

Abstract
Lipid mixing between vesicles functionalized with SNAREs and the cytosolic C2AB domain of synaptotagmin-1 recapitulates the basic Ca2+ dependence of neuronal exocytosis. However, in the conventional ensemble lipid mixing assays it is not possible to discriminate whether Ca2+ accelerates the docking or the fusion of vesicles. Here we report a fluorescence microscopy-based assay to monitor SNARE-mediated docking and fusion of individual vesicle pairs. In situ measurement of the concentration of diffusing particles allowed us to quantify docking rates by a maximum-likelihood approach. This analysis showed that C2AB and Ca2+ accelerate vesicle-vesicle docking with more than two orders of magnitude. Comparison of the measured docking rates with ensemble lipid mixing kinetics, however, suggests that in most cases bilayer fusion remains the rate-limiting step. Our single vesicle results show that only ∼60% of the vesicles dock and only ∼6% of docked vesicles fuse. Lipid mixing on single vesicles was fast (tmix < 1 s) while an ensemble assay revealed two slow mixing processes with tmix ∼ 1 min and tmix ∼ 20 min. The presence of several distinct docking and fusion pathways cannot be rationalized at this stage but may be related to intrasample heterogeneities, presumably in the form of lipid and/or protein composition.
Introduction
Synaptic vesicles fuse to the presynaptic membrane when Ca2+ enters the synapse, thereby causing release of neurotransmitters into the synaptic cleft and mediating neuronal signaling (1). The vesicle (v) associated soluble n-ethylmaleimide-sensitive factor attachment protein receptor (v-SNARE) synaptobrevin (syb) and the target (t) membrane-localized t-SNAREs, SNAP-25 and syntaxin (syx), are considered the heart of the neuronal membrane fusion apparatus (2–4). In preparation for fusion, cognate v- and t-SNAREs assemble in trans to form a four-helix bundle that bridges the vesicle and the presynaptic membrane.
The Ca2+ dependence of synaptic vesicle fusion is generally attributed to the vesicle-anchored protein synaptotagmin-1 (5,6). This hypothesis was strongly corroborated by the reproduction of Ca2+-regulated membrane fusion in vitro upon coreconstitution of SNAREs and the cytosolic C2AB domain of synaptotagmin-1 (syt) in lipid vesicles (7). This synaptotagmin-1 construct has been the focus of extensive research in the quest for the molecular mechanism underlying Ca2+-triggered release (8–12). The core experimental technique for monitoring the activity of SNAREs and auxiliary proteins in vitro remains the fluorometry-based assaying of lipid mixing between protein-derivatized vesicles.
It is now well established that syt confers Ca2+ sensitivity to SNARE-driven vesicle fusion in vitro, manifested in increased lipid mixing upon Ca2+ addition. In this system, syt action has been tracked to specific interactions both with the neuronal SNAREs (9,10,13) and the lipid bilayers (8,11,12,14). The lipidic action of syt is caused by penetration of negatively charged membranes by flexible loops situated in each of the C2 domains upon Ca2+ binding (15). This property has been found to bring opposing membranes into close proximity (14,16) and, at some conditions, induce high membrane curvature (8,12,14), thereby lowering the energy barrier for membrane fusion.
A vesicle fusion reaction is composed of two fundamental steps: vesicle docking and subsequent membrane fusion. These steps are, however, not resolved in the conventional ensemble lipid mixing assay that, due to spatiotemporal averaging, only probes the rate-limiting step of the process. This shortcoming has motivated the development of single vesicle-vesicle (17–21) and vesicle-supported bilayer (22–26) assays that can follow the time course of individual fusion events. In addition, single vesicle assaying can reveal heterogeneous behavior that is masked in ensemble measurements.
In ensemble assays, lipid mixing takes place on a timescale of minutes. On the contrary, single vesicle experiments have consistently reported fusion events with subsecond docking to fusion transitions (17,18,21–25). These fast events were clearly diffusion-limited and, if present in bulk, they would not be resolved in ensemble assays that instead would probe the rate-limiting docking step. Thus, it has been questioned whether the trends produced by different fusion regulators, e.g., syt and complexin, in lipid mixing assays are indicative of changes in the efficiency of vesicle docking, fusion, or both (11,18). The rate constants for vesicle docking provide a guide for determining the rate-limiting step of the fusion reaction and thereby allow a more detailed interpretation of ensemble fusion kinetics that, to this end, comprise the bulk of the knowledge on the subject. However, despite the recent interest in single vesicle assays, docking rate constants for SNARE-derivatized vesicles have been measured on surprisingly few occasions (19,22) and not for reconstitutions including auxiliary proteins. At this stage, it therefore remains unclear whether the well-documented accelerating effect of syt-Ca2+ in the lipid mixing assay reflects accelerated vesicle docking or fusion.
Here we developed a single-vesicle docking and fusion assay based on fluorescence microscopy and performed a quantitative analysis of SNARE-syt-Ca2+-mediated fusion reactions. In contrast to the majority of single vesicle fusion assays that are performed between vesicles and planar bilayers, we report here single vesicle-vesicle fusion to best mimic the conditions found in ensemble assays. We found that Ca2+ addition accelerated docking of SNARE-syt reconstituted vesicles by more than two orders of magnitude. Comparison of typical lipid mixing rates observed in ensemble assays and docking rates measured by us, however, suggests that, in most cases, membrane fusion should be the rate-limiting step of the observed kinetics.
The single vesicle assay, surprisingly, unveiled that with syt and Ca2+ only a fraction of the total vesicle population (comprising roughly 60%) docked. Six-percent of the docked vesicles underwent fast lipid mixing with subsecond kinetics. Some of the mixing events showed biphasic kinetics with an initial fast component (tmix < 1 s) and a secondary slow component (tmix ∼ 1 min). An ensemble version of the single vesicle assay revealed slow lipid mixing kinetics with double-exponential character, indicative of two distinct lipid-mixing processes.
