

Abstract
Bacteriorhodopsin is a model system for membrane proteins. This seven transmembrane helical protein is embedded within a membrane structure called purple membrane. Its structural stability against mechanical stress was recently investigated by atomic force microscopy experiments, in which single proteins were extracted from the purple membrane. Here, we study this process by all-atom molecular dynamics simulations, in which single bacteriorhodopsin molecules were extracted and unfolded from an atomistic purple membrane model. In our simulations, key features from the experiments like force profiles and location of key residues that resist mechanical unfolding were reproduced. These key residues were seen to be stabilized by a dynamic network of intramolecular interactions. Further, the unfolding pathway was found to be velocity-dependent. Simulations in which the mechanical stress was released during unfolding revealed relaxation motions that allowed characterization of the nonequilibrium processes during fast extraction.
Introduction
Membrane proteins mediate a broad range of biochemical functions. Although ∼20–30% of all proteins are membrane proteins (1), only little is known about their energetics and their stability against mechanical stress. One important class of membrane proteins are G-coupled protein receptors (GPCRs) (2), which consist of seven transmembrane helices and mediate signals from the extracellular to the cytoplasmic side of a membrane, e.g., light, hormones, or neurotransmitters, by conformational changes. Therefore, knowledge of the mechanical stability and energetics of these proteins is crucial.
The light-driven proton-pump bacteriorhodopsin (BR) is a model system for seven-transmembrane helical proteins and shares many similarities to GPCRs (3,4). Photoinduced isomerization of the chromophore retinal drives a reaction cascade in which the protein undergoes several conformational changes. In vivo, BR trimers and lipid molecules arrange into a remarkably stable two-dimensional hexagonal structure called “purple membrane”.
One versatile tool to study single proteins and their structural stability is atomic force microscopy (AFM) (5), which served, e.g., to study the mechanical properties of biological macromolecules such as polysaccharides (6), DNA (7), and heparin (8). Furthermore, AFM experiments provided insight into the strength of ligand-receptor bonds (9) and the stability of soluble proteins such as titin (10) and fibronectin (11). Also, membrane bound proteins such as sodium-proton antiporters (12) and aquaporin (13) were studied.
The underlying intramolecular forces that govern macromolecular mechanical stability and unfolding have been studied by force probe molecular dynamics (FPMD) simulations (14,15), e.g., for the unfolding of titin domains (16), folding intermediates (17), the function of titin kinase (18), and the elastic properties of ankyrin (19). Also, the unfolding of membrane proteins was studied using this method (20–22).
In recent AFM experiments (23–32), single BR molecules were mechanically extracted and unfolded from the purple membrane. The forces obtained during extraction revealed a characteristic sawtooth pattern. Using the wormlike chain polymer model, these force peaks were linked to the unfolding of single helices. Contrary to former AFM experiments on soluble multidomain proteins, the order of unfolding was not from the weakest to the strongest link, but along the amino-acid sequence. Also from wormlike chain fits to the force profiles, anchor residues that exhibit considerable resistance against mechanical unfolding were located. These relatively few residues are obviously important for stabilizing the protein.
Although the forces that counteract mechanical unfolding have been precisely measured by AFM, the atomistic origin of the quite diverse behavior of the individual residues is unclear. In this work, the extraction and unfolding of single BR molecules is therefore simulated at atomic level using FPMD simulations. To validate the results from the simulations with the experiments, peak forces and anchor residues were compared. Subsequently, the simulations enabled us to characterize the interactions that provide the anchor residues with such remarkable stability.
To assess a possible influence of the loading rate, which is much larger in the simulations than in the experiments, the extraction simulations were carried out at various loading rates. Simulations in which the extraction process was stopped provided further insight into extraction at very slow velocities.
Methods
Simulation system and details
As starting structure, the BR x-ray structure 1QHJ (33) from the Protein DataBank was chosen. Residue side-chain atoms not resolved in the x-ray structure (Met163, Arg227, and Glu232) were modeled using WHAT IF (34). Terminal residues not resolved in the x-ray structure (1–4 and 233–248) were omitted from the model. BR trimers were created by applying the appropriate symmetry operations to the protein.
To obtain a full membrane system, the proteins were embedded within a hydrated POPC lipid bilayer (provided by Peter Tieleman (35)) using the method described in Faraldo-Gómez et al. (36) and employing the program MSMS (37). Nine lipid molecules per monomer (four at the cytoplasmic and five at the extracellular side) were placed in the simulation box (33). We consider POPC a reasonable mimic for the purple membrane, because BR is known to fold into its native functional state within a POPC bilayer (38). Details are provided in the Supporting Material.
A water layer of 5 nm with sodium and chloride ion concentration of 300 mM, similar to the experiments (23), was added to the hydrated system at the cytoplasmic side of the purple membrane model.
Next, energy minimization of 150 steps was performed using steepest descent. Three equilibration runs with increasingly relaxed harmonic position restraints were performed. First, restraints with a force constant of k = 1000 kJ mol−1 nm−2 were applied to all nonhydrogen atoms of protein and lipid molecules for 200 ps. Second, only the protein restraints were kept for 1 ns. Finally, a 3.5-ns equilibration run was performed without any position restraints.
To provide sufficient space for extracted polypeptides, one more 5-nm water layer was appended at the cytoplasmic side of the system, yielding a water layer of 10 nm in total (Fig. 1 A). This enlarged system was equilibrated for 500 ps.
Figure 1.
