 17-35 that is found in the TSP1 binding site for CRT, and the β3 strand is adjacent to β2 strand (17). The β1 strand is connected to the β2 strand through the α1 helix, and the β14 strand is adjacent to the β1 strand (17). The significantly decreased hydrogen bond occupancy between the β2 and β3 strands and between the β1 and β14 strands caused by TSP1 K24A and K32A mutations (Fig. 3 C and Table 1S) could affect the conformational stability of the TSP1 binding site for CRT and the overall conformational stability of TSP1.
Analyses of the β strand formations over the last 20ns MD simulation trajectories showed that TSP1 K24A and K32A mutations resulted in the shortened β2 strand (Fig. 3 D). The residues in β2 strand included amino acids of R28, R29, L30 and V31. The occupancy of the four residues in β2 strand was 98%, 100%, 100% and 96% for wild-type TSP1, and was 9%, 100%, 100% and 93% for TSP1 K24A and K32A mutant. The amino acid of R28 was rarely in β2 strand by TSP1 K24A and K32A mutations, resulting in the shortened β2 strand. The shortened β2 strand could contribute to the decrease of hydrogen bond occupancy between β2 and β3 by TSP1 K24A and K32A mutations as observed in Fig. 3 C and Table 1S. The changed hydrogen bond and β strand formation by TSP1 K24A and K32A mutations could contribute to the changes of TSP1 conformation as observed in RMSF and dynamical cross-correlation analyses and also provide the structural and molecular basis of the observed loss of protein binding and cell function upon mutation of TSP1 Lys 24 and 32 to Ala (3, 16).
Conformational and structural changes in CRT caused by the mutations of amino acids 24-26 and 32-34 to Ala
Conformational changes in CRT
The RMSD of CRT and the CRT mutant over the 30 ns MD simulations showed that the system reached initial equilibration after 10 ns of MD simulations (Fig. 4S). The TSP1 binding site in CRT has been localized to the sequence of residues 19-36 in the N domain of CRT (4), and CRT mutated at residues 24-26 and 32-34 to Ala is unable to mediate anoikis resistance or focal adhesion disassembly upon stimulation with TSP1(7). The structure of CRT showed that the CRT binding site for TSP1 covers regions of the β2 strand in CRT (19). RMSF calculated over the last 20 ns MD simulation trajectories for both the wildtype CRT and CRT mutant showed that the mutations of CRT residues 24-26 and 32-34 to Ala resulted in the increased conformational fluctuation of the CRT binding site for TSP1 and its adjacent regions, and resulted in the increased conformational fluctuation of the middle region of CRT P-domain (Fig. 4 A), which could affect CRT binding to TSP1 as that observed in the experiments (7). The dynamical cross-correlation maps for CRT and its mutant was shown in Fig. 4 B, in which the top left half represents wild type CRT and the bottom right half, the CRT mutant. Results showed that mutations of CRT residues 24-26 and 32-34 to Ala resulted in an increased correlated movement (red) within the N-domain of CRT (residues (res) 1-180) and an increased anti-correlated motion (blue) between N-domain of CRT (res 1-180) and P-domain of CRT (res 181-290) as compared to those for wildtype CRT. The changed degree of correlated motion by the mutations of residue 24-26 and 32-34 to Ala was also observed for the CRT binding site for TSP1 relative to the other residues of CRT (circled in Fig. 4 B). These changes in the degree of the correlated motion for the residues in CRT and in the conformational flexibility of CRT TSP1 by the mutations of residue 24-26 and 32-34 to Ala could directly result in the changes in the electrostatic energy, van der Waals energy, polar solvation energy, and entropy for CRT interactions with TSP1, affecting TSP1 and CRT binding as observed in our previous thermodynamics study (20) and in cell-based experiments (7).
FIGURE 4.
