

Abstract
There are few examples of host signals that are beneficial to bacteria during infection. Here we found that 31 out of 42 host immunoregulatory chemokines were able to induce release of the virulence factor protein A (SPA) from a strain of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). Detailed study of chemokine CXCL9 revealed that SPA release occurred through a post-translational mechanism and was inversely proportional to bacterial density. CXCL9 bound specifically to the cell membrane of CA-MRSA, and the related SPA-releasing chemokine CXCL10 bound to both cell wall and cell membrane. Clinical samples from patients infected with S. aureus and samples from a mouse model of CA-MRSA skin abscess all contained extracellular SPA. Further, SPA-releasing chemokines were present in mouse skin lesions infected with CA-MRSA. Our data identify a potential new mode of immune evasion, in which the pathogen exploits a host defense factor to release a virulence factor; moreover, chemokine binding may serve a scavenging function in immune evasion by S. aureus.
Keywords: Antimicrobial Peptides, Bacteria, Chemokines, Immunology, Subcellular Fractionation, Host Defense, Immune Evasion
Introduction
Staphylococcus aureus causes a broad range of human disease, including skin and soft tissue infections, osteomyelitis, endocarditis, pneumonia, and septicemia (1). Mortality attributed to S. aureus in the United States is estimated to be ∼18,000 per year (2), and the organism appears to be increasing in virulence due to multiple adaptations, including increasing resistance to antibiotics.
Although many virulence factors have been identified in S. aureus, little is known about how they are regulated in the host environment. Quorum sensing is a general mechanism for inter- and intra-species communication among bacteria (3–6), however, there are few examples of bacterial pathogens that can detect and respond to host-derived signals directly (7, 8). Chemokines are cationic peptides that the host uses as signals for recruitment of immune cells to sites of infection; in addition, some chemokines have been reported to have antimicrobial activity, including anti-staphylococcal activity (9, 10), suggesting that direct interactions with microbes may occur. Because chemokines are characteristically found in high concentrations at sites of infection in vivo, chemokine recognition could be exploited by bacteria to sense host responses and regulate deployment of virulence factors.
While evaluating the mechanisms responsible for chemokine antistaphylococcal activity, we made the serendipitous finding that a large subset of them is able to induce release of the virulence factor S. aureus protein A (SPA)2 from community-associated methicillin-resistant S. aureus (CA-MRSA), and in the present report we characterize this activity.
EXPERIMENTAL PROCEDURES
Reagents
Recombinant human chemokines were obtained from either Peprotech (Rocky Hill, NJ) or R&D (Minneapolis, MN). The CA-MRSA strain FPR3757 was purchased from American Type Culture Collection (Manassas, VA). This is a USA300 LAC epidemic strain whose genome has been completely sequenced, that was originally isolated from an HIV patient (11). Primary rabbit polyclonal antibodies to human CXCL9 were a gift from Dr. Joshua Farber (NIAID, NIH). Antibodies to CXCL10 (rabbit polyclonal ab9807), antibodies to protein A conjugated with HRP (goat polyclonal ab7245), and secondary antibodies were purchased from Abcam (Cambridge, MA).
SPA Release Assay
Bacteria were grown overnight from frozen permanent stock. Fresh trypticase soy broth (TSB) was inoculated with this overnight culture at a 1:100 dilution and grown to midlog phase, A600 = 0.5–1.0. Bacteria were resuspended into phosphate buffered saline (PBS) and brought to a final target A600 indicated in the figure legends by mixing with an appropriate concentration of chemokine diluted in PBS. All incubations were carried out at room temperature (∼24 °C) with shaking (250 rpm). Separate reaction tubes were used for each time point. At designated time points, reactions were sampled for assay of colony forming units (CFU) and then spun at 15,000 rpm for 5 min. Before Western blot analysis, protein concentration in the supernatants was measured by micro BCA assay (Thermo Fisher Scientific, Rockford, IL) to ensure that equal amounts of protein were loaded onto SDS-PAGE gels. Protein A EIA kit (Enzo Life Sciences, Plymouth Meeting, PA) was used to determine SPA concentrations in samples according to the manufacturer's instructions. For total SPA concentration, a 10-fold dilution was used. An equal amount of protein was loaded in the stimulated and unstimulated lanes. To test whether SPA release relied on protein synthesis, we modified a previously published assay showing that chloramphenicol at 10 mg/ml could inhibit 100% amino acid assimilation in S. aureus (12). Briefly, 100 μl of bacteria at A600 0.1–0.2 in PBS were incubated with 100 μl of 32 mg/ml chloramphenicol ± 1000 nm CXCL9 in PBS at room temperature. The low optical density is to ensure no spontaneous SPA release in the negative control tubes. Eight tubes were set up for each assay condition: four for analysis of SPA in supernatant and four for total protein. Both zero and overnight time (20 h) points were analyzed, and each time point was performed in duplicate. For the zero time point, reactions were processed immediately and stored at −80 °C. To harvest SPA released into the supernatant, tubes were centrifuged at 15,000 rpm for 10 min, and 150 μl of supernatant were removed. To determine total SPA amount, 200 μl of SMM buffer (0.5 m sucrose, 0.02 m maleate, pH 6.5, 0.02 m MgCl2) containing 100 mg/ml of lysostaphin were added to reaction tubes and incubated for 1–2 h at 37 °C to release cell wall bound SPA. The total reaction was then precipitated with 100 μl of 100% trichloroacetic acid (TCA). Tubes were incubated on ice and then centrifuged at 15,000 rpm for 10 min. Supernatant was removed, and the pellet was washed once with cold acetone. Dried pellet was resuspended in 200 μl of PBS and stored at −80 °C.
Anti-staphylococcal Growth Assay
Anti-staphylococcal activity was determined as previously described (9). CA-MRSA was diluted 1:100 in TSB and grown to mid-log phase before testing in 10 mm sodium phosphate buffer or phosphate buffer saline (137 mm sodium) containing 1% TSB. Bacteria were washed once and resuspended in buffer in the absence or presence of 1 μm final concentration of chemokine. The reactions were incubated at 37 °C for 2 h and serial dilution was used to determine CFU. Percent of inhibition was determined by the formula [(CFU without chemokine-CFU with chemokine)/CFU without chemokine] × 100.
