

Abstract
N-Deacetylase-N-sulfotransferase 1 (Ndst1) catalyzes the initial modification of heparan sulfate and heparin during their biosynthesis by removal of acetyl groups from subsets of N-acetylglucosamine units and subsequent sulfation of the resulting free amino groups. In this study, we used a phage display library to select peptides that interact with Ndst1, with the aim of finding inhibitors of the enzyme. The phage library consisted of cyclic random 10-mer peptides expressed in the phage capsid protein pIII. Selection was based on the ability of engineered phage to bind to recombinant murine Ndst1 (mNdst1) and displacement with heparin. Peptides that were enriched through multiple cycles of binding and disassociation displayed two specific sequences, CRGWRGEKIGNC and CNMQALSMPVTC. Both peptides inhibited mNdst1 activity in vitro, however, by distinct mechanisms. The peptide CRGWRGEKIGNC presents a chemokine-like repeat motif (BXX, where B represents a basic amino acid and X is a noncharged amino acid) and binds to heparan sulfate, thus blocking the binding of substrate to the enzyme. The peptide NMQALSMPVT inhibits mNdst1 activity by direct interaction with the enzyme near the active site. The discovery of inhibitory peptides in this way suggests a method for developing peptide inhibitors of heparan sulfate biosynthesis.
Keywords: Enzyme Inhibitors, Heparan Sulfate, Heparin, Membrane Enzymes, Peptide Interactions, Glycosaminoglycan Biosynthesis, Molecular Modeling, PAPS, Phage Display, Sulfotransferase Inhibitors
Introduction
Heparan sulfate proteoglycans are found on the cell surface and in the extracellular matrix where they influence a variety of biological processes by interacting with physiologically important proteins, such as growth factors, chemokines and cytokines, extracellular matrix proteins, enzymes, and enzyme inhibitors (1, 2). Their activity is due in large part to the pattern of sulfated sugar residues along the heparan sulfate chains covalently bound to the core proteins of the proteoglycans. Heterogeneity arises during biosynthesis of the chains, which is mediated by several families of glycosyltransferases and sulfotransferases (3, 4). The chains can be further processed after reaching the plasma membrane by endo-6-sulfatases and heparanase.
The first modification reaction is catalyzed by one or more members of a family of bifunctional enzymes known as the N-deacetylase-N-sulfotransferases (Ndsts).2 Four mammalian Ndsts have been identified with high sequence similarity (4). The N-sulfotransferase activity has been assigned to the C-terminal domain of Ndst1 (5–7), and a similar domain structure appears to be conserved in the other Ndsts (8, 9). N-Sulfation of glucosamine residues is prerequisite for the downstream modifications, which include C5 epimerization and 2-O-sulfation of uronic acids and 6-O-sulfation and 3-O-sulfation of the glucosamine units (3).
Much interest exists in finding ways to block the formation of heparan sulfate as a way to explore the relationship of heparan sulfate structure to its biological function. Toward this end, a number of mutant cell lines and model organisms with defects in heparan sulfate biosynthesis have been described (10–12). Additionally, genistein and rhodamine B, nonspecific inhibitors of heparan sulfate biosynthesis, and β-d-xylosides, which act as metabolic decoys, have been exploited in cell culture and in animal models (13–16). Antagonists of heparan sulfate-protein interactions have also been described (17). Specific biologically active inhibitors of the glycosyltransferases and sulfotransferases have not yet been described.
Phage display technology was first reported in 1985 (18). In this technique, recombinant peptides are expressed as part of the bacteriophage capsid protein pIII or pVIII. Libraries of random peptide sequences attached to the capsid proteins are subsequently generated during the production of the bacteriophage particles (19). The peptides expressed on the phage capsid proteins have 109–1012 permutations and can be selected based on the specific binding to a target molecule. The success of this methodology has been demonstrated by the identification of peptides that interact with various proteins (20–24), carbohydrates (25), cultured cells (26), as well as peptides that home to specific tissues (27, 28).
In the present study, we used phage display technology to select peptides capable of interacting with Ndst1. Two peptides were identified that bound to purified recombinant enzyme and inhibited sulfotransferase activity, demonstrating the utility of this approach for finding potential inhibitors of heparan sulfate biosynthesis.
EXPERIMENTAL PROCEDURES
Cell Culture
Wild type CHO-K1 cells and the xylosyltransferase-deficient mutant (pgsA-745) (29) were grown at 37 °C under an atmosphere of 5% CO2 and 100% relative humidity in Ham's F-12 growth medium (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (Cultilab, Campinas, Brazil), 100 μg/ml streptomycin sulfate, and 100 units/ml penicillin.