The fast fusion events observed on single vesicles were not apparent in the ensemble assay. The time constant (tmix ∼ 1 min) of the initial rise of the ensemble kinetics was, however, comparable to the slow component of the biphasic events observed on single vesicles. The second component of the ensemble kinetics showed that an additional 10% of the vesicles underwent very slow lipid mixing with tmix ∼ 20 min. Even over the course of an hour, the majority of vesicle complexes did not fuse. These observations led us to conclude that the model system for neuronal membrane fusion displays pronounced heterogeneous behavior with several distinct modes of both docking and fusion.
Materials and Methods
Materials
POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]), PO (3-ethyl), PC (1-palmitoyl-2-oleoyl-sn-glycero-3-ethyl-phosphocholine), DOPE-Biot (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-n-cap biotinyl), PC (L-a-phosphatidylcholine), PS (L-A-phosphatidylserine), and PIP2 (cholesterol and phosphatidylinositol) were from Avanti (Alabaster, AL).
DiI (1,1′-dioctadecyl-3,3,3′,3′ tetramethylindocarbocyanine perchlorate), i.e., DiIC18 (3), DiO (3,3′ dioctadecyloxacarbocyanine perchlorate), i.e., DiOC18 (3), and neutravidin were from Invitrogen (Tåstrup, Denmark).
Octyl-β-d-glucopyranoside and DL-dithiothreitol were from Sigma (Brøndby, Denmark).
PLL-g-PEG (PLL(20)-g[3.5]-polyethylene glycol(2)) and PLL-g-PEG-Biot (PLL(20)-g[3.5]-PEG(2)/PEG(3.4)-biotin (18%)) were purchased from Surface Solutions (Zürich, Switzerland).
Protein purification
Full-length SNAREs and synaptotagmin-1 C2AB (amino acids 96–421) from rat were received in purified form from the McMahon lab, Cambridge, UK (see (8)).
Vesicles
v-(PC/PS/Cholesterol//DiO-C18 73:15:10:2)) and t-vesicles (PC/PS/Cholesterol/PiP2/DOPE-Biot/DiI-C18 58:25:10:5:0.1:2) were prepared by mixing the lipids in chloroform in a glass vial. Chloroform was evaporated under nitrogen flow followed by 15-min incubation under vacuum. Vesicles were formed by addition of 0.5 mL hydration buffer (50 mM Tris pH 8, 150 mM NaCl, 2 mM DTT). SNARE proteins were reconstituted exactly as previously described (8). See Extended Methods in the Supporting Material for a detailed description.
Surfaces
Glass coverslips were cleaned by copious sonication cycles in 2% (v/v) Hellmanex and water, plasma-etched and mounted in microscope chambers followed by 30-min incubation with 1 g/L PLL-g-PEG and 0.01 g/L PLL-g-PEG-Biot in 15 mM HEPES pH 5–6. The chambers were then washed repeatedly and incubated with 0.1 g/L neutravidin for 10 min followed by wash. Experiments were performed in chamber buffer (25 mM HEPES pH 7.5, 100 mM KCl, 5% Glycerol, 2 mM DTT).
Microscopy
For the fusion assay, a 458-nm laser was used for excitation and fluorescence emission was acquired in two channels: 480–530 nm (donor) and 590–650 nm (acceptor) using a TCS SP5 confocal microscope (Leica, Wetzlar, Germany). For imaging of the t-vesicles, the fluorescence resonance energy transfer (FRET) acceptor was excited directly with a 543-nm laser. Additional details are available in Extended Methods in the Supporting Material.
Data treatment
Software written in Igor Pro Ver. 5.01 (Wavemetrics, Tigard, OR) generated the fluorescence traces of single t-vesicles by region-of-interest (ROI) integration of the fusion movies and cropped events from the traces, corrected for crosstalk, and calculated the apparent FRET efficiency (E) according to
where I denotes the integrated intensity of donor and acceptor dyes. For data recorded with 5 Hz resolution, the cropped event traces were smoothed for the lipid mixing analysis by averaging every four consecutive data points. Events were categorized via a custom-made graphical user interface.
Results and Discussion
Single vesicle docking and fusion assay
The single vesicle assay is sketched in Fig. 1 a. We reconstituted recombinant full-length v- and t-SNAREs (syb, syx, and SNAP25) in two populations of lipid vesicles, following verbatim the previously published protocol by Martens et al. (8). Protein-derivatized vesicles were formed by co-micellization of proteins and lipids followed by rapid dilution and dialysis to remove the detergent. The lipid/protein ratio was 200:1 for tSNARE vesicles (t-vesicles) and 50:1 for vSNARE vesicles (v-vesicles). The reconstitution included the cytosolic C2AB domain of syt.
Figure 1.
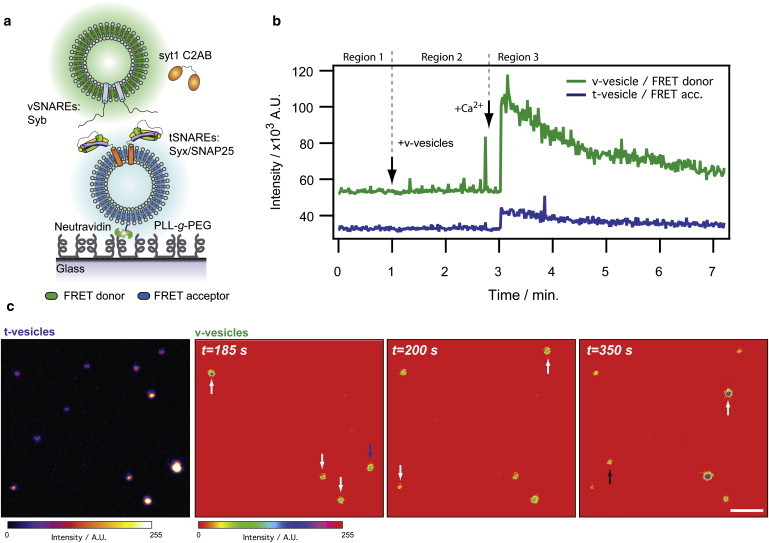
Single vesicle docking and fusion assay. (a) tSNARE vesicles (t-vesicles) were immobilized via biotin-neutravidin coupling onto a PEG-coated glass surface. Complexation of individual vSNARE vesicles (v-vesicles) and t-vesicles was followed by fluorescence microscopy. Lipid mixing was reported as FRET from an energy donor in the v-vesicle membrane to an energy acceptor in the t-vesicle membrane. The reconstitution included the cytosolic C2AB domain of synaptotagmin-1. (b) Docking and fusion kinetics were tracked in fusion movies by integrating the fluorescence intensity in the donor and acceptor channel in an ROI enclosing each t-vesicle. Each experiment was divided into three regions corresponding to 1), time series initiation; 2), addition of v-vesicles and syt; and 3), addition of Ca2+. The trace shows (crosstalk-corrected) data for a single docking event distinguished by an abrupt increase in donor fluorescence. In this case there is a small FRET footprint observed as an increase in acceptor emission upon docking, indicating that the bilayers of the docked vesicles have come into close proximity without fusing. (c) Sample micrographs. An image of t-vesicles and corresponding snapshots of v-vesicles from a fusion movie. The arrows mark docking events (white), a transient docking event (blue), and nonspecific binding (black). Bar: 1 μm.