Western Blot Analysis
Samples with equal amounts of protein in buffer were boiled for 10 min before loading onto 4–15% Tris-HCl polyacrylamide gels in SDS-PAGE. Proteins were transferred onto PVDF membrane and blocked with 5% dry milk in PBS/Tween 20 (0.05%) for at least 30 min. Blots were then incubated with the indicated antibodies. For detecting CXCL9 and CXCL10, rabbit polyclonal antisera directed against CXCL9 or CXCL10 were incubated at 1:5000 dilution in 5% dry milk in PBS/Tween 20 with blots for 1 h. Blots were then washed with PBS/Tween 20 four times and then incubated with secondary goat anti-rabbit IgG antibodies conjugated with horseradish peroxidase (HRP) at a 1:5000 dilution. After 4 additional washes with PBS/Tween 20, blots were then developed with SuperSignal West Pico Chemiluminescent Substrate according to the manufacturer's instructions (Thermo Fisher Scientific). For detection of SPA, goat anti-protein A conjugated to HRP (Abcam ab7245) at 1:5000 dilution was used. Densitometry of band signals was determined using NIH ImageJ software.
Subcellular Fractionation of S. aureus
Subcellular fractionation was performed according to a published protocol for SPA (13). Bacteria at A600 = 1.6 were incubated with or without 500 nm CXCL9 or CXCL10 for 2 h at room temperature, shaking (250 rpm) in PBS. Bacteria were centrifuged at 15,000 rpm for 5 min, and the supernatant fraction was saved. Then the pellet was resuspended with SMM buffer containing lysostaphin [0.5 M sucrose, 0.02 m maleate, 0.02 m MgCl2 (pH 6.5), 100 mg/ml lysostaphin] to remove cell wall proteins at 37 °C for 30 min with mixing. Then the reaction was centrifuged at 15,000 rpm for 10 min. The supernatant contains the cell wall proteins. The pellet containing protoplasts was washed once with SMM buffer. Protoplast was resuspended in membrane buffer (0.1 m NaCl, 0.1 m Tris-HCl, 0.01 m MgCl2, pH 7.5) and lysed by 5 freeze-thaw cycles in a dry ice/ethanol bath. Cell membrane and cytoplasmic proteins were separated by ultracentrifugation at 100,000 × g for 30 min at 4 °C. All fractions were precipitated with TCA and washed with acetone before SDS-PAGE/Western blot analysis.
Clinical Samples and Mouse Skin Abscess Model
Sixteen anonymized clinical samples were obtained from the Dept. of Pathology of George Washington University Hospital. Only the anatomical site of the sample and the microbiology culture report were provided. Samples included sputum, skin wound, bronchoalveolar lavage, and joint fluid. Ten samples were culture-positive for either MSSA or MRSA, and six were culture-negative for both MSSA and MRSA. The latter samples grew either normal flora or non-staphylococcal pathogens, or had no growth, and served as negative controls. Occasionally, 1 ml of PBS was added to samples before centrifugation to reduce viscosity. Samples were centrifuged at 15,000 rpm for 10 min to remove cellular debris and bacteria. Supernatants were boiled to disrupt SPA and immunoglobulin binding, and were then recentrifuged at 15,000 rpm for 10 min. The resulting fluid samples were saved for extracellular SPA analysis using the protein A EIA kit (Enzo Life Sciences) according to manufacturer's protocol. For the mouse skin abscess model, mice were infected with MRSA strain FPR3757 according to a previously published protocol (14). All animal procedures adhered to a protocol approved by the NIAID Animal Care and Use Committee. Briefly, C57BL/6 mice were injected subcutaneously with 100 μl of fluid containing 50 μl of OD600 0.95 MRSA and 50 μl of dextran beads in PBS. As negative controls, mice were injected with dextran beads in PBS only. Animals were euthanized, and skin lesions were harvested by cutting with surgical scissors at post-infection day 1, 4, 7, and 14. Tissues were weighed and transferred to 15 ml centrifuge tubes with 2 ml of HBSS containing a mixture of protease inhibitors (Roche, Indianapolis, IN). Then, each tissue was homogenized with a handheld tissue homogenizer equipped with a hard tissue Omni tip (Omni International). Tubes were kept on ice during the homogenization process. Once homogenized, fluid was transferred to 1.6 ml tubes, flash frozen in a dry ice bath, and stored at −80 °C. To test for extracellular SPA, samples were boiled for 10 min, and spun at 15,000 rpm for 10 min. The supernatant was analyzed for extracellular SPA with the protein A EIA kit. The amount of SPA detected was normalized to the amount of tissue tested. To test mouse skin lesions for KC, Cxcl9, and Cxcl10, samples were analyzed with commercial ELISA kits according to the manufacturer's protocol (R&D).
RESULTS
CXCL9 Stimulates Release of a 50 kDa Protein from S. aureus
CXCL9 has been reported to have direct antistaphylococcal activity for MSSA strains at 10 mm Na+ concentration, and to be present at sites of S. aureus infection in a rat pneumonia model (15). To extend these findings and to investigate mechanisms we incubated MSSA and MRSA strains with CXCL9 at room temperature overnight in PBS containing either 10 mm or 137 mm (physiological) Na+ concentration. The reason to perform reactions at room temperature instead of 37 °C is because >99% of the organisms died when incubated at 37 °C, confirming a previously published observation (16). This effect is Na+ concentration-dependent and chemokine-independent.