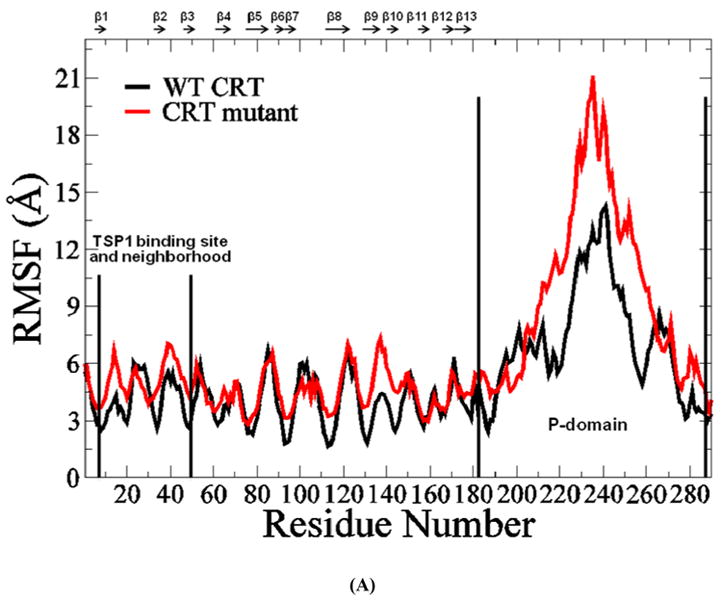
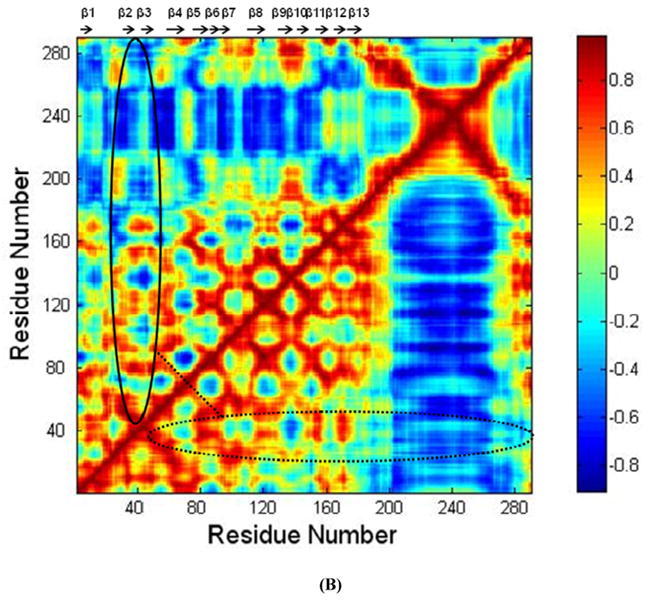
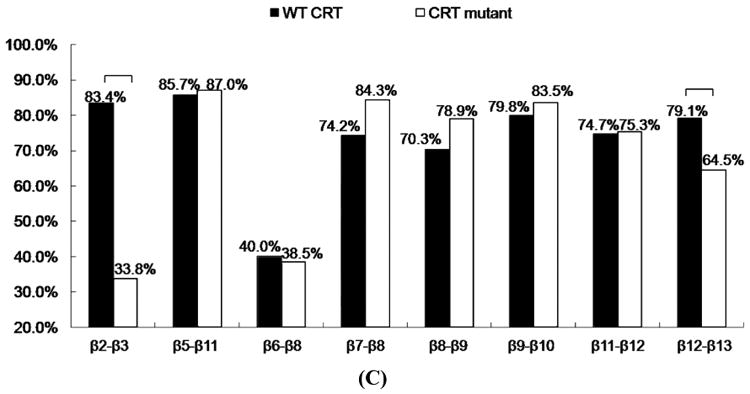
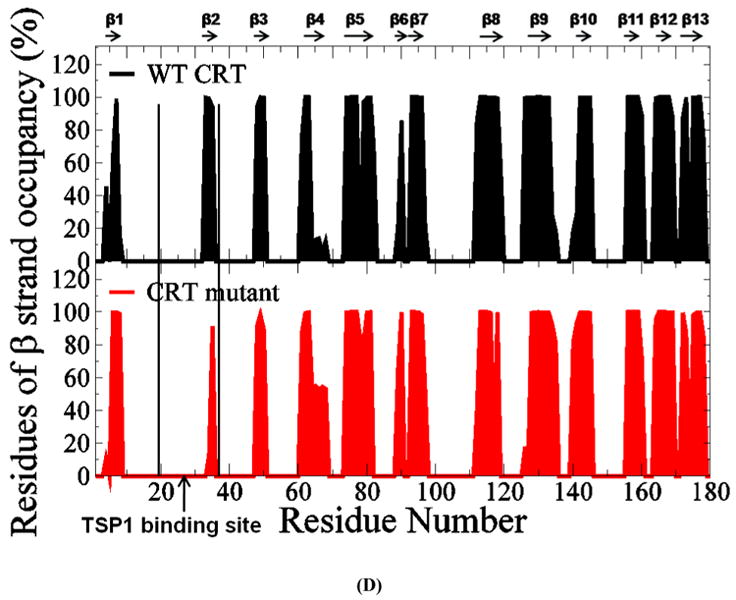
(A) RMSF of CRT. β strands corresponding to the residues in CRT were marked on the top of RMSF figure. (B) Dynamical cross-correlation map for the degree of correlated motion of the residues in CRT (red: correlated motion; blue: anti-correlated motion; circled region: correlated motion between the residues in the binding site of CRT for TSP1 and other residues of CRT). Wild type CRT (top left) compared to CRT mutant (res 24-26 & 32-34 mutated to Ala) (right bottom). β strands corresponding to the residues in CRT were marked on the top of the dynamical cross-correlation map. (C) Hydrogen bond occupancy of each β strand pair in CRT. (D) Residues of β strand occupancy in CRT.