We immobilized t-vesicles, labeled with lipophilic FRET acceptor dye (DiI-C18), at dilute densities at a PLL-g-PEG-coated glass surface via biotin-neutravidin coupling (27), a procedure shown to prevent deformation of surface-tethered vesicles (28). To assay single-vesicle docking and fusion, the stationary t-vesicles were allowed to react with freely diffusing v-vesicles labeled with lipophilic FRET donor dye (DiO-C18). Movies were recorded by time-resolved confocal microscopy with a frame rate of 1 or 5 Hz.
By assigning each t-vesicle at the surface a ROI and integrating the fluorescence intensity in an acquired image series, we obtained traces of donor and acceptor emission as that shown in Fig. 1 b. We divided each experimental run into three regions corresponding to
-
1.
Imaging of t-vesicles.
-
2.
Addition of v-vesicles and syt (Csyt = 7.5 μM).
-
3.
Addition of Ca2+ (0.5 mM).
Docking of a v-vesicle manifested in an abrupt increase in donor fluorescence. The time domain allowed us to track single vesicle-vesicle encounters and characterize them on individual basis. For each event we extracted the docking time (td) defined as the time lapsed from vesicle (region 1) or Ca2+ (region 2) addition to the onset of donor fluorescence. We characterized lipid mixing by evaluating the apparent FRET efficiency (E) using the formula (17)
where Iacc and Idon indicate acceptor and donor intensities. All signals were corrected for crosstalk before analysis (see Section S1 in the Supporting Material).
Fig. 1 c shows an image of the immobilized t-vesicles and snapshots of the v-vesicles from a time series. Docking events (colocalization of v- and t-vesicles) are highlighted by the white arrows. The blue arrow marks a transient docking event where a v-vesicle detaches from the t-vesicle after staying bound for several frames. The black arrow indicates a nonspecifically adsorbed v-vesicle. A sample movie from an experiment is supplied as Movie S1 in the Supporting Material.
The polyethylene glycol (PEG) coating of the surface was implemented to prevent nonspecific binding of the v-vesicles. To evaluate the potency of the PEG we counted the amount of nonspecifically adsorbed v-vesicles. In a typical time series, approximately five events were detected on an area of 2621 μm2. Given the ROI dimensions used for fluorescence integration (∼0.25 μm2) and the typical number of tSNARE vesicles in a movie (∼50), the probability of recording a single false docking event in a given time series is ∼2% and thus highly insignificant.
Morphometric characterization
We applied transmission electron microscopy at cryogenic temperatures (cryoTEM) to quantify size distributions and percentages of multilamellar vesicles (see Fig. 2, a and b). The SNARE-reconstituted vesicles had a rather narrow size distribution peaking at a diameter of 75 nm for v- and 52 nm for t-vesicles. Multilamellarity was low for both populations, 11.7% (v) and 2.6% (t), as compared to a value of 8% previously measured in a study of protein free vesicles prepared by standard freeze-thawing and extrusion procedures (29).
Figure 2.
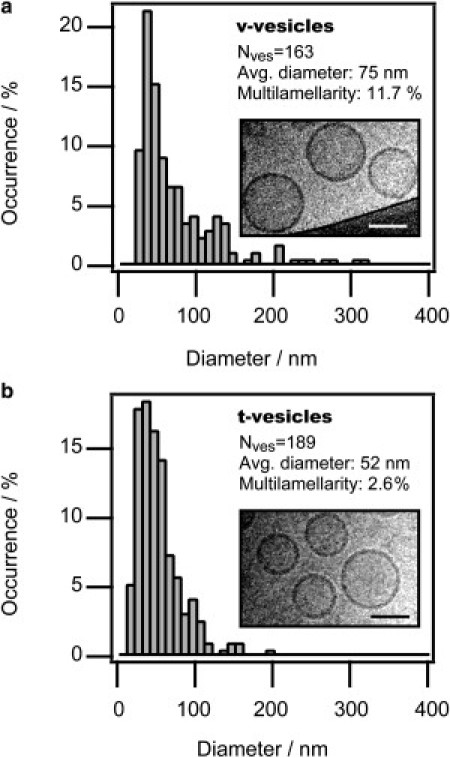
Vesicle morphology. (a and b) Vesicle preparations characterized by cryoTEM. Bars: 50 nm.
Quantification of docking probabilities
We pursued a maximum-likelihood approach that allowed us to quantify the intrinsic docking probability, pd, upon collision of a diffusing v- and a stationary t-vesicle. First, we constructed curves of the accumulated number of docking events from the collected docking times for each t-vesicle (Fig. 3 a). During the incubation without Ca2+ we only observed docking on ∼4% of the t-vesicles. Addition of Ca2+ triggered a rapid acceleration of docking but, surprisingly, only on ∼60% of the t-vesicles. Approximately half of the t-vesicles that docked in response to Ca2+ exhibited consecutive events. Because subsequent events reflect binding to already docked vesicle complexes, we based the quantification of pd on the first events.
Figure 3.