Supernatants were analyzed by Western blot to determine the fate of CXCL9 in the presence of MRSA. Full-length CXCL9 has a molecular mass of ∼11 kDa (Fig. 1, black arrow); MRSA appeared to process this to a truncated form. In addition, less CXCL9 appeared in the supernatant suggesting that MRSA was capable of depleting CXCL9 either by degradation, or by binding, or by both mechanisms. However, the immunoblot analysis revealed a second and unexpected finding, the appearance of a 50 kDa protein from MRSA that was induced in a CXCL9-dependent manner (Fig. 1, white arrow). This 50 kDa protein can also be seen faintly when MRSA was cultured in the absence of chemokines (Fig. 1) indicating it is bacterial in origin. At higher bacterial density, CXCL9 could also induce SPA release at 37 °C within 2 h, whereas no release was observed under these conditions when the organisms was cultured under these conditions in the absence of chemokine (data not shown).
FIGURE 1.

CXCL9 induces release of a 50 kDa protein from MRSA. CXCL9 was incubated at concentrations indicated at the top of each lane with and without MRSA overnight in PBS at room temperature. Supernatant proteins were separated by SDS-PAGE and revealed by immunoblotting with anti-CXCL9 antibody plus secondary antibody. Black arrow, size of full-length CXCL9; white arrow, unexpected 50 kDa protein induced in the system by CXCL9. Data are representative of three independent experiments.
The 50 kDa Protein Is Protein A of S. aureus
Because the 50 kDa protein required antibody reagents for visualization, it could potentially represent a multimeric form of CXCL9, or a cross-reactive protein, or a protein able to bind antibodies nonspecifically. We favored the third possibility, and in particular considered SPA which can bind specifically to the Fc portion of IgG and has a molecular mass of 50 kDa. In support of this hypothesis, specific anti-SPA antibodies conjugated with HRP were able to detect induction of a 50 kDa protein in supernatants of MRSA after overnight incubation with CXCL9. Induction was CXCL9 concentration-dependent (Fig. 2A), and required a threshold of ∼5 nm (or 50 ng/ml) CXCL9, which is close to the Kd for binding of CXCL9 to its receptor CXCR3 (17). The increase in amount of this protein was not due to differences in CFU or total protein content induced by the different conditions tested (Fig. 2B and data not shown). In the staphylococcus literature, there are no acceptable housekeeping protein controls, like actin in mammalian systems. Together our results suggest that CXCL9 can directly induce SPA release into the supernatant. We next analyzed the kinetics of SPA release from MRSA (Fig. 2C). In the absence of chemokines, only a low level of spontaneous SPA release was observed and not until 5 h after the start of incubation, whereas when MRSA was incubated with CXCL9 (not shown) or with 500 nm of the related chemokine CXCL10 (Fig. 2C), SPA release could be observed as early as 2 h after incubation, and attained much higher levels.
FIGURE 2.

The 50 kDa protein induced by CXCL9 incubation with MRSA is SPA. MRSA was incubated with the concentration of CXCL9 indicated on the x axis in each panel overnight at room temperature. Supernatant proteins were separated by SDS-PAGE and revealed by Western blotting using anti-SPA antibody conjugated to horseradish peroxidase (A, top panel); SPA protein content was quantitated by NIH ImageJ software (A, bottom panel). CXCL9 did not affect the viability of MRSA in the system (B). Data are representative of three independent experiments. MRSA in the absence or presence of 500 nm CXCL10 was assayed for SPA release at different time points by Western blot with anti-protein A antibodies (C).
CXCL9 and CXCL10 Induction of SPA Release from MRSA Is Bacterial Density-dependent
Because MRSA releases SPA spontaneously, we analyzed the effect of bacterial density on this process. We found that spontaneous release of SPA was directly dependent on bacterial density whereas chemokine-dependent release was inversely dependent on bacterial density (Fig. 3, A and B). Thus, CXCL9 was most effective at inducing SPA release at low bacterial density. CXCL10 showed the same bacterial density dependence for induction of SPA release from MRSA (Fig. 3, C and D).
FIGURE 3.

Chemokine induction of SPA release from MRSA is dependent on bacterial density. Bacteria at the indicated density were incubated with or without 500 nm CXCL9 or CXCL10 (plus and minus signs, respectively) overnight at room temperature, and supernatants were then analyzed for SPA content by SDS-PAGE/Western blot using anti-protein A antibody (A, C). SPA content was quantified by NIH ImageJ software, and the ratio of SPA signal in the presence and absence of chemokine was plotted (B, D).
CXCL9 Acts at a Post-translational Step to Induce Release of SPA from MRSA
To determine whether SPA release by CXCL9 is dependent on new protein synthesis, we used chloramphenicol, a ribosomal inhibitor, to block translation (12). For this assay, the density of bacteria was purposely kept low to prevent spontaneous SPA release by the organism (Fig. 4A). Under these conditions, 500 nm CXCL9 was able to promote SPA release, and this was not affected by incubation with chloramphenicol at a concentration known to inhibit S. aureus protein synthesis (Fig. 4A). The concentration of chloramphenicol used did not affect bacterial viability in PBS, as PBS contains no nutrients, but it was active in inhibiting MRSA growth in trypticase soy agar plates (data not shown). Thus, induction of SPA release from MRSA by CXCL9 is independent of new protein synthesis.
FIGURE 4.

Chemokine induction of SPA release from MRSA does not require new protein synthesis. MRSA was incubated in the presence or absence of 500 nm CXCL9 and 16 mg/ml chloramphenicol (Cm) for the time indicated on the x axis at room temperature, then extracellular SPA concentration (A) and total SPA content (B) were determined by ELISA as described under “Experimental Procedures.” The experiment was performed using low bacterial density (A600 = 0.1) to optimize the induced SPA signal. Data are from a single experiment representative of two experiments performed.
The total amount of SPA in the system was also determined for each reaction by treatment with lysostaphin (Fig. 4B). SPA is normally covalently linked to the bacterial cell wall, and this enzyme specifically cleaves it from this site quantitatively. The results indicated that total SPA remains constant throughout the experiment, and is unaffected by CXCL9 or chloramphenicol. In addition, the results indicated that the amount of SPA released from the cell in response to CXCL9 represented only a small fraction of total SPA.