Changes of hydrogen bond and β strand formations in CRT
The CRT structural model shows that CRT is mainly composed of a structurally stable globular N-domain formed by antiparallel β sheets and a flexible P-domain with an extended arm structure (19). The CRT binding site for TSP1 (res 19-36) is located within the CRT N-domain and spans through β2 strand (4, 19). We analyzed the hydrogen bonds and their occupancy between β strands over the last 20ns MD simulations for wildtype CRT and CRT mutant (res 24-26&32-34 mutated to Ala). Almost 50% decreased hydrogen bond occupancy between β2 and β3 strands and more than 10% decreased hydrogen bond occupancy between β12 and β13 strands were resulted from the mutations of CRT res 24-26&32-34 to Ala (Fig. 4 C and Table 2S).
Analyses of the β strand formations over the last 20ns MD simulation trajectories showed that mutations of CRT residues 24-26 and 32-34 to Ala resulted in the shortened β2 strand (Fig. 4 D). The residues in β2 strand included amino acids of V33, L34 and S35. The occupancy of the three residues in β2 strand was 100%, 99%, and 95% for wild-type CRT, and was 0%, 13%, and 91% for CRT mutant. The amino acids both V33 and L34 were rarely in β2 strand by the mutations of CRT res 24-26&32-34 to Ala, causing the shortened β2 strand. The shortened β2 strand could contribute to the decreased hydrogen bond occupancy between β2 and β3 strands by mutations of residue 24-26 and 32-34 to Ala in CRT as observed in Fig. 4 C and Table 2S. β2 strand (res 33-35) is located within the CRT binding site for TSP1 and β12 and β13 strands were located near the CRT binding site for TSP1 (19). The changed β2 strand and significantly decreased hydrogen bond occupancy between β2 and β3 strands and between β12 and β13 strands by the mutations of CRT residues 24-26 and 32-34 to Ala could change the conformational stability of the CRT binding site for TSP1 and the overall conformational stability of CRT as observed in RMSF and dynamical cross-correlation analyses, which could reduce binding of TSP1 and CRT. These results provide a molecular and structural basis for the experimental observation that the mutations of CRT res 24-26 and 32-34 to Ala decrease TSP1-CRT binding and reduce signaling of anoikis resistance (7).
Conclusions
In this study, we used simulation results examining the effect of mutation of key residues in TSP1 and CRT on TSP1-CRT binding to validate experimental biochemical observations. Using MD simulations, we elucidated the molecular and structural basis for the critical role of residues 24 and 32 in TSP1 and residues 24-26 and 32-34 in CRT for TSP1-CRT binding as observed in biochemical studies. Mutations of residues 24 and 32 to Ala in TSP1 and mutations of residues 24-26 and 32-34 to Ala in CRT all resulted in increased conformational flexibility of the binding site and an overall changed conformational flexibility of the protein, a changed degree of dynamical correlated motion between the residues in the binding site and other residues in protein, and a changed degree of overall correlated motions between the residues in protein. Mutation of the key residues in TSP1 and CRT also caused shortening of the β strand in the binding site and decreased hydrogen bond occupancy for β strand pairs within or near its binding site for the other protein. These changes could directly result in alterations in the electrostatic energy, van der Waals energy, polar solvation energy, and entropy for TSP1 interactions with CRT, contributing to the loss of or reduced binding of the TSP1-CRT complex with resultant attenuation of TSP1-CRT binding induced cellular activities. These studies provide new knowledge regarding the structural basis of these interactions with the potential for developing drug candidates and other strategies for the allosteric regulation of TSP1-CRT binding and its induced cellular activities.