Recombinant Ndst1 and Ndst3
cDNAs for mouse Ndst1 (mNdst1; amino acids 42–882) and human Ndst3 (hNdst3; amino acids 48–873) lacking their cytoplasmic and transmembrane domains were cloned in frame with the C-terminal domain of protein A (8). Either wild type or pgsA745 cells were transfected with the recombinant plasmids using FuGENE 6 reagent (Roche Applied Science, Basel, Switzerland) as indicated in the legends. The conditioned culture medium was collected daily for 5 days, and mNdst1 and hNdst3 were purified by stirring with IgG-Sepharose beads (10 μl/ml of medium for 24 h at 4 °C). The beads were washed three times with 50 mm phosphate buffer, pH 7.0. For kinetic experiments, mNdst1 or hNdst3 was eluted from the IgG-Sepharose beads using 0.03 m glycine, pH 3.0, and the sample was adjusted to pH 7.4 by the addition of a solution containing 0.5 m HEPES, pH 9.0, 1% Triton X-100, 10 mm MgCl2, and 1 mm MnCl2. All of the other assays were performed with enzyme bound to IgG-Sepharose beads. Recombinant mHs3st-1 was a generous gift from Dr. Jian Liu (University of North Carolina).
Endogenous Heparan Sulfate in mNdst1 Preparation
To investigate whether endogenous heparan sulfate copurified with mNdst1, recombinant protein was generated in wild type cells cultured in the presence of 150 μCi of [35S]sulfate/ml of growth medium. [35S]Glycosaminoglycans were identified by agarose gel electrophoresis (30) and by enzymatic digestion with chondroitinase ABC (Sigma) and heparitinases I and II (31).
Phage Peptide Libraries and Selection
The screening library used in this study consisted of decamers bound by cysteine residues, CX10C, where C and X represent cysteine and other amino acid residues, respectively. The peptide phage library was constructed as described previously (19). The library titer was ∼1012 transforming units/ml.
Biopanning was performed by mixing 109 phage with IgG-bound mNdst1 (5 μg) prepared from wild type CHO cells in a solution composed of 3% bovine serum albumin in phosphate-buffered saline (137 mm NaCl, 15.2 mm Na2HPO4, 1.5 mm KH2PO4, 2.7 mm KCl, pH 7.5). After 1 h at room temperature, the beads were sedimented by centrifugation and washed 10 times with buffer. Bound bacteriophage were dislodged with heparin (10 μg/ml, 30 min at room temperature) and used to infect K91kan Escherichia coli. After 1 h at room temperature, the cells were diluted in 20 ml of Luria broth medium and precipitated with 6% polyethylene glycol in the presence of 0.32 m NaCl. The precipitated bacteriophage pool was then used for the next round of biopanning. Five rounds of biopanning were performed, and in each round an aliquot of infected K91kan E. coli was grown on agar plates in the presence of kanamycin (100 μg/ml) and tetracycline (20 μg/ml). The plates were incubated for 18 h at 37 °C, and individual plaques were selected for DNA sequencing.
DNA Sequencing
DNA from the phage plaques was amplified by PCR (25 cycles of 95 °C for 20 s, 50 °C for 15 s, and 60 °C for 1 min) using primers that flanked the peptide insertion sites (5′-GGTCTAGAATTCGCCCCAGCGGCCCC-3′ and 5′-AGGCTCGAGGATCCTCGGCCACGGGGC-3′) and submitted for sequencing.
Recombinant GST Peptides
cDNA sequences encoding the peptides CNMQALSMPVTC and CRGWRGEKIGNC were cloned into the vector pGEX-KG (GE Healthcare, Waukesha, WI) between EcoRI and XhoI sites to generate fusion peptides with GST. The recombinant GST peptides were purified according to the manufacturer's instructions, and a final concentration of 20 μm was used for enzymatic assays.
Synthesis of Linear Peptides
An automated bench top solid phase peptide synthesizer (PSSM 8 system; Shimadzu, Columbia, MD) was used for the synthesis of all the peptides using the N-(9-fluorenyl)methoxycarbonyl (Fmoc) procedure. After synthesis the peptides were purified by C18 reversed phase HPLC chromatography, and their purity was confirmed by mass spectrometry. The linear peptides were synthesized without the flanking cysteine residues present in the phage and GST constructs.