CXCL9 and CXCL10 Differentially Bind to MRSA Cell Wall and Cell Membrane
Preliminary data indicated that CXCL9 was being removed from the supernatant in the system (Fig. 1) in part due to association with the bacterial pellet (data not shown). To determine where CXCL9 binds to MRSA, a subcellular fractionation analysis was performed using a procedure originally developed to identify the subcellular location of SPA (13). MRSA was incubated with or without chemokine in PBS for 2 h before subcellular fractionation. As expected, SPA was detected in the cell wall fraction (Fig. 5A). In contrast, CXCL9 was associated with the cell membrane fraction of MRSA (Fig. 5B). The CXCL9 in the supernatant fraction represented unbound CXCL9. The same procedure was also performed using CXCL10. Again, SPA was detected in the cell wall fraction (Fig. 5C). However, unlike CXCL9, CXCL10 was associated with both cell wall and cell membrane fractions of MRSA (Fig. 5D). The CXCL10 in the supernatant represented unbound CXCL10.
FIGURE 5.
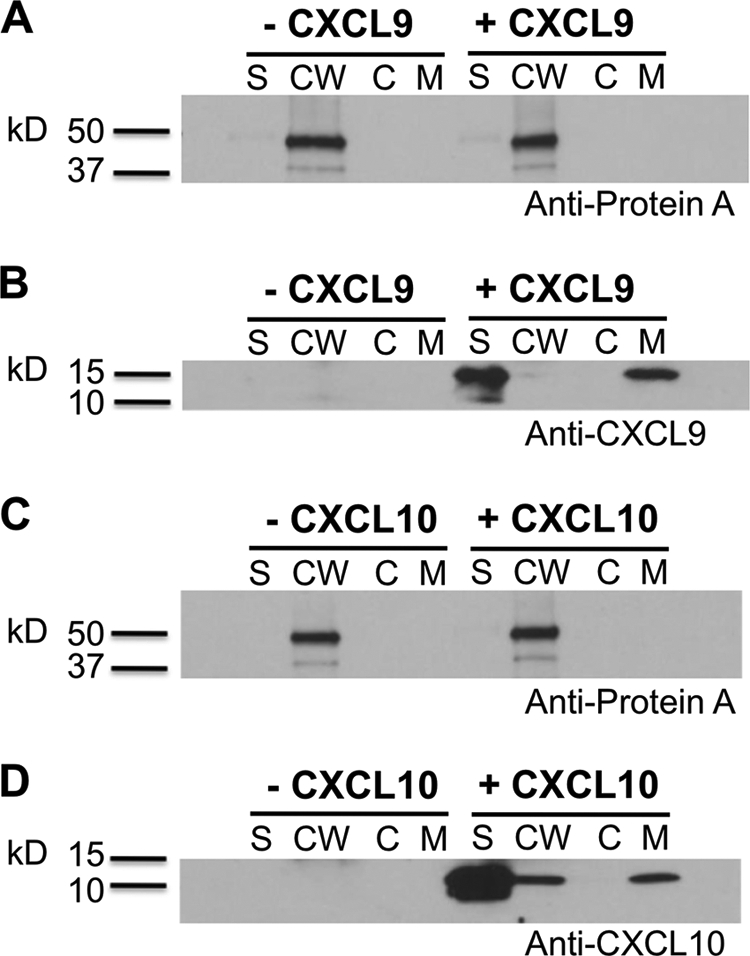
CXCL9 and CXCL10 bind directly to MRSA. MRSA was incubated in the presence or absence of 500 nm CXCL9 or CXCL10 for 2 h at room temperature, and then subjected to subcellular fractionation. Supernatant (S), cell wall (CW), cytoplasm (C), and cell membrane (CM) fractions were analyzed by SDS-PAGE/Western blot with antibodies specific for SPA (A, C) or for chemokines CXCL9 (B) or CXCL10 (D). Data are from a single experiment representative of two experiments performed.
Many but Not All Chemokines Stimulate Release of SPA from MRSA
To determine the chemokine specificity of SPA release from MRSA, all available human chemokines were screened at 500 nm concentration; the antimicrobial properties of chemokines against MSSA and MRSA were also determined (Table 1). Protein content and CFUs were determined for each reaction. Consistent with previous reports, in 10 mm Na+ all ELR- CXC chemokines strongly inhibited bacterial viability. However, this antimicrobial effect was diminished or absent at 137 mm Na+ concentration for both MSSA and MRSA. In general, this trend of diminished or absent antimicrobial activity at 137 mm Na+ concentration was observed for all anti-staphylococcal chemokines. In contrast, thirty-one of forty two chemokines tested were able to induce SPA release from MRSA in 137 mm Na+ concentration. All of these were basic chemokines (pI >8.0). In addition, all chemokines with antimicrobial properties in low salt were able to induce SPA release at 137 mm Na+ concentration. However, there were chemokines that induced SPA release that lacked antimicrobial activity. Interestingly, these included the ELR+ CXC chemokines that attract neutrophils.
TABLE 1.
Direct effects of chemokines on S. aureus: antibacterial and SPA-releasing activities
Survey of two direct effects of chemokines on S. aureus: antibacterial activity versus SPA-releasing activity. All commercially available human chemokines (42) were tested for their ability to induce SPA release in 137 mm Na+ concentration. Antimicrobial properties of chemokines against MSSA strain ATCC 29213 and MRSA strain FPR3757 were also tested, at both 10 mm and 137 mm Na+ concentration, as described under “Experimental Procedures.” Symbols denote percent inhibition of bacterial growth compared with control: ++++, >80%; +++, 60–79%; ++, 40–59%; +, 20–39%; −, <20%, ND; not done. Only MRSA strain FPR 3757 was tested for SPA release. Symbols + and − represent positive and negative SPA release, respectively. Both ELISA and Western blots for SPA were performed to generate this table. Chemokines in parentheses are found in mouse but not human. The pI of each chemokine is also shown.