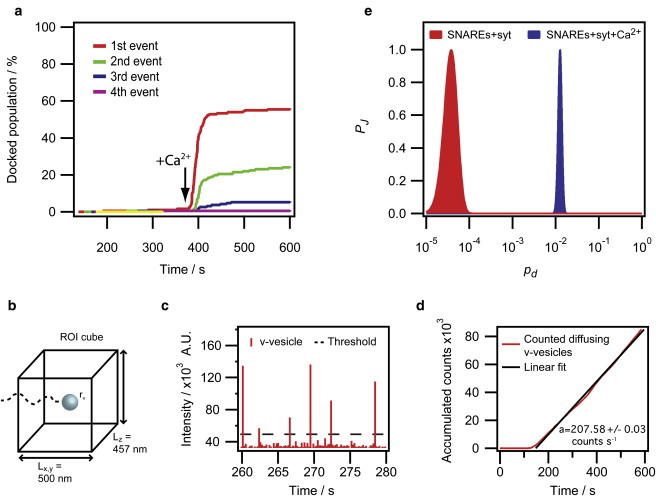
Quantitative docking analysis. (a) Accumulated docking counts as a function of time. Ca2+ was added half-way through the series. (b) Schematic illustrating the ROI approach for counting diffusing v-vesicles. The effective dimensions of the applied ROIs are indicated. (c) Trace of diffusing v-vesicles obtained by ROI integration throughout a fusion movie in a region of the surface having no t-vesicles. Diffusing vesicles were counted using a threshold (dashed line). (d) Accumulated count of diffusing vesicles constructed from 676 ROIs. The data were fitted with a straight line to extract the average v-vesicle count. (e) Joint probability, PJ (normalized to 1 to ease comparison), as a function of the intrinsic docking probability, pd, calculated from the data in panel a for the cases without and with Ca2+.
The number of docking attempts before successful binding on a t-vesicle can be calculated from the concentration of the freely diffusing v-vesicles. We decided to measure the v-vesicle concentration in situ by counting the number of diffusing vesicles present within the field of view. Importantly, the in situ concentration measurement allowed us to control for and exclude potential depletion of the v-vesicles near the surface due to docking.
To achieve this, we parceled the fusion movies into an array of ROIs (Fig. 3 b) and integrated the fluorescence of the v-vesicles throughout the movie resulting in traces of the form Fig. 3 c. The presence of a v-vesicle inside the ROI is observed as a spike in the trace. We quantified the number of diffusing vesicles using a threshold. The threshold was systematically assigned by quantifying the background noise observed before addition of v-vesicles to the chamber and enforcing a confidence of 0.01% in v-vesicle detection (see Section S2 in the Supporting Material). Using this threshold approach, we constructed curves of the accumulated number of counted v-vesicles versus time (Fig. 3 d). A total of 676 ROIs was applied per movie. The linear trend of the accumulated curve proves the absence of depletion.
A linear fit of the accumulated number of v-vesicles versus time revealed the average number of counted v-vesicles per frame which we converted to concentration via the volume of the applied ROIs. Due to the limited speed of confocal line-scanning, only a fraction of the diffusing vesicles (22%) is captured by this approach and it is necessary to correct to obtain the true concentration. We estimated the fraction of detectable vesicles from Einstein's diffusion equation and with the assumption that a vesicle gives enough signal to be detected only if it remains within the ROI during the time it takes to image it by confocal raster scanning. This assumption was justified by successful measurement of the concentration of fluorescent beads with a known concentration (see Section S2.1 in the Supporting Material for details).
It is important to note that although a single ROI is scanned in ∼10 ms it takes considerably longer before the ROI is scanned again in the next frame (0.2 s or 1 s depending on the applied settings). Thus, the observed count frequency in the ROIs reflects only a small fraction of the vesicles that actually visited this part of the surface during the experiment. Furthermore, it should be emphasized that the effective volume of the applied ROIs deviates from the size of the immobilized t-vesicles. For these reasons, the count frequency is not directly equivalent to the frequency of docking attempts on the t-vesicles. Standard diffusion theory, however, provides us a relation between the v-vesicle concentration and the average frequency of docking attempts on the t-vesicles (see Berg (30) and Section S2.2 in the Supporting Material),
| (1) |
where D is the diffusion coefficient, rv and rt denote the radius of v- and t-vesicles, respectively, and C is the v-vesicle concentration. The value D was calculated as 5.71 μm2/s using the Stokes-Einstein equation
with viscosity (h) 1.002 × 10−3 Pa s and applying the average v-vesicle radius from the cryoTEM data (rv = 37.5 nm). For calculating I we used the average vesicle radii from the cryoTEM data (rt = 26 nm). For the data shown in Fig. 3 d, we obtain an average v-vesicle count per frame of
We applied 676 ROIs in total and the volume of a single ROI was
(see Section S2.1 in the Supporting Material). Taking into account that only 22% of the v-vesicles could be detected, we arrive at a concentration of 4.1 nM. According to Eq. 1, this amounts to a docking attempt frequency of ∼I = 5.6 hits s−1. The expected v-vesicle concentration, calculated with the assumption that all lipids initially mixed end up in vesicles, is 4.3 nM and thus in good agreement with the measured value of 4.1 nM (see Extended Methods in the Supporting Material). It should be noted, however, that this is not a trivial result given the quite complex protocol for vesicle preparation. The described procedure was used to convert the recorded docking times into the number of docking attempts before binding.
We then determined the intrinsic docking probability upon collision of a v- and a t-vesicle, pd, by the principle of maximum-likelihood as the value that optimized the joint probability, PJ, of the data (31):
| (2) |
Here, Pdocking is the probability to observe docking on a t-vesicle after na attempts and Pno docking denotes the probability for observing a t-vesicle that did not dock after na attempts. The latter factor is necessary for conditions where docking does not saturate within the time of the experiment.
Fig. 3 e shows PJ as a function of pd for the data presented in Fig. 3 a separated into without and with Ca2+. From the saturation of the curve in Fig. 3 a, it is clear that only a fraction of the vesicles underwent Ca2+-triggered docking. For extraction of pd, we treated the vesicles that docked after addition of Ca2+ as the total docking capable population (i.e., the ∼40% t-vesicles that never was observed to dock were excluded in the evaluation of PJ). The standard deviation, σ, of pd was obtained by fitting the normalized PJ with the normal distribution. A more detailed description of the procedure is supplied in Section S3 in the Supporting Material together with a verification of the method based on simulated data.