[35S]PAPS
[35S]PAPS was prepared using yeast enzymes. Approximately 100 g of frozen baker's yeast was thawed in the presence of 50 mm phosphate buffer and homogenized at 4 °C. The supernatant was collected after centrifugation (6,760 × g for 20 min at 4 °C) and precipitated by slow addition of saturated ammonia sulfate with stirring (4 °C for 18 h). The precipitated material was centrifuged as described above and solubilized using 0.02 m Tris-HCl, pH 8.0. An aliquot of 0.3 ml of this enzyme preparation was incubated with 1 mCi of carrier-free [35S] (IPEN, São Paulo, Brazil) in the presence of 5 mm ATP, 10 mm MgCl2, and 40 mm KCl, in Tris-HCl buffer (0.1 m, pH 8.5). The mixture was centrifuged (10,000 × g, 4 °C, 10 min), and the supernatant was separated by preparative electrophoresis on Whatman 3MM paper in a buffer composed of 0.01 m citrate phosphate and 0.01 m Tris acetate, pH 6.5 (500 V, 90 min). Under these conditions PAPS separates from inorganic sulfate and adenyl-5′-phospho[35S]sulfate and PAP. The [35S]PAPS was eluted from the paper, lyophilized, suspended in 0.02 m, filtered, and stored in Tris-HCl, pH 8.0, at −70 °C.
Enzymatic Assay of mNdst1, hNdst3, and mHs3st1
Sulfotransferase activity of mNdst1 was assayed for 1 h at 37 °C with N-desulfoheparin as substrate (32) (0.5 μm; apparent Km of 13 ± 2 μm), carrier-free [35S]PAPS (105 cpm/reaction), 50 mm HEPES buffer, pH 7.0, 1% Triton X-100, 10 mm MgCl2, and 1 mm MnCl2 in a volume of 50 μl. The reaction was stopped by the addition of 0.5 m EDTA (2 μl) and chondroitin sulfate (2 mg) as carrier. The sample was applied to a 1-ml column of DEAE-Sephacel (GE Healthcare, Waukesha, WI), pre-equilibrated with 20 mm sodium acetate buffer, pH 6.0, containing 0.2 m NaCl. Labeled chains were eluted using 20 mm sodium acetate, pH 6.0, containing 1 m NaCl. 35S counts incorporated into N-desulfoheparin were quantified by scintillation counting using Ultima Gold. Sulfotransferase activity of Hs3st1 was assayed for 1 h at 37 °C with heparan sulfate isolated from CHO culture medium in the presence of [35S]PAPS (105cpm/reaction) in 50 mm HEPES buffer, pH 7.0, 1% Triton X-100, 10 mm MgCl2, and 1 mm MnCl2 at a final volume of 50 μl. The reaction products were quantitated essentially as described above.
The inhibitory activity of the peptides on sulfotransferase activity in CHO cell homogenates was measured essentially as described previously (33). CHO cells were grown to confluence in 10-cm culture dishes in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum, 50 units/ml penicillin, and 50 μg/ml streptomycin at 37 °C. Cell layers were washed thrice with phosphate-buffered saline, detached with a rubber policeman in 1 ml of ice-cold buffer consisting of 10 mm Tris, pH 7.2, 2 mm CaCl2, 10 mm MgCl2, 150 mm NaCl, 0.5% Triton X-100, 20% glycerol and a mixture of protease inhibitors (5 μm N-α-tosyl-l-lysine chloromethyl ketone, 3 μm l-1-p-tosylamino-2-phenylethyl chloromethyl ketone, 30 μm phenylmethylsulfonyl fluoride, 3 μm pepstatin), and sonicated with three 1-s pulses. The standard reaction mixture contained 2.5 μmol of imidazole HCl, pH 6.8, 3.75 μg of protamine chloride, 25 nmol of N-desulfoheparin (Neoparin, San Leandro, CA), 50 pmol of [35S]PAPS, and 20 μg of cell protein as described above in a final volume of 50 μl. The reaction mixtures were incubated at 37 °C for 30 min, and the reaction was stopped by immersing the reaction tubes in ice. Radioactive heparan sulfate chains were purified using DEAE-Sephacel columns as described above, and the 35S counts incorporated into the sugar chains were quantified.