| Chemokine |
Inhibition assay |
SPA release |
|||||
|---|---|---|---|---|---|---|---|
| MSSA |
MRSA |
MRSA |
|||||
| [Na+] |
[Na+] |
[Na+] |
|||||
| Family | Member | pI | 10 mm | 137 mm | 10 mm | 137 mm | 137 mm |
| CXC ELR+ | CXCL1 | 10.09 | − | − | − | − | + |
| CXCL2 | 10.45 | + | ++ | + | − | + | |
| CXCL3 | 10.15 | − | − | + | − | − | |
| CXC ELR- | CXCL4 | 8.71 | − | − | − | − | + |
| CXC ELR+ | CXCL5 | 11.4 | − | − | − | − | + |
| CXCL6 | 10.44 | − | + | ++ | − | + | |
| CXCL7 | 8.73 | − | − | − | − | − | |
| CXCL8 | 9.85 | + | − | + | − | + | |
| CXC ELR- | CXCL9 | 11.1 | ++++ | ++ | ++++ | − | + |
| CXCL10 | 10.77 | ++++ | − | ++++ | − | + | |
| CXCL11 | 11.1 | ++++ | ++ | ++++ | + | + | |
| CXCL12 | 10.48 | ++++ | − | ++++ | + | + | |
| CXCL13 | 10.92 | ++++ | ++ | ++++ | + | + | |
| CXCL14 | 10.47 | − | − | − | − | + | |
| CXC ELR+ | (CXCL15) | 7.07 | ND | ||||
| CXC ELR+ | CXCL16 | 8.75 | − | − | + | − | + |
| CX3C | CX3CL1 | 10.24 | − | − | − | + | + |
| C | XCL1 | 11.28 | − | − | − | − | + |
| XCL2 | 11.20 | ND | |||||
| CC | CCL1 | 10.48 | + | − | ++ | − | + |
| CCL2 | 9.74 | + | + | − | − | − | |
| CCL3 | 4.48 | − | − | − | − | − | |
| CCL3L1 | 4.47 | − | − | − | − | − | |
| CCL4 | 4.47 | − | ++ | − | + | − | |
| CCL5 | 8.61 | − | − | − | − | − | |
| (CCL6) | 9.22 | ND | |||||
| CCL7 | 10.33 | − | − | − | + | − | |
| CCL8 | 9.88 | − | − | − | − | + | |
| (CCL9/10) | 8.64 | ND | |||||
| CCL11 | 10.74 | − | − | − | − | + | |
| (CCL12) | 9.27 | ND | |||||
| CCL13 | 10.62 | + | + | +++ | ++ | + | |
| CCL14 | 8.25 | − | ++ | + | − | − | |
| CCL15 | 8.01 | − | − | − | − | − | |
| CCL16 | 9.86 | − | − | − | − | + | |
| CCL17 | 9.81 | ++ | − | − | + | + | |
| CCL18 | 9.47 | − | − | + | − | + | |
| CCL19 | 10.31 | − | − | +++ | − | + | |
| CCL20 | 10.26 | +++ | + | +++ | − | + | |
| CCL21 | 10.71 | + | + | + | + | + | |
| CCL22 | 8.98 | − | − | + | − | + | |
| CCL23 | 9.09 | − | − | + | + | + | |
| CCL24 | 10.76 | − | − | − | − | − | |
| CCL25 | 10.92 | ++++ | ++ | +++ | + | + | |
| CCL26 | 10.85 | ++++ | − | ++++ | + | + | |
| CCL27 | 9.07 | − | − | − | − | + | |
| CCL28 | 10.88 | ++++ | ++ | ++++ | − | + | |
Extracellular SPA and SPA-inducing Chemokines Are Present in Human and Mouse Tissue Samples Infected with S. aureus
To determine whether chemokine induction of SPA release from S. aureus may occur in the context of actual infection of mammalian hosts, we measured SPA levels and chemokine content in clinical samples from patients known to be infected with the organism. Four of ten clinical samples analyzed were positive for extracellular SPA (Fig. 6A). Of these, one contained MSSA and three contained MRSA. The control samples, which grew normal flora or non-S. aureus pathogens, or had no growth, were all negative for extracellular SPA. Consistent with this, we also detected extracellular SPA in skin samples from mice challenged with MRSA strain FPR3757 in a skin abscess model on days 1, 4, and 7 post-infection (Fig. 6B). Negative control mice injected with PBS had no detectable extracellular SPA (dotted line in Fig. 6B). Thus, extracellular SPA can be found in the context of staphylococcal infections in vivo.
FIGURE 6.

Extracellular SPA is present in both human and mouse tissue sites infected with S. aureus. A, human. Clinical samples known to be infected with MSSA or MRSA were tested for extracellular SPA by ELISA. As controls, samples infected with other pathogens, or that grew only normal flora or that had no growth were also tested for SPA. B, mouse. Skin lesions of C57Bl/6 mice infected subcutaneously with MRSA were harvested and tested for extracellular SPA on the days indicated post-infection. Dotted line represents signal of control dextran beads in PBS injected skin lesions. C, MRSA skin abscesses in mice contain chemokines able to induce SPA release from MRSA. Skin lesions of mice infected with MRSA were harvested and assayed by ELISA for the indicated chemokine at the indicated times post-infection. Each data point represents the average of duplicate determinations minus the control dextran beads in PBS-injected skin lesion values. The S.E. is <17% of the mean and is not plotted.
Skin lesions infected with MRSA also contained at the protein level three out of three SPA-releasing chemokines that were assayed: KC, Cxcl9, and Cxcl10 (Fig. 6C). Of the three chemokines tested, Cxcl9 was present at the highest level, followed by KC; Cxcl10 was present at very low levels. As controls, recombinant SPA (from 10 ng/ml to 10 mg/ml) and mice injected with dextran beads in PBS did not interfere with the ELISA conditions (data not shown).