Docking results
We employed the described method for extracting the docking probabilities for the SNARE-syt-derivatized vesicles. Addition of Ca2+ enhanced pd by more than two orders of magnitude from
(see Table 1). Notably, the acceleration of docking upon Ca2+ addition is much more pronounced than the ∼2–10 fold increase in lipid mixing rates typically observed in ensemble assays.
Table 1.
Results of the quantitative docking analysis
| Sample | Number of t-vesicles | Docking counts | pd × 10−4 | kdock × 105 [s−1 M−1] |
|---|---|---|---|---|
| SNAREs + syt | 191 | 8 | 0.7 ± 0.5 | 5 ± 4 |
| SNAREs + syt + Ca2+ | 191 | 99 | 130 ± 16 | 874 ± 108 |
| SNAREs | 150 | 13 | 0.7 ± 0.4 | 5 ± 3 |
| SNAREs + Ca2+ | 150 | 6 | 0.9 ± 0. 8 | 6 ± 5 |
| SNARE free vesicles + syt | 78 | 0 | — | — |
| SNARE free vesicles + syt + Ca2+ | 78 | 60 | 500 ± 8 | 3362 ± 538 |
| SNARE free vesicles, no syt | 102 | 0 | — | — |
| SNARE free vesicles, no syt + Ca2+ | 102 | 0 | — | — |
| SNAREs + syt (direct meth.) | 202 | 2 | 4 ± 3 | 27 ± 20 |
| SNAREs + syt + Ca2+ (direct meth.) | 202 | 117 | 260 ± 40 | 1748 ± 269 |
The reported kdock indicates the rate constant for vesicle-vesicle docking in solution calculated using the pd from the single vesicle assay.
To test whether the Ca2+-specific increase in pd was indeed caused by syt, we omitted it from the reconstitution. This resulted in the loss of the Ca2+ sensitivity of docking:
and
Surprisingly, when we excluded the SNAREs but retained syt-Ca2+, pd increased to
From this, we conclude that syt-Ca2+ docks SNARE-free vesicles four times more efficiently than vesicles containing SNAREs. This result suggests that syt-Ca2+ stimulated docking is a result of syt's ability to bridge apposed bilayers (14) and not a result of interactions with the SNAREs. SNAREs in fact attenuated syt-Ca2+-mediated docking which could be explained by the SNAREs sterically reducing the density of syt on the membranes or by sterically inhibiting interbilayer bridging. Without Ca2+, syt had no detectable effect on docking. Without any proteins, no docking was detected, regardless of the presence of Ca2+.
To allow comparison of the measured docking probabilities and the kinetics recorded in ensemble fusion assays, we converted the measured docking probabilities to rate constants (kdock), according to the relation
where the diffusion-limited docking rate for vesicles interacting in solution is given by
(see Table 1). The values r and D denote v- and t-vesicle radii and diffusion coefficients.
A previous estimation of
for SNARE-bearing vesicles interacting freely in solution (without syt-Ca2+) obtained from fluorescence correlation spectroscopy (19) is in good agreement with our result of
indicating that pd measured on one reconstitution provides a reasonable guide for comparable experimental conditions. Liu et al. (22), however, reported a significantly larger docking probability of pd = 0.02 for vSNARE vesicles docking to a supported bilayer decorated with tSNAREs (the value of pd from Liu et al. (22) represents the protein densities that best compare to ours). It would be interesting to determine whether this apparent difference in pd between vesicle-vesicle and vesicle-supported bilayer docking is a result of the different bilayer geometries in the two systems.
Direct reconstitution
It has previously been suggested that reconstitution of proteins by incubation of vesicles with SNAREs in detergent below the CMC, named the direct reconstitution method, diminishes the fusogenicity of the vesicles due to an increase in vesicle size (32). To test whether this protocol had an effect in our assay, we prepared vesicles using the direct approach. Under our conditions, however, the size distributions were largely unchanged (see Section S4 in the Supporting Material). Without Ca2+, docking was still inefficient, at
though sixfold higher than for the indirect method. In the presence of syt and Ca2+, the direct reconstitution doubled the docking efficiency to
compared to the standard reconstitution.
Lipid mixing assay
To characterize lipid mixing, we cropped single vesicle encounters from the movies and calculated the apparent FRET between the donor labeled v-vesicles and the acceptor labeled t-vesicles. With the present format of the assay it was not meaningful to analyze lipid mixing for the configurations with inefficient docking (pd ∼ 10−4) due to insignificant statistics (i.e., <10 events per condition). However, the exact same reconstitution has recently been investigated by Martens et al. (8) using an ensemble lipid mixing assay. From these experiments, it was concluded that both SNAREs, syt and Ca2+, are required to obtain significant lipid mixing. Consequently, we only analyzed lipid mixing on single vesicles for this condition.
Fig. 4, a–d, shows characteristic vesicle-vesicle interactions (additional traces are supplied in Section S5 in the Supporting Material). We identified four discernable types of events. The majority of the events (78%) were of the type presented in Fig. 4 a. Clearly, these correspond to stable docked states, where the membranes are still too far apart to produce FRET. The decay of donor and acceptor fluorescence is solely caused by bleaching. Next, we observed transient docking events (10%), Fig. 4 b, where a v-vesicle docks, stays bound for several frames, and thereafter dissociates without any lipid mixing. We did not encounter kiss-and-run-like events (transient events exhibiting partial mixing) as observed by others for fusion mediated by yeast SNAREs (17).
Figure 4.