Enzymatic N-Deacetylase Assay
N-Acetyl glucosamine N-deacetylase activity of recombinant mNdst1 was assayed as described previously (8). In short, activity was measured using [acetyl-3H]heparosan prepared from E. coli K5 capsular polysaccharide (33, 34). The assay was performed in 50 mm MES buffer, pH 6.5, containing 1% Triton X-100, 10 mm MnCl2, and 1 × 105 cpm [3H]heparosan. The reaction was stopped after 30 min at 37 °C by the addition of 0.5 volumes of 0.2 m HCl, 1 volume of 0.1 m acetic acid, and 1 volume of water. [3H]Acetic acid was recovered by extracting the sample three times with 1 volume of ethyl acetate. An aliquot of the pooled extracts (0.5 ml) was analyzed by liquid scintillation counting.
Peptide Competition Assays
Competition assays using the peptides are described in the text and figure legends. The following equation was used to calculate the apparent Ki values,
 |
where Vo is the velocity of the reaction without the inhibitor, Vi is the velocity in the presence of the inhibitor, and [I] is molar inhibitor concentration. The plot of
 |
versus [I] was used to calculate the Kiapp, where the slope represents 1/Kiapp.
Peptide Binding to Heparin-Sepharose
A heparin-Sepharose column (2 ml; GE Healthcare) was prewashed with 10 mm HEPES, pH 7.0. Selected peptides (20 μm) were applied to the column and eluted using 10 mm HEPES, pH 7.0, containing different NaCl concentrations (0.05–1.0 m). Ten bed volumes were used in each step, and the elution profile was monitored by the absorption of the peptide bond at 214 nm.
Competition Binding Assay
mNdst1 was purified from wild type CHO cells radiolabeled with [35S]sulfate. A sample of immobilized enzyme (20 μg/ml) was incubated for 1 h at 4 °C with different concentrations of peptide and 4 μm PAPS (Sigma). The beads were then washed six times by centrifugation with buffer containing 50 mm HEPES, pH 7.0, 0.1% Triton X-100, 10 mm MgCl2, and 1 mm MnCl2. The remaining heparan sulfate associated with the enzyme was collected by sedimenting the beads by centrifugation (3,000 × g for 1 min). The sample was suspended in 10 μl of buffer containing carrier heparan sulfate (Sigma, St. Louis, MO) and subjected to agarose gel electrophoresis using 0.05 m 1,3 diaminopropane acetate buffer, pH 9.0. After electrophoresis heparan sulfate was precipitated in the gel with 0.1% cetyltrimethyl ammonium bromide (Sigma). The dried gel was stained with 0.1% toluidine blue as described to visualize the heparan sulfate, and the radioactive heparan sulfate was detected by autoradiography (30).
Fluorescence Assays
Binding of peptides was measured by determining the change in intrinsic fluorescence of tryptophan residues in recombinant mNdst1. mNdst1 was purified from conditioned culture medium using IgG-Sepharose beads as described above and removed from the beads by thrombin cleavage at 22 °C for 2 h with 10 units of thrombin (GE Healthcare)/20 μl of resin. Soluble mNdst1 was purified by binding to PAP-agarose beads (Sigma). The beads were washed twice with 50 mm phosphate buffer, and mNdst1 was eluted using 100 μl of 0.5 m HEPES, pH 9.0, 1% Triton X-100, 10 mm MgCl2, and 1 mm MnCl2.
Intrinsic tryptophan fluorescence was monitored by measuring the fluorescence emission (λem) at 350 nm (5-nm slit) after excitation at λex = 285 nm (5-nm slit) (Shimadzu RF-5301-PC, Tokyo, Japan). Binding affinities of the peptides for mNdst1 (20 μg/ml) were determined by measuring the change in intrinsic fluorescence observed at increasing peptide concentrations in 10 mm HEPES, pH 7.0, at 37 °C. Signal from buffer alone was subtracted from the readings to eliminate the background. The hyperbolic curves obtained could be best fitted according to the equation,
where ΔF is the variation in emission intensity induced by bound peptide, ΔFmax is the maximum of fluorescence variation at infinite peptide concentration, and Kd is the apparent dissociation constant of the peptide-mNdst1 complex. The interaction of the peptide CRGWRGEKIGNC with heparan sulfate was analyzed using intrinsic tryptophan florescence of the single tryptophan residue in the peptide.