DISCUSSION
The present work has identified an unexpected direct pro-microbial activity for human chemokines: the ability to induce release of the virulence factor protein A (SPA) from the human pathogen S. aureus. In addition, we have confirmed that many chemokines have direct, leukocyte-independent anti-staphylococcal activity, and we have extended this by defining anti-staphylococcal activity against clinical isolates of both MSSA and MRSA for all 42 commercially available human chemokines in both high and low concentrations of Na+. In contrast to the anti-staphylococcal activity, which requires extremely high concentrations of chemokine and low concentration of Na+, we found that SPA-inducing activity occurs at low nanomolar concentrations of chemokine and at physiologic concentrations of Na+.
Consistent with these in vitro findings, in vivo we detected SPA in an extracellular soluble form in tissue and body fluids of patients infected naturally with MSSA or MRSA, as well as in mice infected experimentally with MRSA; we also found SPA-releasing chemokines in the context of experimental MRSA skin abscess in mice. Together these results imply that S. aureus may exploit chemokines, which are normally used in host defense to recruit leukocytes to sites of infection, as a trigger for deploying a virulence factor that acts through immune evasion. More generally, our work implies that bacteria can detect and respond to host-derived signals. Sensing chemokines may provide S. aureus, and potentially other bacteria, with knowledge of an impending antimicrobial response by the host. To date, there is only one other report of bacteria sensing a host immune activation factor: Pseudomonas aeruginosa binding to interferon γ via its outer membrane protein F and responding by expressing quorum sensing virulence genes (8). Taken together, the data suggest that outcome of S. aureus infection may be determined in part by a balance of antimicrobial and promicrobial chemokine-dependent activities: 1) recruitment of leukocytes to the site, 2) direct antimicrobial activity and 3) pro-microbial induction of SPA release.
Host factors that control staphylococcal infection mainly involve the innate immune system. In particular, at the cellular level, phagocytes, especially neutrophils, are critical (18, 19). Consistent with this, at the molecular level, pattern recognition receptors such as TLR2 & its downstream transducer MyD88 are important for control of S. aureus infection in both mice and humans (20, 21). However, little is known in S. aureus infection about the role of the chemokines that subserve innate immunity. CXCR2, which is preferentially expressed on neutrophils, is the only chemokine receptor reported to be important in clearing S. aureus, as shown using Cxcr2 knock-out mice in a brain abscess model (22).
Nevertheless, several groups have reported that many chemokines and chemokine receptors can be strongly up-regulated by S. aureus infection in both pneumonia and skin abscess models (23–25). In the pneumonia model, 20 of 25 chemokines tested were found to be up-regulated (23). Of these 20, we initially focused on CXCL9 and CXCL10, since they were reported to be highly expressed at infected sites, yet are highly selective for Th1 effector T cells, which paradoxically are not present at these sites. Thus, we hypothesized that CXCL9 and CXCL10 may have an unconventional role, unrelated to leukocyte recruitment and their specific receptor CXCR3. Both chemokines are highly inducible by the signature Th1 cytokine interferon-γ. Because interferon-γ administration is detrimental in S. aureus bacteremia, and since both IFN-γ knock-out mice and mice administered neutralizing antibodies directed against IFN-γ are all protected from lethality in S. aureus bacteremia (26, 27), IFN-γ or possibly factors induced by IFN-γ, such as CXCL9 and/or CXCL10, must enhance staphylococcal virulence. Our data showing that these chemokines induce SPA release provide a potential mechanism consistent with this hypothesis.
Protein A is a well-established virulence factor, as determined by experiments in vivo showing that Protein A mutants of S. aureus have attenuated virulence in pneumonia and bacteremia models (28–30), and that protein A could function as a vaccine target for MRSA (31). Although a number of potential mechanisms have been identified, which of these accounts for virulence in vivo has not been clearly delineated. In particular, SPA is best known for its ability to bind IgG (32), but can also bind other host factors, including von Willebrand's factor (33) and the VH3 receptor of B-cells (34). SPA also possesses antiphagocytic activity (35, 36), and the ability to activate TNFR1 (37) and to induce apoptosis of naïve B cells (38). Purified protein A has been reported to be sufficient to stimulate CXCL8 production by lung epithelial cells and cause neutrophil recruitment in vivo. Recent data also suggest that extracellular protein A contributes to biofilm formation by S. aureus (39). The ability of chemokines to induce SPA release could simply serve to enlarge a protective shield around the organism through one or more of these activities, defending it from the immune response. Transcriptional analysis has revealed that both Cxcl9 and Cxcl10 are up-regulated in a mouse model of wound healing (40). According to our current data on SPA release, the presence of these chemokines could enhance the vulnerability of wounds to S. aureus infections. In addition to wounds, CXCL9 and CXCL10 levels are also up-regulated during influenza infections (41), and many secondary bacterial pneumonias after influenza are caused by S. aureus, including community-associated MRSA strains (42, 43). However, it is unclear if the presence of chemokines directly affects MRSA susceptibility in this setting.
SPA is covalently linked to the cell wall of S. aureus by action of the enzyme sortase (44). However, a small portion of SPA can be released into the extracellular environment during exponential growth in vitro (45–47). Some strains of MRSA produce only extracellular protein A (48). The release mechanism is unknown and the significance has not been previously evaluated in vivo.
Additional work will be needed to delineate the structural basis for chemokine induction of SPA release. Our survey identified 31 of 42 human chemokines that possessed this activity. Most chemokines are basic proteins, and all chemokines that induced SPA release had a pI greater than 8 (Table 1). However, not all basic chemokines induced SPA release. There was a stronger association of this activity with the presence of a cationic C terminus, a feature that has also been associated with the antimicrobial properties of chemokines. Thus, chemokines that possess antimicrobial properties at low sodium concentration tend to induce SPA release at 137 mm sodium concentration.
We also analyzed the three-dimensional structures of chemokines that promote protein A release for similarity. In particular, the electrostatic potentials of chemokines with solved structures were calculated and visualized by PDB 2PQR and Jmol (49, 50). Both active and inactive chemokines for protein A release had areas of cationic patches, with no clear differences between the two groups.