Supplementary Material
Acknowledgments
We acknowledge Dr. Marek Michalak for providing PDB file of calreticulin. The authors would like to thank the anonymous reviewers for their helpful remarks.
This work was supported by TeraGrid supercomputer allocation (National Science Foundation Grant MCB090009) to Y.S. and National Institutes of Health Grant HL79644 to J.E.M.-U.
Abbreviations
- TSP1
thrombospondin 1
- CRT
calreticulin
- LRP1
LDL receptor-related protein complex
- MD
molecular dynamics
- SMD
steered molecular dynamics
- RMSD
root mean square deviation
- RMSF
root mean square fluctuation
Footnotes
Supporting Information Available: The constructed TSP1-CRT complex in our previous study was shown as Figure 1S. The simulation protocol and the results of constant force SMD for TSP1-CRT complex, TSP1 K24A&K32A mutant-CRT complex and the TSP1-CRT mutant complex were described (Figure 2S). RMSD of TSP1 and its mutant and RMSD of CRT and its mutant over the MD simulations showed that that the system reached the initial equilibration after 10 ns of MD simulations (Figure 3S and Figure 4S). Structures of wild-type TSP1 N-domain and TSP1 N-domain K24A & K32A mutant were shown to compare the β1-14 and β2-3 strands of the wild-type and mutant TSP1 (Figure 5S and Figure 6S). Occupancy of the hydrogen bond formed between residues in the β strands of TSP1 and its mutant over the last 20ns MD simulation trajectories was shown in Table 1S. Occupancy of the hydrogen bond formed between residues in the β strands of CRT and its mutant over the last 20ns MD simulation trajectories was shown in Table 2S. The supporting material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Murphy-Ullrich JE. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest. 2001;107:785–790. doi: 10.1172/JCI12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenwood JA, Murphy-Ullrich JE. Signaling of de-adhesion in cellular regulation and motility. Microsc Res Tech. 1998;43:420–432. doi: 10.1002/(SICI)1097-0029(19981201)43:5<420::AID-JEMT8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 3.Goicoechea S, Orr AW, Pallero MA, Eggleton P, Murphy-Ullrich JE. Thrombospondin mediates focal adhesion disassembly through interactions with cell surface calreticulin. J Biol Chem. 2000;275:36358–36368. doi: 10.1074/jbc.M005951200. [DOI] [PubMed] [Google Scholar]
- 4.Goicoechea S, Pallero MA, Eggleton P, Michalak M, Murphy-Ullrich JE. The anti-adhesive activity of thrombospondin is mediated by the N-terminal domain of cell surface calreticulin. J Biol Chem. 2002;277:37219–37228. doi: 10.1074/jbc.M202200200. [DOI] [PubMed] [Google Scholar]
- 5.Orr AW, Elzie CA, Kucik DF, Murphy-Ullrich JE. Thrombospondin signaling through the calreticulin/LDL receptor-related protein co-complex stimulates random and directed cell migration. J Cell Sci. 2003;116:2917–2927. doi: 10.1242/jcs.00600. [DOI] [PubMed] [Google Scholar]
- 6.Orr AW, Pedraza CE, Pallero MA, Elzie CA, Goicoechea S, Strickland DK, Murphy-Ullrich JE. Low density lipoprotein receptor-related protein is a calreticulin coreceptor that signals focal adhesion disassembly. J Cell Biol. 2003;161:1179–1189. doi: 10.1083/jcb.200302069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pallero MA, Elzie CA, Chen J, Mosher DF, Murphy-Ullrich JE. Thrombospondin 1 binding to calreticulin-LRP1 signals resistance to anoikis. Faseb J. 2008;22:3968–3979. doi: 10.1096/fj.07-104802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elzie CA, Murphy-Ullrich JE. The N-terminus of thrombospondin: the domain stands apart. Int J Biochem Cell Biol. 2004;36:1090–1101. doi: 10.1016/j.biocel.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Sweetwyne MT, Pallero MA, Lu A, Van Duyn Graham L, Murphy-Ullrich JE. The calreticulin-binding sequence of thrombospondin 1 regulates collagen expression and organization during tissue remodeling. Am J Pathol. 2010;177:1710–1724. doi: 10.2353/ajpath.2010.090903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bornstein P. Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol. 1995;130:503–506. doi: 10.1083/jcb.130.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlson CB, Bernstein DA, Annis DS, Misenheimer TM, Hannah BL, Mosher DF, Keck JL. Structure of the calcium-rich signature domain of human thrombospondin-2. Nat Struct Mol Biol. 2005;12:910–914. doi: 10.1038/nsmb997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norgaard Toft K, Larsen N, Steen Jorgensen F, Hojrup P, Houen G, Vestergaard B. Small angle X-ray scattering study of calreticulin reveals conformational plasticity. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbapap.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Ellgaard L, Riek R, Braun D, Herrmann T, Helenius A, Wuthrich K. Three-dimensional structure topology of the calreticulin P-domain based on NMR assignment. FEBS Lett. 2001;488:69–73. doi: 10.1016/s0014-5793(00)02382-6. [DOI] [PubMed] [Google Scholar]