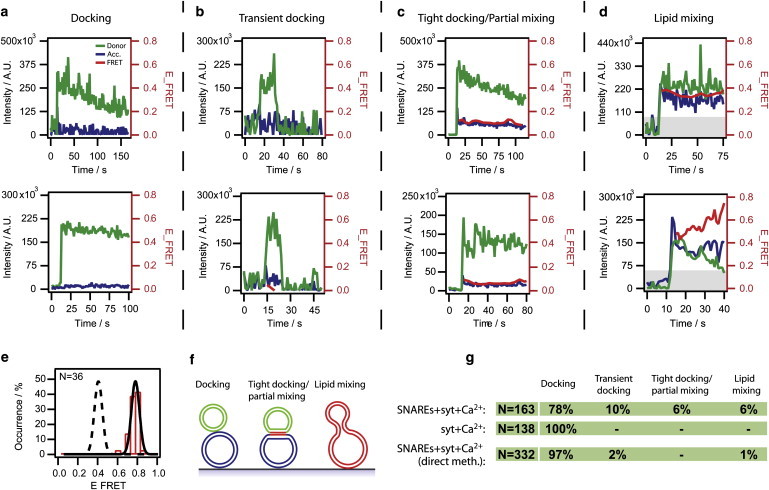
Lipid mixing. (a–d) Characteristic events. (a) v-vesicle docking to an immobilized t-vesicle. (b) v-vesicle docking transiently and dissociating without any lipid mixing. (c) Tight docking/partial mixing characterized by a small FRET footprint. (d) v- and t-vesicle undergoing lipid mixing resulting in a FRET signature above the threshold (shaded area). For all events the time axis was adjusted relative to the onset of donor fluorescence. (e) Histogram of E measured on a reference sample premixed with donor and acceptor and fitted with a Gaussian distribution (solid line). To simulate the event of hemifusion, i.e., half-lipid mixing, the fit was offset to one-half the peak value (dashed line). (f) Sketch of interactions. (g) Distribution of events. N indicates the number of analyzed events.
Twelve-percent of the events displayed FRET (see Fig. 4, c and d). To further categorize this class of events we used a vesicle sample simulating the product of a full fusion event prepared with a 1:1 mixture of donor and acceptor dyes at 1 mol % each (17,18). We constructed a histogram of single-vesicle E values and fitted the data with a Gaussian yielding a peak at E = 0.75 with a width of 0.06 (see Fig. 4 e). The event of full lipid mixing should result in an E within this distribution while the event of hemifusion (outer leaflet mixing) should produce approximately one-half this value.
To simulate the latter scenario, we offset the fit of E to half the peak value (see dashed line on Fig. 4 e), and based on this curve we fixed a lower threshold of E = 0.2 for lipid mixing. Six-percent of the events qualified for this category. Some of these events exhibited fast transitions (t < 1 s) to a stable E above the threshold (Fig. 4 d, top), while others showed a biphasic behavior with an initial abrupt jump to an intermediary E above the threshold followed by a slowly increasing E (Fig. 4 d, bottom).
Six-percent of the events displayed FRET insignificant to be considered lipid mixing (see Fig. 4 c). We reasoned that these events most likely reflect docked states where the vesicles have deformed to produce a zone of tight contact giving rise to a small FRET signature (28) (Fig. 4 f). However, we cannot exclude the possibility that these events represent partial mixing. Accordingly, we classified this class as “tight docking/partial mixing” events. These events were always irreversible.
As a control, we tested whether lipid mixing was SNARE-specific by examining syt-Ca2+-mediated interactions between SNARE-free vesicles. In these experiments we only observed stable docked vesicle complexes without any degree of FRET. We did not observe transient docking events for this condition indicating that this property is linked to the SNAREs. Our data thus confirm that syt-Ca2+-mediated membrane apposition is insufficient to obtain lipid mixing without SNAREs (8,14). Furthermore, our data suggest that SNAREs are necessary to obtain tight docking.
As discussed above, the vesicles prepared by the direct reconstitution exhibited more efficient docking. However, they produced only a modest 1% of lipid mixing, thus complying with previous ensemble results (32), though our data indicate that this low degree of fusion is not due to an increase in vesicle sizes. Fig. 4 g summarizes the distributions of the four event types.
Ensemble lipid mixing
To compare single and ensemble measurements, we conducted a bulk replica of the single-vesicle FRET assay using a cuvette fluorometer (see Extended Methods in the Supporting Material). This also allowed us to follow the fusion reaction over a longer period of time than what was possible on single vesicles due to bleaching.
In the presence of Ca2+, ensemble lipid mixing increased slowly over the course of 1 h both without and with syt (see Fig. 5). The kinetics could not be fitted with mono-exponentials whereas double-exponentials described the data well (see Table 2). From the fits it is evident that addition of syt indeed enhanced both the rate and yield of lipid mixing, thus recapitulating previously published results (see, e.g., (7–9)). Both samples exhibited a fast (kmix = 11 ± 1 × 10−3 s−1 without and kmix = 15 ± 1 × 10−3 s−1 with syt) and a slow component (kmix = 0.32 ± 0.01 × 10−3 s−1 without and kmix = 0.84 ± 0.01 × 10−3 s−1 with syt). Syt also had an effect on the overall yield of mixing, which more than doubled from 6.8% to 15.2% (evaluated after 1 h of reaction).
Figure 5.
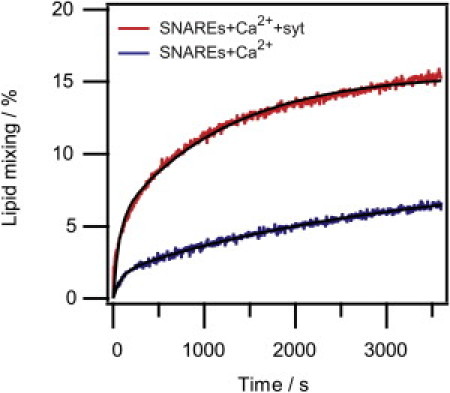
Ensemble lipid mixing FRET assay. (Solid lines) Double-exponential fits of the data (see Table 2).
Table 2.