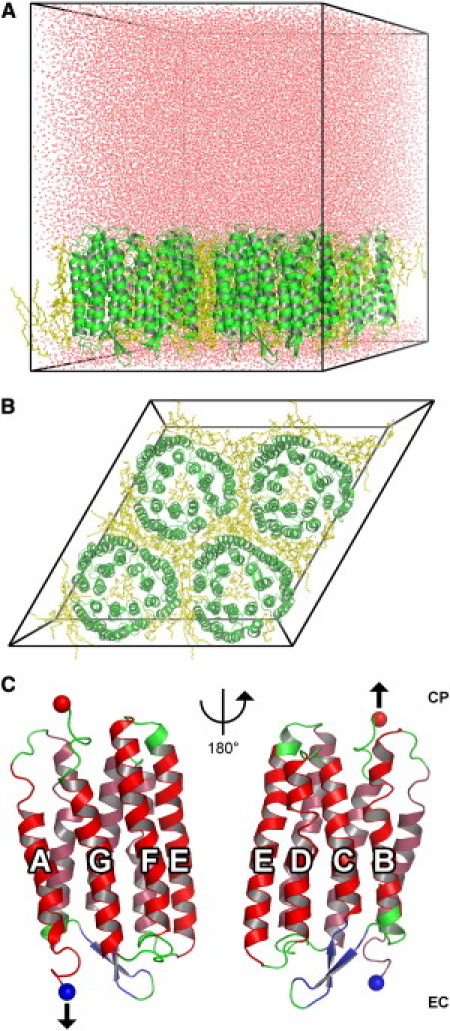
Simulation system setup. (A, Side view) The expanded volume at the cytoplasmic side provides sufficient space for extracted polypeptides. BR molecules are shown in green, lipid molecules in yellow, and water molecules in red. (Black lines) Simulation box. (B, Top view) Four unit cells, each containing a BR trimer, form a hexagonal two-dimensional crystal. Colors as before. (C) Mechanical unfolding setup. Either the C- or the N-termini (red and blue sphere, respectively) were subjected to a pulling potential as described in Methods (arrow), enforcing extraction toward the cytoplasmic and extracellular side, respectively. Colors represent the secondary structure of the crystal structure (33). Helices A–G are indicated.
To avoid periodic boundary artifacts during the extraction of one monomer, the system was replicated to construct a system of four trimers according to the crystal symmetry of a hexagonal lattice. The full simulation box thus contained 12 BR monomers (Fig. 1 B). Periodic boundary conditions were used for all simulations.
The system was equilibrated for 500 ps and used as starting structure for all subsequent cytoplasmic extraction simulations described below. For extraction simulations toward the extracellular side, the 10-nm water layer was placed at the extracellular side of the purple membrane model, and the N-terminal was subjected to a force acting downwards in Fig. 1 A. Both simulation systems comprised 236,124 atoms, with a box size of 12.16 × 12.16 × 15.32 nm3.
All simulations were carried out using the software package GROMACS 3.3 (39). Proteins and ions were described by the OPLS all-atom force field (40). The TIP4P water model (41) was employed. Lipid molecules were described with a unified atom model (42). Partial charges for the retinal were taken from Kandt et al. (43). All other force-field parameters were converted into the OPLS force field as described.
All simulations were run in the NPT ensemble. The system was coupled to a constant temperature of 300 K using a Berendsen thermostat (44) with a relaxation time constant of τ = 0.1 ps. A Berendsen barostat with a relaxation time constant of τ = 1.0 ps and a compressibility of 4.5 · 10−5 bar−1 was used to keep the pressure at 1 bar. Semiisotropic pressure coupling was used; no coupling was applied in x and y direction, whereas in z direction the box was free to adapt to pressure changes.
Long-range electrostatic interactions beyond 1.0 nm were calculated using particle mesh Ewald summation (45,46). A grid dimension of 0.12 nm and fourth-order b-spline interpolation was used. Lennard-Jones interactions were truncated at 1.0 nm. The length of bonds involving hydrogen atoms were constrained using LINCS (47). An integration time step of 2 fs was used.
FPMD simulations with various extraction velocities were carried out for extraction toward both the cytoplasmic and the extracellular side. In each simulation, the Cα atom of the C-terminus (N-terminus, respectively) (Fig. 1 C) was subjected to a harmonic pulling potential Vpull, which was moved with constant velocity in positive (negative, respectively) z direction away from the membrane, parallel to the membrane normal,
| (1) |
where k = 500 kJ mol−1 nm−2 is the spring constant, the z position of the respective Cα atom, and v the extraction velocity. An offset zoffset was introduced to allow for nonzero initial forces, as will be described below. The center of mass of the nonextracted proteins was kept fixed. No position restraints were applied.
Aiming at extracting and unfolding a complete BR monomer, the fully extended polypeptide chain would be too long to fit into the chosen simulation box. To circumvent this problem and to keep the system computationally tractable, we repeatedly cut off unfolded parts of the protein that had moved sufficiently far away from the membrane to render interactions, in particular electrostatic ones, with the membrane negligible. Accordingly, whenever the pulled Cα atom approached the simulation box boundary to below 1 nm, the unfolding simulation was stopped, extracted residues that reside >1 nm above the membrane in z direction were removed, and the voids caused by peptide removal were filled with water molecules. Ions were added or removed to keep the system neutral. New charged termini were built and the system was energy-minimized and equilibrated at 300 K for 20 ps with position restraints on heavy atoms of proteins and lipid molecules.
The extraction simulation was then resumed, with the new terminal Cα atom subjected to the new Vpull and appropriately chosen zoffset to match the initial force of the new simulation to the final force of the previous one. Specifically, the new force
was required to equal the force
applied before removal of the residues. Accordingly, and using Eq. 1 and with F = −▿V, the new offset position zoffset, new was chosen as
| (2) |
The spring constant was kept unchanged during the simulations. This procedure was iterated until complete protein extraction.
Anchor points
As observed in the AFM experiments (23), the unfolding of BR proceeds stepwise. Apparently, some residues referred to as anchor points are able to withstand a much higher force against mechanical unfolding than others. In the experiments, anchor points have been located from peaks in the force curves by wormlike chain polymer model fits (48). To facilitate direct comparison to AFM results, anchor points were also determined from our simulations as described below. In contrast to the experiments, detailed structural information is here available, which therefore has been used to determine anchor points without referring to the wormlike chain model.
We proceeded in two steps (Fig. 2). First, for each frame of an extraction simulation a transition point (Fig. 2 A, arrows) between the unfolded (red) and folded (gray) part of the protein was determined. To this end, unfolded parts of the protein were identified via their markedly increased deviation Δz of Cα positions from the x-ray structure in z direction (Fig. 2 B).
Figure 2.