To our knowledge, this is the first report identifying extracellular protein A in clinical samples from humans. However, extracellular protein A was not detected in every clinical sample infected with S. aureus. Several factors might explain this. First, the bacterial load in the infected site might be below the threshold necessary for detection of SPA release. In this regard, one of the S. aureus culture-positive samples grew only in a liquid culture medium and not directly on the culture plates. Second, most (∼97%) but not all strains of S. aureus produce protein A, and the amount expressed varies among strains (46, 51, 52). Finally, samples obtained by lavage may dilute extracellular SPA below the threshold for detection.
In the mouse skin abscess model of MRSA infection, we detected both extracellular protein A and several chemokines capable of inducing protein A release. We cannot conclude from this that chemokines are responsible for the release of protein A from the organism in vivo, only that bacteria and chemokines are in the same environment where they may interact. Proving that chemokines induce SPA release in vivo will be difficult since there are so many chemokines with this activity at the infected site.
The ability of MRSA to bind CXCL9 and CXCL10, and presumably other chemokines, could also attenuate the immune response independent of any functional response by the organism, by a simple scavenging mechanism that would limit the amount of chemokine available to bind to leukocyte chemokine receptors for promoting leukocyte chemotaxis. Host chemokine-binding proteins have been identified that are thought to limit inflammation through the same type of mechanism (53–55). For CXCL9, less protein was detected after incubation with MRSA by Western blot. It remains to be seen how many other chemokines besides CXCL9 and CXCL10 bind to MRSA, and whether protein A release correlates with binding.
In conclusion, our data show that chemokines can bind to MRSA and that this is associated with both direct antimicrobial and SPA-releasing activity in vitro. These activities have very different requirements for chemokine and sodium concentration such that SPA-releasing activity may be more likely to occur in vivo than anti-staphylococcal activity. Defining S. aureus-encoded chemokine-binding factors will be needed to further delineate the mechanism underlying this activity, and such factors may provide targets for development of novel anti-staphylococcal antibiotics, an important goal given the increasing resistance of this organism to currently available agents.
Acknowledgment
We thank Dr. John Keiser for help in providing clinical samples.
This work was supported by the Division of Intramural Research of the NIAID, National Institutes of Health.
- SPA
- S. aureus protein A
- CA-MRSA
- community-associated methicillin-resistant S. aureus.
REFERENCES
- 1. Moreillon P., Que Y.-A., Glauser M. P. (2005) in Principles and Practice of Infectious Diseases (Mandell G. L., Bennett J. E., Dolin R. eds), pp. 2321–2351, Sixth Ed., Elsevier, Philadelphia, Pennsylvania [Google Scholar]
- 2. Klevens R. M., Morrison M. A., Nadle J., Petit S., Gershman K., Ray S., Harrison L. H., Lynfield R., Dumyati G., Townes J. M., Craig A. S., Zell E. R., Fosheim G. E., McDougal L. K., Carey R. B., Fridkin S. K. (2007) JAMA 298, 1763–1771 [DOI] [PubMed] [Google Scholar]
- 3. Ji G., Beavis R. C., Novick R. P. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 12055–12059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bassler B. L., Losick R. (2006) Cell 125, 237–246 [DOI] [PubMed] [Google Scholar]
- 5. Ji G., Beavis R., Novick R. P. (1997) Science 276, 2027–2030 [DOI] [PubMed] [Google Scholar]
- 6. Otto M. (2001) Peptides 22, 1603–1608 [DOI] [PubMed] [Google Scholar]
- 7. Coburn P. S., Pillar C. M., Jett B. D., Haas W., Gilmore M. S. (2004) Science 306, 2270–2272 [DOI] [PubMed] [Google Scholar]
- 8. Wu L., Estrada O., Zaborina O., Bains M., Shen L., Kohler J. E., Patel N., Musch M. W., Chang E. B., Fu Y. X., Jacobs M. A., Nishimura M. I., Hancock R. E., Turner J. R., Alverdy J. C. (2005) Science 309, 774–777 [DOI] [PubMed] [Google Scholar]
- 9. Yang D., Chen Q., Hoover D. M., Staley P., Tucker K. D., Lubkowski J., Oppenheim J. J. (2003) J. Leukoc Biol. 74, 448–455 [DOI] [PubMed] [Google Scholar]
- 10. Cole A. M., Ganz T., Liese A. M., Burdick M. D., Liu L., Strieter R. M. (2001) J. Immunol. 167, 623–627 [DOI] [PubMed] [Google Scholar]
- 11. Diep B. A., Gill S. R., Chang R. F., Phan T. H., Chen J. H., Davidson M. G., Lin F., Lin J., Carleton H. A., Mongodin E. F., Sensabaugh G. F., Perdreau-Remington F. (2006) Lancet 367, 731–739 [DOI] [PubMed] [Google Scholar]
- 12. Gale E. F., Folkes J. P. (1953) Biochem. J. 53, 493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schneewind O., Mihaylova-Petkov D., Model P. (1993) EMBO J. 12, 4803–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bunce C., Wheeler L., Reed G., Musser J., Barg N. (1992) Infect. Immun. 60, 2636–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montgomery C. P., Daum R. S. (2009) Infect. Immun. 77, 2159–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parfentjev I. A., Catelli A. R. (1964) J. Bacteriol. 88, 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liao F., Rabin R. L., Yannelli J. R., Koniaris L. G., Vanguri P., Farber J. M. (1995) J. Exp. Med. 182, 1301–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mölne L., Verdrengh M., Tarkowski A. (2000) Infect. Immun. 68, 6162–6167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verdrengh M., Tarkowski A. (1997) Infect. Immun. 65, 2517–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takeuchi O., Hoshino K., Akira S. (2000) J. Immunol. 165, 5392–5396 [DOI] [PubMed] [Google Scholar]