Molecular Modeling
Three-dimensional coordinates of the binary complex of the N-sulfotransferase domain with PAP were retrieved from the Protein Data Bank (35, 36) (Protein Data Bank code 1NST; resolution, 2.3 Å). The peptides were built using PRODRG2 (37). AutoDock4.0 was used as a grid-based docking procedure (38, 39). To perform docking experiments, the missing side chain atoms in the Nst1 were added, PAP was removed, and the hydrogens and partial charges were added to the peptide ligands and Nst1. For the ligand, Gasteiger charges (40) were calculated using Autodock tools. The enzyme model was also prepared with Autodock Tools, deleting all water molecules, adding polar hydrogens, and loading Kollman United Atoms charges (41). The AutoGrid settings with 76 × 64 × 68 grid size and a grid spacing of 0.462 Å were used for preparing the grid. The genetic algorithm with local search options as implemented in AutoDock was used to dock the flexible peptide. For the “flexible” docking experiments, backbone (ϕ/ψ) and side chain torsions were allowed to rotate a total of 32 torsions (42). The Lamarckian genetic algorithm, with 2.5 × 108 energy evaluations (high) and 200 generations with step sizes of 0.2 for translation and 5.0 for quaternion and torsion, respectively, was employed. The best docked conformers with the lowest free energies (−12 kcal/mol) conformations were taken for further analysis.
Statistical Analysis
The average values ± standard deviation are provided for all of the experiments in which n ≥ 3 replicates were analyzed, as indicated in the figure and table legends. Significance was determined by single-tailed Student's t test with significance taken as p < 0.05.
RESULTS
Biopanning Assay
A library of bacteriophage was constructed containing peptides fused to the pIII coat protein, which is expressed in five copies/bacteriophage. The library was biopanned against a chimera of mNdst1 consisting of the full-length protein minus the cytoplasmic tail and transmembrane domain fused to protein A. mNdst1 was immobilized on IgG beads (“Experimental Procedures”), and heparin was used to displace bound phage. In the third and subsequent rounds of selection, phage containing two structurally distinct peptides were greatly enriched, specifically CRGWRGEKIGNC and CNMQALSMPVTC. These two peptides have distinctive amino acid sequence and vary in charge, suggesting that the peptides might have unique binding properties.
Peptide Inhibition of mNdst1 and hNdst3 Sulfotransferase Activity
Sulfotransferase activity was determined in the presence of bacteriophage containing the two identified peptides at different phage titers (104 and 108 transforming units/ml) (Fig. 1A). Both peptides, presented as phages, reduced sulfotransferase activity, with CNMQALSMPVTC somewhat more potent at both concentrations tested. Both peptides also inhibited Ndst1 when presented as GST chimeras, with CNMQALSMPVTC again exhibiting greater activity (Fig. 1B). When displayed on the phage, the peptides assume a cyclic conformation because of the disulfide bond formation (19). Whether the peptides are cyclic when fused to GST is unknown. However, the inhibitory activity appeared to be independent of cyclization, because synthetic linear peptides lacking the cysteine residues also inhibited sulfotransferase activity (Fig. 1, C and D). Analysis of the inhibitory pattern showed an apparent inhibition constant (Ki app) of 140 ± 20 μm for NMQALSMPVT and 480 ± 12 μm for RGWRGEKIGN. We attempted unsuccessfully to inhibit N-sulfotransferase activity in crude CHO cell extracts using linear peptides possibly caused by rapid degradation of these molecules, as previously discussed by others (43). However, the peptide-GST fusion CNMQALSMPVTC inhibited sulfotransferase activity by 25 and 40% at 10 and 100 μm, respectively.
FIGURE 1.

Inhibition of mNdst1 sulfotransferase activity by phage selected peptides. mNdst1 produced in mutant pgsA-745 was assayed in the presence or absence of phage, GST chimeras, or synthetic peptides as indicated. A, inhibition by the peptides by two different concentrations of phage (transforming units/ml). B, inhibition by 20 μm of peptide-GST fusion. C and D, inhibition by synthetic linear peptides expressed as [(Vo/Vi) − 1] versus different concentrations of the peptides. The experiments were performed in triplicate at least three times. The error bars represent the standard deviation from the mean.
In contrast to these findings, linear and peptide-GST fusion forms of the peptides did not affect the activity of recombinant murine Hs3st1 (33 pmol/min control versus 32 pmol/min at 100 μm of peptide CNMQALSMPVTC), even when assayed at a higher concentrations (32 pmol/min at 1 mm). Linear and cyclic peptides also had no effect on the N-deacetylase activity of mNdst (409 cpm/min control versus 415 cpm/min at 100 μm of peptide CNMQALSMPVTC). Thus, the selected peptides selectively affect N-sulfotransferase activity under the conditions used to measure enzyme activity in vitro and N-deacetylase activity. As shown below, docking studies also indicate selective binding of one of the peptides to Ndst1.