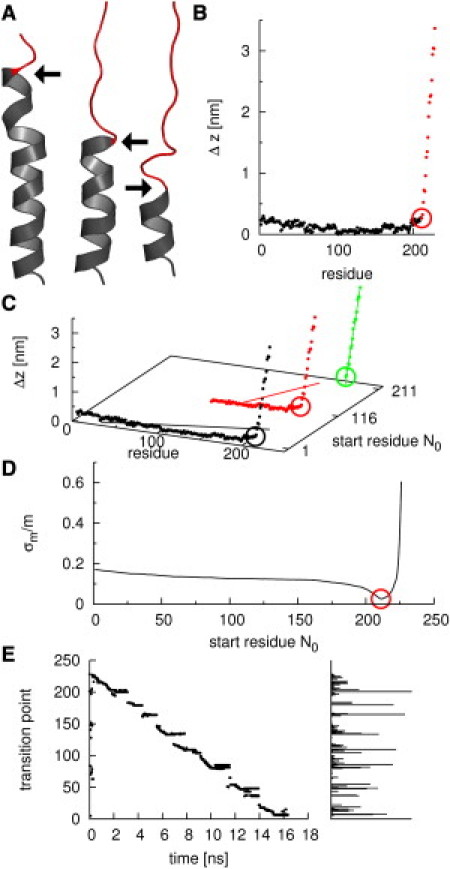
Determination of anchor points. (A) Snapshots of an unfolding helix. Black and red regions indicate folded and unfolded parts, respectively, with arrows depicting transition points. (B) Displacement Δz of residues from the crystal structure in z direction; colors as in panel A. (Circle) Transition point. (C) Transition points are determined via a line fit. Example data sets with line fits are shown. (Black) Full data set. (Red) Data set without the first 115 residues. (Green) Data set with only unfolded residues. (D) Relative error σm/m of the slope m of the line fits as a function of the first residue in the data sets. The minimal relative error was used to define the transition point (red circle). (E) Time development (left) and frequency (right) of transition points determined from an extraction simulation.
Transition points were defined in an automated manner by using the fact that Δz values of the unfolded part of the protein increase nearly linearly with residue number, in contrast to the folded part. Accordingly, transition points were determined as the start of this linear range. To that end, the quality of fits to varying ranges (Fig. 2 C, different colors) with varying start residues N0 but the same end residue was used. (For mechanical extraction toward the cytoplasmic side, conversely, ranges with the same start residues but different end residues were used.)
Accordingly, a linear function with slope m and intercept b was fitted to the Δzi values for all residues i in each of the considered ranges with length n (Fig. 2 C) using
| (3) |
and
| (4) |
The fit quality was quantified by the relative error σm/m of the slope, with
| (5) |
Fig. 2 D shows the fit quality as a function of start residue number N0 for the chosen example. Transition points were then defined as the minimum of σm/m (red circle).
The left part of Fig. 2 E shows transition points determined from an extraction simulation as described above. As can be seen—and as quantified by the histogram (right side)—some transition points occur markedly more often than others. This histogram served to determine the location of the anchor points for comparison with the AFM data.
Due to the stochastic nature of the unfolding process, histograms obtained from separate trajectories will differ from each other. To improve statistics, all histograms for a given extraction velocity were merged and filtered using a Gaussian filter with a one-residue half width.
Results and Discussion
After equilibration, 27 extraction simulations were carried out, both toward the cytoplasmic and the extracellular side. To study to which extent the unfolding path and anchor points are velocity-dependent, unfolding simulations were carried out at five different extraction velocities ranging from 50 m/s to 1 m/s. Each simulation was terminated after complete extraction of the respective BR monomer. As also observed in the AFM experiments (23), adjacent proteins were essentially unperturbed, leaving a stable water-filled hole at the former position of the extracted protein. Except otherwise noted, we will focus at those observables that were similar for all simulations and velocities.
Force profiles
Fig. 3, A and B, shows typical force profiles obtained from simulations at different extraction velocities for extraction toward the cytoplasmic (A) and the extracellular side (B). All force profiles show four main force peaks labeled 1, 2, 3, and 4 at similar spring positions. Their overall shape and position is similar to the force peaks obtained by AFM. Some of the main force peaks split up into subpeaks, e.g., the first and third peak of the cytoplasmic trajectories. Overall, the force pattern does not depend on the extraction velocity.
Figure 3.
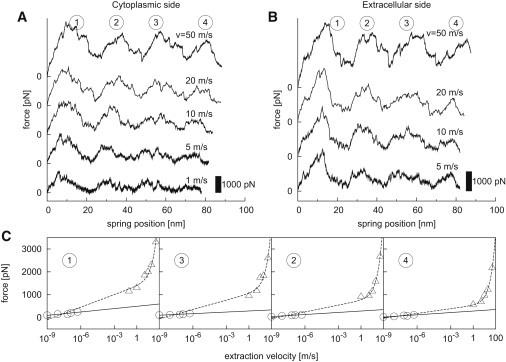
(A and B) Sample force profiles from extraction simulations for different extraction velocities toward the cytoplasmic (A) and the extracellular side (B). All force profiles start at 0 pN. (C) Comparison between peak forces from AFM experiments (circles) and extraction simulations (triangles) for extraction toward the cytoplasmic side. Two fits are shown for each peak; a logarithmic fit to only the AFM data (solid lines) and a fit to both AFM and MD data using Eq. 6 (dashed lines).
For high extraction velocities (20 m/s and 50 m/s), abrupt force jumps are occasionally observed. These drops occur when peptides are removed from the system as described in Methods and are due to transiently reduced frictional drag of the unfolded peptide after cutting.
As in the experiments, the force peaks observed in the simulations decreased from peak 1 to peak 4. We attribute this effect to the decreasing number and strength of interactions within the nonextracted protein due to the extraction of unfolded parts during the later parts of the simulations. Force minima reach 0 pN for extraction velocities below 5 m/s. This indicates that at these velocities, as also reported in Gräter et al. (18), frictional forces become small with respect to unfolding forces.
Comparison to AFM forces
Fig. 3 C compares the heights of the force peaks between simulation (triangles) and experiment (26) (circles) for different extraction velocities. In accordance to the AFM force peaks, the MD force peaks increase with faster extraction velocities. To compare the unfolding forces observed in the simulations with those measured by AFM at much slower extraction velocities, a simple model for the logarithmic velocity dependence of activated barrier crossing is used (18,49),
| (6) |
Here, γ denotes a friction coefficient and a and v0 are fitting parameters.