Coefficient values of the double-exponential fits of the ensemble lipid mixing kinetics according to f(t) = A1[1−exp(−k1 t)] +A2[1−exp(−k2 t)]
| Sample | A1 (%) | k1 (10−2 × s−1) | A2 (%) | k2 (10−4 × s−1) |
|---|---|---|---|---|
| SNAREs + Ca2+ + syt | 5.2 ± 0.1 | 1.5 ± 0.1 | 10.4 ± 0.1 | 8.4 ± 0.1 |
| SNAREs + Ca2+ | 1.8 ± 0.1 | 1.1 ± 0.1 | 6.8 ± 0.1 | 3.2 ± 0.1 |
With the knowledge of kdock obtained from the single vesicle assay (Table 1) we can estimate the effective docking rate of the vesicles in solution. Assuming the docking reaction follows pseudo-first-order kinetics, which is reasonable considering that v-vesicles can bind to already docked t-vesicles (see Fig. 3 a) and in this way the concentration of binding sites stays approximately constant, we can estimate the effective docking rate as
where V0 is the initial concentration of t-vesicles. In this experiment we had V0 = 18 nM (calculated from the lipid concentration and the average t-vesicle size) yielding
and
The observed fast kmix with syt in bulk is 105-fold slower than the estimated docking-limited rate, strongly indicating that membrane fusion is the rate-limiting step of the observed kinetics. In the case without syt the observed fast kmix corresponds to the estimated docking-limited rate. Thus, the fast component of this reaction is likely limited by diffusion whereas the slow (dominant) component is not. Clearly, the >100-fold increase in kdock upon addition of syt observed on single vesicles is not recapitulated in the ensemble lipid mixing kinetics. Thus, the accelerated lipid mixing with syt in the ensemble assay cannot be attributed to faster docking alone.
The double-exponential lipid mixing traces are indicative of two distinct processes. The fast component with syt in bulk (kmix = 15 ± 1 × 10−3 s−1) agrees qualitatively with the secondary minute scale increase in E observed for the biphasic single vesicle mixing events (see Fig. 4 d, bottom). The slow component of the bulk data shows that a population of vesicles follows a very slow route to fusion. This slow component may represent the progression of the partially mixed/tightly docked single vesicle states into full fusion.
The probability to fuse is by definition related to the rate of fusion. Indeed, syt enhanced the bulk lipid mixing rates by a factor of 1.4 for the fast and 2.6 for the slow components. Syt also triggered an increase in the amplitudes of both the fast (2.9-fold) and slow (1.5-fold) mixing reactions. The amplitudes do not reflect an increase in the probability to fuse but shows that our samples are composed of fusion-competent and fusion-incompetent vesicles. Surprisingly, syt seems to be changing the ratios of these populations.
Conclusions
Here we studied SNARE-syt driven membrane docking and fusion reactions using a single vesicle assay. By recording interactions of single surface-tethered t-vesicles and freely diffusing v-vesicles using fluorescence microscopy, we extracted intrinsic vesicle-vesicle docking probabilities. We found that syt-Ca2+ increased the docking probability per vesicle-vesicle encounter, and therefore the docking rate, with more than two orders of magnitude. This syt-Ca2+-specific 100-fold acceleration is not recapitulated in conventional ensemble lipid mixing assays, including ours, that report ∼2–10-fold acceleration of lipid mixing kinetics.
If we use the single vesicle data to estimate the effective rates of vesicle docking in ensemble assays, we find that they are generally faster (kdock-eff ∼ 10−2 s−1 without and kdock-eff ∼ 1 s−1 with syt-Ca2+) than typically recorded lipid mixing rates that are in the range kmix ∼ 10−4 s−1 without and kmix ∼ 10−4–10−2 s−1 with syt-Ca2+ (7–12). The effective docking rates were calculated with the assumption of pseudo-first-order kinetics, using our values for kdock and 10 nM t-vesicles (which is a lower estimate for the typically applied vesicle concentration). These findings suggest that the accelerating effect of syt-Ca2+ in the ensemble assays cannot be attributed solely to an acceleration of vesicle docking. It should be noted, however, that although our docking data should provide a reasonable guide for comparable reconstitution conditions there may be configurations that exhibit significantly different kinetics.
Our single vesicle analysis revealed that syt-Ca2+ triggered docking was restricted to a subpopulation of the t-vesicles comprising roughly 60%. The origin of this behavior remains to be established, but it might provide an explanation why most ensemble assays do not converge toward 100% (7,8,10–12) of the theoretically possible lipid mixing because not all vesicles dock.
From the single vesicle data we found that 6% of the vesicle complexes exhibited rapid transitions to lipid mixed states upon docking (kmix > 1 s−1). Interestingly, all fusion events we recorded were simultaneous (within our temporal resolution of ∼1 s) to docking. Within our observation time of ∼1 min before bleaching, we never observed a docked or tightly docked state being converted to fusion. The observation of a subpopulation of fast-fusing vesicles for which docking comprises the rate-limiting step is consistent among the single vesicle studies with neuronal SNAREs (17,18,22–25). Such fast fusion events should, in principle, be present also in ensemble assays where they should appear as a lipid mixing component with kinetics matching the docking rate. As we demonstrated here, this is, however, not generally the case. This discrepancy could have several possible explanations, e.g., the limited temporal resolution in bulk assays due to slow sample mixing or temperature equilibration for predocked samples.
Comparison of the single vesicle results and the kinetics observed in an equivalent ensemble fusion assay highlighted the presence of two additional slow lipid mixing processes in our system in addition to the fast fusion events observed on single vesicles. Indeed, several reports of double-exponential ensemble lipid mixing kinetics can be found in the literature (9,11,12).
Our results show that, in the model system for neuronal exocytosis, not all vesicles dock, and that only a subpopulation of vesicles fuse, and the vesicles that fuse follow at least three kinetically distinct paths. The presence of several distinct docking and fusion pathways is likely related to intrasample heterogeneities presumably in the form of lipid and/or protein composition.
The molecular origins of the heterogeneous behavior among individual vesicles in the in vitro system are not known at this stage; interestingly, however, heterogeneous release probability among synaptic vesicles is well documented in cells and is believed to be a defining feature in the transmission characteristics of synapses (33,34), though it remains to be resolved whether there is any mechanistic connection between these two observations. Of course, considering the molecular complexity of synaptic vesicles (35) and their small size, it would indeed not be entirely surprising if they are not produced with an identical protein composition. If intravesicle compositional heterogeneities exist in vivo, it might be interesting to consider whether they contribute to the system as a source of noise or as means to increase the compositional, and hence, functional diversity in synaptic transmission.
Acknowledgments
We thank Sascha Martens and Harvey T. McMahon, Laboratory of Molecular Biology, Cambridge, UK, for supplying purified proteins and protocols and for helpful discussions. We thank Anna Carnerup and Tommy Nylander, Center for Chemistry and Chemical Engineering, Lund University, Sweden, for assistance with cryoTEM. Finally, we thank Andreas H. Kunding for helpful discussions.