- 15.Tan Y, Chen M, Li Z, Mabuchi K, Bouvier M. The calcium- and zinc-responsive regions of calreticulin reside strictly in the N-/C-domain. Biochim Biophys Acta. 2006;1760:745–753. doi: 10.1016/j.bbagen.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Murphy-Ullrich JE, Gurusiddappa S, Frazier WA, Hook M. Heparin-binding peptides from thrombospondins 1 and 2 contain focal adhesion-labilizing activity. J Biol Chem. 1993;268:26784–26789. [PubMed] [Google Scholar]
- 17.Tan K, Duquette M, Liu JH, Zhang R, Joachimiak A, Wang JH, Lawler J. The structures of the thrombospondin-1 N-terminal domain and its complex with a synthetic pentameric heparin. Structure. 2006;14:33–42. doi: 10.1016/j.str.2005.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schrag JD, Bergeron JJ, Li Y, Borisova S, Hahn M, Thomas DY, Cygler M. The Structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol Cell. 2001;8:633–644. doi: 10.1016/s1097-2765(01)00318-5. [DOI] [PubMed] [Google Scholar]
- 19.Guo L, Groenendyk J, Papp S, Dabrowska M, Knoblach B, Kay C, Parker JM, Opas M, Michalak M. Identification of an N-domain histidine essential for chaperone function in calreticulin. J Biol Chem. 2003;278:50645–50653. doi: 10.1074/jbc.M309497200. [DOI] [PubMed] [Google Scholar]
- 20.Yan Q, Murphy-Ullrich JE, Song Y. Structural insight into the role of thrombospondin-1 binding to calreticulin in calreticulin-induced focal adhesion disassembly. Biochemistry. 49:3685–3694. doi: 10.1021/bi902067f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 23.Yan Q, Murphy-Ullrich JE, Song Y. Structural insight into the role of thrombospondin-1 binding to calreticulin in calreticulin-induced focal adhesion disassembly. Biochemistry. 2010;49:3685–3694. doi: 10.1021/bi902067f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan D, Song Y. Role of Altered Sialylation of the I-like Domain of β1 Integrin in the Binding of Fibronectin to β1 Integrin: Thermodynamics and Conformational Analyses. Biophys J. 2010;99:208–217. doi: 10.1016/j.bpj.2010.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suever JD, Chen Y, McDonald JM, Song Y. Conformation and free energy analyses of the complex of calcium-bound calmodulin and the Fas death domain. Biophys J. 2008;95:5913–5921. doi: 10.1529/biophysj.108.130542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Pan D, Bellis SL, Song Y. Effect of altered glycosylation on the structure of the I-like domain of beta1 integrin: A molecular dynamics study. Proteins. 2008;73:989–1000. doi: 10.1002/prot.22126. [DOI] [PubMed] [Google Scholar]
- 27.Song Y, Guallar V, Baker NA. Molecular dynamics simulations of salicylate effects on the micro- and mesoscopic properties of a dipalmitoylphosphatidylcholine bilayer. Biochemistry. 2005;44:13425–13438. doi: 10.1021/bi0506829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys J. 1997;72:1568–1581. doi: 10.1016/S0006-3495(97)78804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu H, Isralewitz B, Krammer A, Vogel V, Schulten K. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys J. 1998;75:662–671. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao M, Craig D, Vogel V, Schulten K. Identifying unfolding intermediates of FN-III(10) by steered molecular dynamics. J Mol Biol. 2002;323:939–950. doi: 10.1016/s0022-2836(02)01001-x. [DOI] [PubMed] [Google Scholar]
- 31.Hytonen VP, Vogel V. How force might activate talin's vinculin binding sites: SMD reveals a structural mechanism. PLoS Comput Biol. 2008;4:e24. doi: 10.1371/journal.pcbi.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darden T, York D, Pedersen LG. Particle mesh Ewald: An Nâlog(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.