Fits to both AFM and MD values (Fig. 3 C, dashed lines) yield a good description for all MD force peaks and for AFM force peak 4. For AFM force peaks 1, 2, and 3, the logarithmic slope of the AFM force peaks (solid lines) is smaller than that of the fit to both AFM and MD, indicating that for these cases the unfolding pathway between both methods may differ.
Remarkably, this discrepancy decreases, and eventually vanishes, for the later stages of the unfolding process, where fewer sterical hindrances obstruct further unfolding. This indicates that, besides the intrinsic unfolding forces of the helices, interactions to the remaining protein contribute to the overall forces. The additional velocity dependence can be explained by structural rearrangements that may occur during AFM extraction but that are too slow for MD timescales.
To examine the causes of the force peaks during extraction, the unfolding pathway of the protein during the extraction simulations will be analyzed in more detail and compared to the unfolding pathway proposed by the AFM experiments.
Unfolding pathway
Fig. 4 shows unfolding snapshots of a BR monomer. As example, the unfolding and extraction of helices G and F toward the cytoplasmic side at an extraction velocity of 5 m/s is shown. The snapshots show a continuous and sequential unfolding and extraction of the two helices. As in the experiments, the order of the extraction on the helix scale is given by the extraction direction: For extraction toward the cytoplasmic side, helix G unfolds first and helix A last. This order is reversed for extraction toward the extracellular side.
Figure 4.
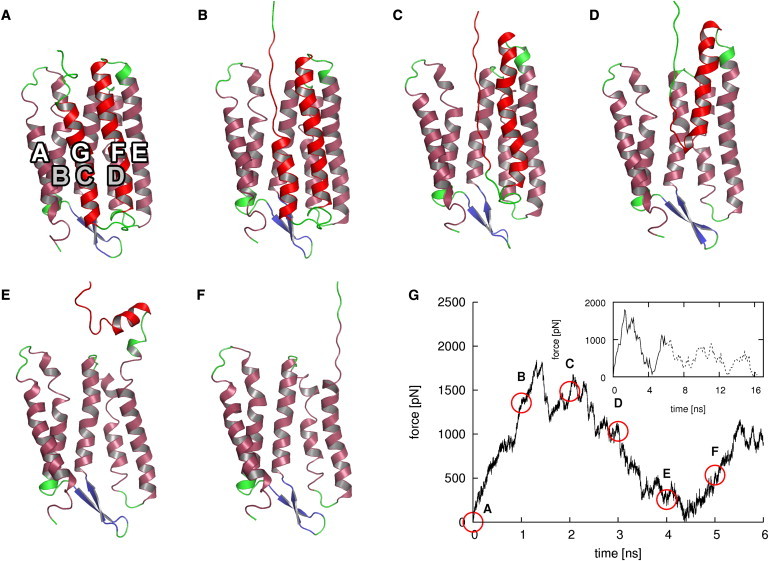
Unfolding pathway of helices G and F of BR (A–F) and corresponding forces (G). Snapshots were taken at 1 ns intervals. Colors as in Fig. 1C. Bold letters denote transmembrane helices. Gray letters belong to helices that are only partly visible. Red circles in the force profile denote snapshot times. (Inset) Complete force profile. The solid part corresponds to the main plot.
On the level of the individual helices, a sequential unfolding of individual helix turns is seen (Fig. 4, A–F). During unfolding of part of each helix, only small structural changes are seen in its remaining folded part and the remaining helices. One exception is helix E, part of which unfolds together with helix F (Fig. 4 E). Most unfolding events occur within the membrane, with the unfolding helix nearly in place and oriented parallel to the remaining helices (Fig. 4, A–D). An exception are the last parts of helices F, D, and B, which are dragged out of the membrane partly intact, then kink with respect to their original alignment, and unfold in bulk water parallel to the membrane surface.
The rise and fall of the force during extraction correlates with the unfolding of single helices (Fig. 4 G). During the unfolding of helix G and F the force increases and decreases, respectively. Upon complete unfolding of helix G and F, the force reaches its maximum and minimum, respectively. The extraction and unfolding of helix pairs E and D as well as C and B creates a force peak in a similar manner. The last peak in the force spectrum is caused by the unfolding and extraction of helix A. Extraction toward the extracellular side proceeds in a similar manner.
Anchor points
One main result of the experiments was that at certain residues, referred to as anchor points, larger forces were required for unfolding to proceed. Apparently, these residues resist mechanical unfolding particularly strongly, which indicates strong interactions relevant for the general stability of the protein. In the experiments, anchor point locations were determined via wormlike chain fits to the force curves (48). To locate anchor points in the extraction simulations, all trajectories were subjected a systematic search protocol.
To that aim, transition points between folded and unfolded parts of the protein were determined during the course of the simulations. Because anchor points are expected to delay unfolding, transition points should occur more frequently at their locations, and should therefore show up as peaks in transition point histograms.
Indeed, transition point histograms reveal a noncontinuous unfolding in the simulations (Fig. 5, A and B, light gray areas). Maxima from this distribution can be clearly identified and define anchor points. Anchor points from the extraction simulations show no velocity-dependent locations and nearly all are seen for all extraction velocities at each respective extraction direction.
Figure 5.