The authors thank the Danish Research Councils for Independent and Strategic Research, the Lundbeck Foundation, and the University of Copenhagen programs of excellence “BioScart,” “Single Molecule Nanoscience,” and “UNIK-Synthetic Biology” for financial support.
Supporting Material
References
- 1.Sudhof T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 2.Weber T., Zemelman B.V., Rothman J.E. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 3.Jahn R., Scheller R.H. SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 4.Brunger A.T., Weninger K., Chu S. Single-molecule studies of the neuronal SNARE fusion machinery. Annu. Rev. Biochem. 2009;78:903–928. doi: 10.1146/annurev.biochem.77.070306.103621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernández-Chacón R., Königstorfer A., Südhof T.C. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- 6.Chapman E.R. How does synaptotagmin trigger neurotransmitter release? Annu. Rev. Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- 7.Tucker W.C., Weber T., Chapman E.R. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 2004;304:435–438. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- 8.Martens S., Kozlov M.M., McMahon H.T. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- 9.Chicka M.C., Hui E.F., Chapman E.R. Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+ Nat. Struct. Mol. Biol. 2008;15:827–835. doi: 10.1038/nsmb.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu X.B., Xu Y.B., Shin Y.K. Synaptotagmin I and Ca2+ promote half fusion more than full fusion in SNARE-mediated bilayer fusion. FEBS Lett. 2006;580:2238–2246. doi: 10.1016/j.febslet.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 11.Stein A., Radhakrishnan A., Jahn R. Synaptotagmin activates membrane fusion through a Ca2+-dependent trans interaction with phospholipids. Nat. Struct. Mol. Biol. 2007;14:904–911. doi: 10.1038/nsmb1305. [DOI] [PubMed] [Google Scholar]
- 12.Hui E.F., Johnson C.P., Chapman E.R. Synaptotagmin-mediated bending of the target membrane is a critical step in Ca2+-regulated fusion. Cell. 2009;138:709–721. doi: 10.1016/j.cell.2009.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi U.B., Strop P., Weninger K.R. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat. Struct. Mol. Biol. 2010;17:318–324. doi: 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Araç D., Chen X.C., Rizo J. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- 15.Hui E.F., Bai J.H., Chapman E.R. Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys. J. 2006;91:1767–1777. doi: 10.1529/biophysj.105.080325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diao J.J., Yoon T.Y., Ha T. C2AB: a molecular glue for lipid vesicles with a negatively charged surface. Langmuir. 2009;25:7177–7180. doi: 10.1021/la901676e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon T.Y., Okumus B., Ha T. Multiple intermediates in SNARE-induced membrane fusion. Proc. Natl. Acad. Sci. USA. 2006;103:19731–19736. doi: 10.1073/pnas.0606032103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon T.Y., Lu X., Shin Y.K. Complexin and Ca2+ stimulate SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 2008;15:707–713. doi: 10.1038/nsmb.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cypionka A., Stein A., Walla P.J. Discrimination between docking and fusion of liposomes reconstituted with neuronal SNARE-proteins using FCS. Proc. Natl. Acad. Sci. USA. 2009;106:18575–18580. doi: 10.1073/pnas.0906677106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuette C.G., Hatsuzawa K., Jahn R. Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc. Natl. Acad. Sci. USA. 2004;101:2858–2863. doi: 10.1073/pnas.0400044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee H.-K., Yang Y., Yoon T.Y. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science. 2010;328:760–763. doi: 10.1126/science.1187722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T.T., Tucker W.C., Weisshaar J.C. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys. J. 2005;89:2458–2472. doi: 10.1529/biophysj.105.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T.T., Smith E.A., Weisshaar J.C. Lipid mixing and content release in single-vesicle, SNARE-driven fusion assay with 1–5 ms resolution. Biophys. J. 2009;96:4122–4131. doi: 10.1016/j.bpj.2009.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fix M., Melia T.J., Simon S.M. Imaging single membrane fusion events mediated by SNARE proteins. Proc. Natl. Acad. Sci. USA. 2004;101:7311–7316. doi: 10.1073/pnas.0401779101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowen M.E., Weninger K., Chu S. Single molecule observation of liposome-bilayer fusion thermally induced by soluble n-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs) Biophys. J. 2004;87:3569–3584. doi: 10.1529/biophysj.104.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Domanska M.K., Kiessling V., Tamm L.K. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J. Biol. Chem. 2009;284:32158–32166. doi: 10.1074/jbc.M109.047381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christensen S.M., Stamou D. Surface-based lipid vesicle reactor systems: fabrication and applications. Soft Matter. 2007;3:828–836. doi: 10.1039/b702849k. [DOI] [PubMed] [Google Scholar]
- 28.Bendix P.M., Pedersen M.S., Stamou D. Quantification of nano-scale intermembrane contact areas by using fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 2009;106:12341–12346. doi: 10.1073/pnas.0903052106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kunding A.H., Mortensen M.W., Stamou D. A fluorescence-based technique to construct size distributions from single-object measurements: application to the extrusion of lipid vesicles. Biophys. J. 2008;95:1176–1188. doi: 10.1529/biophysj.108.128819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berg H.C. Princeton University Press; Princeton, NJ: 1993. Random Walks in Biology. [Google Scholar]
- 31.Bhatia V.K., Madsen K.L., Stamou D. Amphipathic motifs in BAR domains are essential for membrane curvature sensing. EMBO J. 2009;28:3303–3314. doi: 10.1038/emboj.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X.C., Araç D., Rizo J. SNARE-mediated lipid mixing depends on the physical state of the vesicles. Biophys. J. 2006;90:2062–2074. doi: 10.1529/biophysj.105.071415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neher E., Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 34.Meinrenken C.J., Borst J.G.G., Sakmann B. Local routes revisited: the space and time dependence of the Ca2+ signal for phasic transmitter release at the rat calyx of Held. J. Physiol. 2003;547:665–689. doi: 10.1113/jphysiol.2002.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takamori S., Holt M., Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.