- 21. von Bernuth H., Picard C., Jin Z., Pankla R., Xiao H., Ku C. L., Chrabieh M., Mustapha I. B., Ghandil P., Camcioglu Y., Vasconcelos J., Sirvent N., Guedes M., Vitor A. B., Herrero-Mata M. J., Aróstegui J. I., Rodrigo C., Alsina L., Ruiz-Ortiz E., Juan M., Fortuny C., Yagüe J., Antón J., Pascal M., Chang H. H., Janniere L., Rose Y., Garty B. Z., Chapel H., Issekutz A., Maródi L., Rodriguez-Gallego C., Banchereau J., Abel L., Li X., Chaussabel D., Puel A., Casanova J. L. (2008) Science 321, 691–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kielian T., Barry B., Hickey W. F. (2001) J. Immunol. 166, 4634–4643 [DOI] [PubMed] [Google Scholar]
- 23. Montgomery C. P., Daum R. S. (2009) Infect. Immun. 77, 2159–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McLoughlin R. M., Solinga R. M., Rich J., Zaleski K. J., Cocchiaro J. L., Risley A., Tzianabos A. O., Lee J. C. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 10408–10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McLoughlin R. M., Lee J. C., Kasper D. L., Tzianabos A. O. (2008) J. Immunol. 181, 1323–1332 [DOI] [PubMed] [Google Scholar]
- 26. Sasaki S., Nishikawa S., Miura T., Mizuki M., Yamada K., Madarame H., Tagawa Y. I., Iwakura Y., Nakane A. (2000) Infect. Immun. 68, 2424–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakane A., Okamoto M., Asano M., Kohanawa M., Minagawa T. (1995) Infect. Immun. 63, 1165–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Labandeira-Rey M., Couzon F., Boisset S., Brown E. L., Bes M., Benito Y., Barbu E. M., Vazquez V., Höök M., Etienne J., Vandenesch F., Bowden M. G. (2007) Science 315, 1130–1133 [DOI] [PubMed] [Google Scholar]
- 29. Bubeck Wardenburg J., Patel R. J., Schneewind O. (2007) Infect. Immun. 75, 1040–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Palmqvist N., Foster T., Tarkowski A., Josefsson E. (2002) Microb. Pathog. 33, 239–249 [DOI] [PubMed] [Google Scholar]
- 31. Kim H. K., Cheng A. G., Kim H. Y., Missiakas D. M., Schneewind O. (2010) J. Exp. Med. 207, 1863–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grov A. (1968) Acta Pathol. Microbiol. Scand. 73, 400–406 [PubMed] [Google Scholar]
- 33. Hartleib J., Köhler N., Dickinson R. B., Chhatwal G. S., Sixma J. J., Hartford O. M., Foster T. J., Peters G., Kehrel B. E., Herrmann M. (2000) Blood 96, 2149–2156 [PubMed] [Google Scholar]
- 34. Goodyear C. S., Corr M., Sugiyama F., Boyle D. L., Silverman G. J. (2007) J. Immunol. 178, 2636–2640 [DOI] [PubMed] [Google Scholar]
- 35. Verbrugh H. A., Van Dijk W. C., Peters R., Van Der Tol M. E., Verhoef J. (1979) Immunology 37, 615–621 [PMC free article] [PubMed] [Google Scholar]
- 36. Spika J. S., Verbrugh H. A., Verhoef J. (1981) Infect. Immun. 34, 455–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gómez M. I., Lee A., Reddy B., Muir A., Soong G., Pitt A., Cheung A., Prince A. (2004) Nat. Med. 10, 842–848 [DOI] [PubMed] [Google Scholar]
- 38. Goodyear C. S., Silverman G. J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 11392–11397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merino N., Toledo-Arana A., Vergara-Irigaray M., Valle J., Solano C., Calvo E., Lopez J. A., Foster T. J., Penadés J. R., Lasa I. (2009) J. Bacteriol. 191, 832–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ishida Y., Gao J. L., Murphy P. M. (2008) J. Immunol. 180, 569–579 [DOI] [PubMed] [Google Scholar]
- 41. Kash J. C., Tumpey T. M., Proll S. C., Carter V., Perwitasari O., Thomas M. J., Basler C. F., Palese P., Taubenberger J. K., García-Sastre A., Swayne D. E., Katze M. G. (2006) Nature 443, 578–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. (2009) MMWR Morb. Mortal Wkly. Rep. 58, 1071–1074 [PubMed] [Google Scholar]
- 43. (2009) MMWR Morb. Mortal Wkly. Rep. 58, 941–947 [PubMed] [Google Scholar]
- 44. Mazmanian S. K., Liu G., Ton-That H., Schneewind O. (1999) Science 285, 760–763 [DOI] [PubMed] [Google Scholar]
- 45. Movitz J. (1976) Eur. J. Biochem. 68, 291–299 [DOI] [PubMed] [Google Scholar]
- 46. Forsgren A. (1970) Infect. Immun. 2, 672–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ton-That H., Liu G., Mazmanian S. K., Faull K. F., Schneewind O. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 12424–12429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lindmark R., Movitz J., Sjöquist J. (1977) Eur. J. Biochem. 74, 623–628 [DOI] [PubMed] [Google Scholar]
- 49. Dolinsky T. J., Czodrowski P., Li H., Nielsen J. E., Jensen J. H., Klebe G., Baker N. A. (2007) Nucleic Acids Res. 35, W522–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dolinsky T. J., Nielsen J. E., McCammon J. A., Baker N. A. (2004) Nucleic Acids Res. 32, W665–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fomenko G. A. (1980) Zh Mikrobiol. Epidemiol. Immunobiol., 57–61 [PubMed] [Google Scholar]
- 52. Sanford B. A., Thomas V. L., Ramsay M. A., Jones T. O. (1986) J. Clin. Microbiol. 24, 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Patel M., McInnes I. B., Graham G. (2009) Curr. Mol. Med. 9, 86–93 [DOI] [PubMed] [Google Scholar]
- 54. Pruenster M., Rot A. (2006) Biochem. Soc. Trans. 34, 1005–1008 [DOI] [PubMed] [Google Scholar]
- 55. Horne K., Woolley I. J. (2009) Inflamm. Res. 58, 431–435 [DOI] [PubMed] [Google Scholar]