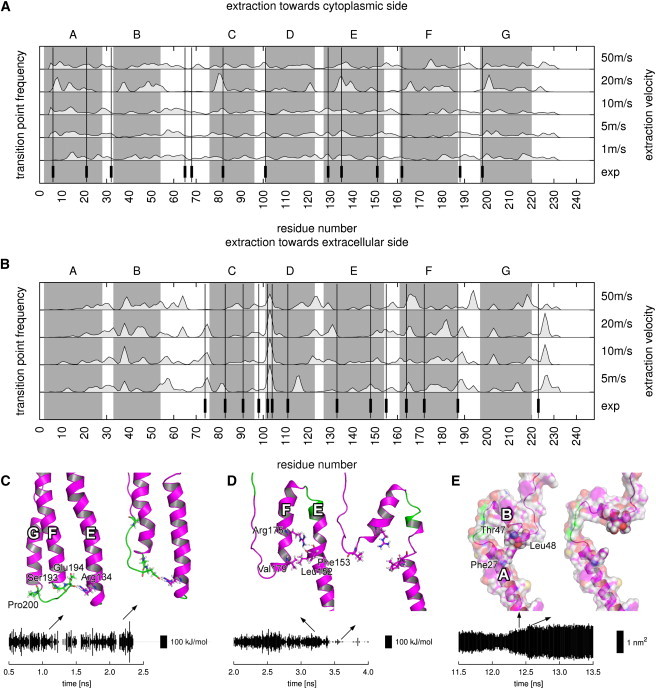
(A and B) Comparison between AFM anchor points (vertical bars and lines) (28) and frequency of transition points determined by MD (light gray areas). Transition point histograms are filtered with a Gaussian filter with a one residue half width. (Dark gray areas) Helices. (C–E) Structural and energetic determinants of anchor points. Each panel shows snapshots before and after rupture of an anchor point. The plots show hydrogen bond energies (C and D) or hydrophobic solvent-accessible surface areas (E) of selected residues. Bold letters denote helices. Key residues are shown as sticks (C and D) or spheres (E). (C) Anchor point Pro200 in a stable conformation and shortly before rupture. The plot shows the summed energy of hydrogen bonds between Arg134 and Ser193 and Glu194. (D) Anchor point Val179 before and after rupture. The plot shows the summed energy of hydrogen bonds belonging to Arg175. (E) Anchor point Leu48 before and after rupture. The plot shows the hydrophobic solvent accessible surface area of Phe27.
To better compare anchor points from AFM and MD, we divided the latter into three groups: The first group consists of MD anchor points that cover AFM anchor points within their respective uncertainties. A second group is defined as MD anchor points occurring close to AFM anchor points. The first two groups cover ∼40% of all anchor points for both extraction directions, showing a good agreement between both methods. For the last group, no match between AFM and MD data is seen.
In the first group, almost all AFM anchor points are also seen in the simulations. In the case of extraction toward the cytoplasmic side, AFM anchor points Gly6, Arg82, Val101, Lys129, and Phe135 are well matched by MD anchor points. For extraction toward the extracellular side, Glu74, Leu100, and Asp102 show a good agreement. Given their occurrence in both AFM and MD, these anchor points seem to be independent of the extraction velocity, and the interactions revealed by the simulations are likely to also dominate the AFM forces.
For the second group, a noticeable deviation of up to three residues between anchor points from MD and from AFM is seen. Notable examples are AFM anchor points Ile198 and Val188 for extraction toward the cytoplasmic side and Arg164 and Leu223 for extraction toward the extracellular side. These deviations may arise either from uncertainties in the experimental determination of the anchor points via the wormlike chain model (28), or, alternatively from a possible velocity dependence of anchor point locations. Like the first group, this group agrees remarkably well with the AFM experiments.
The third group, where no match between AFM and MD is observed, falls into two categories. The first, less frequent category contains anchor points from AFM that are not observed by MD. Only three anchor points fall into this category, Tyr83, Pro91, and Leu111 from the extraction toward the extracellular side. The fact that these anchor points are only seen by AFM indicates a velocity dependence of the underlying reaction paths. The second, more frequent category includes all MD anchor points that are not seen by AFM. Most notably, MD anchor points are observed in helices B, D, F, and G during the extraction toward the cytoplasmic side and in helices A, B, F, and G and the loop region connecting helices D and E during the extraction toward the extracellular side.
We assume that these anchor points are not seen by AFM because they occur during force drops, whereas rising forces are needed to detect anchor points in the analysis of the experiments (48). Because the transition point method used for the simulations does not involve forces, anchor points can also be detected during decreasing forces. This idea is also confirmed by the strongly decreasing forces that accompany the unfolding of helices B, D, and F in both extraction directions (see Fig. 3) and explains almost all MD anchor points not seen by AFM.
Remarkably, anchor points occur more frequently in the extraction simulations toward the cytoplasmic side than in the opposite direction. We attribute this finding to interactions between the retinal and the helices in contact with it that stabilize the protein core. As the retinal is covalently bound to Lys216 located in helix G, it is removed upon extraction of helix G. Because in the extraction simulations toward the cytoplasmic side, helix G is removed from the protein core already very early in each simulation, the stabilization is lost, and a larger number of weaker interactions dominate. In contrast, for extraction toward the extracellular side, the retinal remains within the protein for nearly the complete process, and, due to the interactions with the retinal, more pronounced but possibly fewer anchor points are expected. This is seen in Fig. 5, A and B. In this scenario, one would also expect more details and anchor points in the force profile for bacterioopsin extraction than seen for BR, which indeed has been observed (24).
Energetics causing anchor points
Which are the interactions that stabilize intermediate structures and thus cause the observed anchor points? To address this question, we analyzed the sequence of structural changes and the rupture of intramolecular interactions leading to the occurrence of these points in more detail.
Analysis of interactions between each residue and the remaining protein revealed that the residues lose their interactions in sequential order (see Fig. S1 in the Supporting Material). This behavior even applies to hot-spot residues with higher interaction energies. Fluctuations of residuewise energies suggest a rearrangement of interactions within the protein.
Based on their location within the helices and on their stabilizing energetics, we divided anchor points into three groups. The first group consists of anchor points that are located at the ends of the transmembrane helices and are stabilized by interhelical hydrogen bonds. Anchor points in the second group are also stabilized by interhelical hydrogen bonds but reside in the center of the helices. The third group consists of anchor points in the center of helices that are stabilized by hydrophobic contacts.
Fig. 5 C depicts Pro200 as a typical example of anchor points from the first group. Upon the unfolding of helix G, the extracellular end of the adjacent helix F is held back by hydrogen bonds between backbone atoms from Ser193 and Glu194 from helix F to side-chain atoms of Arg134 from helix E (first snapshot). Before complete unfolding of helix G, a rearrangement of interactions is seen: New hydrogen bonds between side-chain atoms from Arg134 to side-chain atoms from Ser193 and Glu194 are formed (second snapshot). The nearly constant energy of hydrogen bonds belonging to Arg134 (lower plot) shows that this rearrangement preserves the strength of interactions that hold back helix F. To this end, a dynamic network of hydrogen bonds connects terminal regions of adjacent transmembrane helices.
Anchor points from the second group are also stabilized by hydrogen bonds but are located in the center of the helices, like Val179 located in helix F (Fig. 5 D). Upon extraction, hydrogen bonds between side-chain atoms of the nearby Arg175 to backbone atoms from residues from helix E maintain the orientation of helix F (first snapshot). Once these interactions break up, the remaining part of helix F kinks with respect its former orientation (second snapshot).
In contrast, anchor points belonging to the third group, like Leu48 located in helix B (Fig. 5 E), are stabilized by nonpolar interactions rather than hydrogen bonds. Phe27, Thr47, and Leu48 form a hydrophobic core between helices A and B that disintegrates during unfolding of helix B, indicated by the increasing hydrophobic surface area of Phe27 (lower graph). Once the hydrophobic core is completely disrupted (maximal hydrophobic surface), the remaining part of helix B tilts away from its former alignment (second snapshot).
A table of anchor points observed in extraction simulations toward the cytoplasmic side is available in Table S1.
Our analysis of mechanically relevant interactions in BR thus revealed as the main determinants for the observed anchor points a network of hydrogen bonds and hydrophobic contacts. Further, a highly dynamic rearrangement of these interactions during extraction is seen. The competition of quite different kinetics suggests that the observed unfolding pathway depends on the timescale set by the extraction velocity. Thus, the question arises, how slower extraction velocities and correspondingly longer timescales of extraction would influence the unfolding process.
Relaxation processes
Indeed, the different kinetics of interactions described in the previous section suggests a velocity-dependent unfolding pathway. This was also proposed by fits to the peak forces obtained by AFM and MD (see Comparison to AFM Forces). Thus, for sufficiently small extraction velocities, we would expect to observe also in the simulations a simultaneous unfolding of helix pairs rather than the sequential unfolding of single helices seen so far.
To test this hypothesis, we determined how the unfolding pathway of partially unfolded helices proceeds once the extraction velocity is changed from fast MD values (∼10 m/s) to slow AFM values (∼10−9 m/s). Because the difference in velocities is very large, extraction at AFM velocities is quasistatic within MD timescales. To this end, we took snapshots from an extraction simulation with v = 20 m/s as start for simulations with a resting spring, v = 0 m/s. To study an extended unfolding pathway, we chose snapshots where helices G or F were partly unfolded.
In both simulations, forces began to decrease instantaneously (Fig. 6 A). The force development can be described by a biexponential decay
| (7) |
with slow and fast relaxation times τslow and τfast, respectively. Ffinal, F1, and F2 are fit constants (values are given in Table S2). The fast relaxation times ((730 ± 5) ps and (95 ± 2) ps for the simulation with a partially unfolded helix G and F, respectively) can be attributed to the loss of friction built up during the preceding extraction simulation. By assuming that the attachment point for the harmonic potential moves ∼1 nm during the fast relaxation, a minimal velocity of vanishing relaxation can be determined using v < 1 nm/τfast. For a time constant of τfast ∼ 1 ns, one obtains v < 1 m/s. This is in agreement with the previous observation that frictional forces become negligible below extraction velocities of 5 m/s (see Force Profiles).
Figure 6.
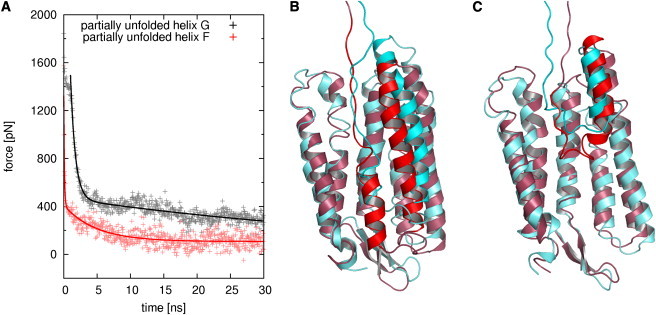
Relaxation simulations with partially unfolded helices G (gray) and F (red), respectively. (A) Forces (symbols) and biexponential fits (lines). The force fit for the first simulation starts after the retinal was extracted from the protein core. (B) Snapshots before (red) and after (cyan) the relaxation simulation with a partially unfolded helix G. (C) Similar, but for a partially unfolded helix F (same colors).
The slow relaxation seen in the later part of the simulations revealed much longer relaxation times (τslow = (55,000 ± 6000) ps and τslow = (4810 ± 60) ps for the first and second simulation, respectively). Because in the slowest extraction simulation (v < 1 m/s) the unfolding of the protein takes ∼80 ns, relaxation times of ∼50 ns can only be covered by much slower extraction velocities not accessible to our simulations. However, it should be possible to observe structural relaxation in the 30-ns relaxation simulations.
In the first relaxation simulation, a shift of both helices G and F in extraction direction was observed (Fig. 6 B). Helix G did not further unfold but remained parallel to helix F during further extraction, which was not observed in the extraction simulations. This clearly indicates mutual extraction and unfolding of helices G and F at long timescales. We conclude that for very small extraction velocities, the unfolding pattern observed by AFM already shows up in the simulations.
The partially unfolded helix F (Fig. 6 C) showed further unfolding during the slow relaxation phase at small forces (∼100 pN) (Fig. 6 C). Because peak forces in the experiments are in a similar range and helix F unfolds after a peak, a spontaneous unfolding of helix F in the experiments can be assumed. Fast unfolding is further corroborated by the short relaxation time of ∼5 ns.
The results from both relaxation simulations show that the unfolding pathway of BR under external force changes with extraction velocity and is therefore timescale-dependent. Based on these observations, the discrepancies in the proposed unfolding pathways can be explained. At slow extraction velocities in AFM experiments (<10−7 m/s), the extraction and unfolding of pairs of helices is visible, whereas at high extraction velocities in MD simulation (>1 m/s), single helix turns unfold. This is also in line with the observation of the unfolding of single helices at higher AFM extraction velocities (>10−7 m/s) (26).
Summary and Conclusion
We presented all-atom FPMD simulations in which we extracted and unfolded single BR monomers from an atomistic purple membrane model. The extraction was carried out at various velocities both toward the cytoplasmic and the extracellular side of the membrane. To maintain a computationally tractable box size, a new protocol was introduced and successfully tested, in which the extracted and unfolded part of the protein was removed regularly and the extraction continued at the cleaved part of the protein, with the respective forces adjusted appropriately.
Force profiles obtained from the simulations were similar to those obtained by AFM in many respects. In both methods, four distinctive peaks with decreasing values were observed, pointing to a sequential unfolding of the protein. For extraction toward the cytoplasmic side, extraction and unfolding of each helix pair GF, DE, and BC, as well as of helix A, caused a force peak. In contrast to the experiments, which suggested a concerted unfolding of helix pairs, in the simulations we observed a sequential unfolding of individual helix turns, although—as in the experiments—only every second helix actually gave rise to a pronounced force peak. This observation suggests a second possible interpretation of the AFM force profiles, according to which the experiments would also be compatible with sequential unfolding of individual helices. In any case, details of the sequence of events during unfolding may depend on the loading rate.
Despite these possible differences, similar anchor points were seen by MD, suggesting that the underlying molecular interactions are largely independent of extraction velocity. MD anchor points fall into three groups. The first group comprises anchor points that fully agree with those seen by AFM, the second group consists of anchor points close to AFM anchor points, and the third group includes anchor points not seen by AFM. Most anchor points of the last group occur after the main force peaks, during force drops, where they are unlikely to be seen in the experiments, which can explain this discrepancy. Overall, remarkably good agreement is seen, with the position of most anchor points being largely unaffected by the quite different timescales of AFM and MD.
As the dominant molecular interactions that stabilize the anchor points and, hence, govern the mechanical stability of BR, we identified hydrogen bonds and hydrophobic contacts. During extraction, these interactions form a highly dynamic network, where transient interactions are established subsequent to the rupture of old ones. Thus, the unfolding of BR does not follow a predefined path; rather, the competition between the kinetics of the interaction network and the unfolding timescale can give rise to different velocity dependent unfolding pathways.
To explore this possibility further, relaxation simulations were carried out to detect slow relaxation motions that are not visible in the fast extraction simulations. To this end, extraction simulations with a partially unfolded protein were taken and continued without further extraction. Indeed, an onset of spontaneous collective unfolding of both helices G and F was seen, which deviated from the fast unfolding pathway, toward the one suggested by AFM carried out at much longer timescales.
The good agreement between the results from the experiments and the simulations suggest that our simulations capture the essential characteristics of BR extraction. Because the simulations were performed with a POPC membrane model for the purple membrane, one might expect slightly changed interactions between the protein and the bilayer. However, as our model fully includes the hexagonal symmetry of the purple membrane and therefore also all intermolecular contacts, our results support the role of this specific arrangement of BR within the bilayer.
It will be interesting to see whether dynamic interaction networks of the type observed in this work also govern the timescale-dependent mechanical properties of other GPCRs or even members of other membrane protein families. Further, this approach may also reveal atomistic details of the origin of recently observed mechanical fingerprints of ligand binding (50,51).
Acknowledgments
The authors thank Daniel Müller, Hermann Gaub, and Max Kessler for helpful discussions, and Ulrike Gerischer and Ulrich Zachariae for carefully reading the manuscript.
Supporting Material
References
- 1.Krogh A., Larsson B., Sonnhammer E.L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 2.Pierce K.L., Premont R.T., Lefkowitz R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 3.Stoeckenius W., Bogomolni R.A. Bacteriorhodopsin and related pigments of halobacteria. Annu. Rev. Biochem. 1982;51:587–616. doi: 10.1146/annurev.bi.51.070182.003103. [DOI] [PubMed] [Google Scholar]
- 4.Haupts U., Tittor J., Oesterhelt D. Closing in on bacteriorhodopsin: progress in understanding the molecule. Annu. Rev. Biophys. Biomol. Struct. 1999;28:367–399. doi: 10.1146/annurev.biophys.28.1.367. [DOI] [PubMed] [Google Scholar]
- 5.Binnig G., Quate C.F., Gerber C. Atomic force microscope. Phys. Rev. Lett. 1986;56:930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- 6.Rief M., Oesterhelt F., Gaub H.E. Single molecule force spectroscopy on polysaccharides by atomic force microscopy. Science. 1997;275:1295–1297. doi: 10.1126/science.275.5304.1295. [DOI] [PubMed] [Google Scholar]
- 7.Lee G.U., Chrisey L.A., Colton R.J. Direct measurement of the forces between complementary strands of DNA. Science. 1994;266:771–773. doi: 10.1126/science.7973628. [DOI] [PubMed] [Google Scholar]
- 8.Marszalek P.E., Oberhauser A.F., Fernandez J.M. The force-driven conformations of heparin studied with single molecule force microscopy. Biophys. J. 2003;85:2696–2704. doi: 10.1016/s0006-3495(03)74692-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Florin E.L., Moy V.T., Gaub H.E. Adhesion forces between individual ligand-receptor pairs. Science. 1994;264:415–417. doi: 10.1126/science.8153628. [DOI] [PubMed] [Google Scholar]
- 10.Rief M., Gautel M., Gaub H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 11.Li L., Huang H.H.-L., Fernandez J.M. Mechanical unfolding intermediates observed by single-molecule force spectroscopy in a fibronectin type III module. J. Mol. Biol. 2005;345:817–826. doi: 10.1016/j.jmb.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 12.Kedrov A., Ziegler C., Müller D.J. Controlled unfolding and refolding of a single sodium-proton antiporter using atomic force microscopy. J. Mol. Biol. 2004;340:1143–1152. doi: 10.1016/j.jmb.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 13.Möller C., Fotiadis D., Müller D.J. Determining molecular forces that stabilize human aquaporin-1. J. Struct. Biol. 2003;142:369–378. doi: 10.1016/s1047-8477(03)00066-2. [DOI] [PubMed] [Google Scholar]
- 14.Grubmüller H., Heymann B., Tavan P. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 1996;271:997–999. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- 15.Izrailev S., Stepaniants S., Schulten K. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 1997;72:1568–1581. doi: 10.1016/S0006-3495(97)78804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu H., Isralewitz B., Schulten K. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys. J. 1998;75:662–671. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gräter F., Grubmüller H. Fluctuations of primary ubiquitin folding intermediates in a force clamp. J. Struct. Biol. 2007;157:557–569. doi: 10.1016/j.jsb.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Gräter F., Shen J., Grubmüller H. Mechanically induced titin kinase activation studied by force-probe molecular dynamics simulations. Biophys. J. 2005;88:790–804. doi: 10.1529/biophysj.104.052423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sotomayor M., Corey D.P., Schulten K. In search of the hair-cell gating spring elastic properties of ankyrin and cadherin repeats. Structure. 2005;13:669–682. doi: 10.1016/j.str.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Cieplak M., Filipek S., Krzyśko K.A. Pulling single bacteriorhodopsin out of a membrane: comparison of simulation and experiment. Biochim. Biophys. Acta. 2006;1758:537–544. doi: 10.1016/j.bbamem.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 21.Seeber M., Fanelli F., Caflisch A. Sequential unfolding of individual helices of bacterioopsin observed in molecular dynamics simulations of extraction from the purple membrane. Biophys. J. 2006;91:3276–3284. doi: 10.1529/biophysj.106.088591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fanelli F., Seeber M. Structural insights into retinitis pigmentosa from unfolding simulations of rhodopsin mutants. FASEB J. 2010;24:3196–3209. doi: 10.1096/fj.09-151084. [DOI] [PubMed] [Google Scholar]
- 23.Oesterhelt F., Oesterhelt D., Müller D.J. Unfolding pathways of individual bacteriorhodopsins. Science. 2000;288:143–146. doi: 10.1126/science.288.5463.143. [DOI] [PubMed] [Google Scholar]
- 24.Müller D.J., Kessler M., Gaub H. Stability of bacteriorhodopsin α-helices and loops analyzed by single-molecule force spectroscopy. Biophys. J. 2002;83:3578–3588. doi: 10.1016/S0006-3495(02)75358-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janovjak H., Kessler M., Müller D.J. Unfolding pathway of native bacteriorhodopsin depend on temperature. EMBO J. 2003;22:5220–5229. doi: 10.1093/emboj/cdg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janovjak H., Struckmeier J., Müller D.J. Probing the energy landscape of the membrane protein bacteriorhodopsin. Structure. 2004;12:871–879. doi: 10.1016/j.str.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Cisneros D.A., Oesterhelt D., Müller D.J. Probing origins of molecular interactions stabilizing the membrane proteins halorhodopsin and bacteriorhodopsin. Structure. 2005;13:235–242. doi: 10.1016/j.str.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Kessler M., Gaub H.E. Unfolding barriers in bacteriorhodopsin probed from the cytoplasmic and the extracellular side by AFM. Structure. 2006;14:521–527. doi: 10.1016/j.str.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 29.Kessler M., Gottschalk K.E., Gaub H.E. Bacteriorhodopsin folds into the membrane against an external force. J. Mol. Biol. 2006;357:644–654. doi: 10.1016/j.jmb.2005.12.065. [DOI] [PubMed] [Google Scholar]
- 30.Sapra K.T., Doehner J., Muller D.J. Role of extracellular glutamic acids in the stability and energy landscape of bacteriorhodopsin. Biophys. J. 2008;95:3407–3418. doi: 10.1529/biophysj.108.131904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sapra K.T., Balasubramanian G.P., Muller D.J. Point mutations in membrane proteins reshape energy landscape and populate different unfolding pathways. J. Mol. Biol. 2008;376:1076–1090. doi: 10.1016/j.jmb.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sapra K.T., Park P.S.-H., Muller D.J. Mechanical properties of bovine rhodopsin and bacteriorhodopsin: possible roles in folding and function. Langmuir. 2008;24:1330–1337. doi: 10.1021/la702299z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belrhali H., Nollert P., Pebay-Peyroula E. Protein, lipid and water organization in bacteriorhodopsin crystals: a molecular view of the purple membrane at 1.9 Å resolution. Structure. 1999;7:909–917. doi: 10.1016/s0969-2126(99)80118-x. [DOI] [PubMed] [Google Scholar]
- 34.Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. 29. [DOI] [PubMed] [Google Scholar]
- 35.Tieleman D.P., Berendsen H.J.C. A molecular dynamics study of the pores formed by E. coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylethanolamine bilayer. Biophys. J. 1998;74:2786–2801. doi: 10.1016/S0006-3495(98)77986-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faraldo-Gómez J.D., Smith G.R., Sansom M.S.P. Setting up and optimization of membrane protein simulations. Eur. Biophys. J. 2002;31:217–227. doi: 10.1007/s00249-002-0207-5. [DOI] [PubMed] [Google Scholar]
- 37.Sanner M.F., Olson A.J., Spehner J.-C. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 38.Curran A.R., Templer R.H., Booth P.J. Modulation of folding and assembly of the membrane protein bacteriorhodopsin by intermolecular forces within the lipid bilayer. Biochemistry. 1999;38:9328–9336. doi: 10.1021/bi982322+. [DOI] [PubMed] [Google Scholar]
- 39.van der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 40.Jorgensen W.L., Tirado-Rives J. The OPLS potential functions for proteins—energy minimizations for crystals of cyclic-peptides and crambin. J. Am. Chem. Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 41.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Comput. Phys. 1983;79:926–935. [Google Scholar]
- 42.Berger O., Edholm O., Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kandt C., Schlitter J., Gerwert K. Dynamics of water molecules in the bacteriorhodopsin trimer in explicit lipid/water environment. Biophys. J. 2004;86:705–717. doi: 10.1016/S0006-3495(04)74149-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 45.Darden T., York D., Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 46.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald potential. J. Chem. Phys. 1995;103:8577–8592. [Google Scholar]
- 47.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 48.Bustamante C., Marko J.F., Smith S. Entropic elasticity of λ-phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 49.Heymann B., Grubmüller H. AN02/DNP-hapten unbinding forces studied by molecular dynamics atomic force microscopy simulations. Chem. Phys. Lett. 1999;303:1–9. [Google Scholar]
- 50.Kedrov A., Ziegler C., Müller D.J. Differentiating ligand and inhibitor interactions of a single antiporter. J. Mol. Biol. 2006;362:925–932. doi: 10.1016/j.jmb.2006.07.049. [DOI] [PubMed] [Google Scholar]
- 51.Cisneros D.A., Oberbarnscheidt L., Muller D.J. Transducer binding establishes localized interactions to tune sensory rhodopsin II. Structure. 2008;16:1206–1213. doi: 10.1016/j.str.2008.04.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.