Binding Studies
The direct interaction of the peptide NMQALSMPVT with mNdst1 was assessed by measuring changes in intrinsic tryptophan fluorescence, which reflects conformational change. Increasing the concentration of peptide resulted in a progressive change in fluorescence and showed saturability with an apparent Kd of 120 ± 15 μm (Fig. 2A). Interestingly, the estimated affinity of the peptide for the mNdst1 was similar to the values of Ki app found for the inhibition of sulfotransferase activity. Fig. 2B shows the changes in the mNdst1 fluorescence emission spectra as a function of peptide concentration. Notably, binding of peptide to the enzyme promoted a red shift from 306 to 317 nm. The red shift indicates that the buried tryptophan residues became exposed to a more polar environment (44). Because Ndst1 contains numerous tryptophan residues, the change in fluorescence could not be ascribed to specific residues. However, the data are consistent with a conformational change induced by peptide binding that affects enzyme activity. The synthetic scrambled peptides QTMVPASNLM and SVNQATLPMM did not induce a change in intrinsic fluorescence (Fig. 2C) or sulfotransferase activity (Fig. 3A). Interestingly, the peptide CNMQALSMPVTC also inhibited the N-sulfotransferase activity of recombinant human Ndst3 (Fig. 3B), suggesting that it bound to a common determinant in both enzymes.
FIGURE 2.

Peptide-induced changes of intrinsic fluorescence of recombinant Ndst1. A, different concentrations of the peptide NMQALSMPVT were mixed with 20 μm mNdst1, and the intrinsic tryptophan fluorescence emission was measured (λex = 285 nm; λem = 350 nm) The change in fluorescence showed saturable behavior consistent with binding of the peptide to mNdst1. B, emission spectra were obtained for 20 μm mNdst1 alone (□) or in the presence of 100 μm (♢), 200 μm (●), or 400 μm NMQALSMPVT (▿). C, scrambled peptides QTMVPASNLM and SVNQATLPMM do not induce changes of intrinsic tryptophan fluorescence of mNdst1. The spectra were obtained for 20 μm mNdst1 alone or in the presence of 20 μm (△) or 400 μm QTMVPASNLM (×), 20 μm (○) or 400 μm SVNQATLPMM (+), or 400 μm NMQASLMPVT (□).
FIGURE 3.

N-Sulfotransferase activity of recombinant isoforms of Ndsts. The sulfotransferase activity of mNdst1 (A) and hNdst3 (B) was measured in the absence of peptides (Control) or in the presence of 20 μm of NMQALSMPVT or the scrambled peptide QTMVPASNLM. The assays were performed in triplicate, and the error bars indicate the standard deviation from the mean (p < 0.05).
The peptide RGWRGEKIGN contains three positively charged amino acids, similar to the spacing of positively charged residues in many heparin-binding proteins (45, 46). Thus, the inhibitory activity of this peptide could be related to its ability to bind to the heparin substrate as well as the enzyme. Indeed, peptide RGWRGEKIGN bound to heparin-Sepharose, whereas NMQALSMPVT did not (Fig. 4). Taking advantage of the presence of a tryptophan residue in the RGWRGEKIGN sequence, we tested the interaction of this peptide with heparan sulfate by monitoring its fluorescence in mixtures. Increasing the concentration of heparan sulfate induced changes in the fluorescence emission of the peptide and the fluorescence spectrum, consistent with binding of the peptide to the heparan sulfate chain (Fig. 5).
FIGURE 4.

Peptide affinity for heparin. Peptides were bound to heparin-Sepharose and eluted using a gradient of sodium chloride (0–1 m NaCl). RGWRGEKIGN bound to heparin-Sepharose, whereas NMQALSMPVT did not bind and eluted in the flowthrough fraction (not shown).
FIGURE 5.

Peptide RGWRGEKIGN binds to heparan sulfate. A, different concentrations of heparan sulfate were added to RGWRGEKIGN, and fluorescence emission of the single tryptophan residue in the peptide was measured. B, emission spectra of 0.5 μm peptide RGWRGEKIGN alone (○) or with 20 μm (■) or 40 μm heparan sulfate (△).
Based on these findings, we wondered whether selection of this peptide in the biopanning experiments might have been due to the presence of endogenous heparan sulfate bound to the enzyme, which had been expressed and purified from wild type CHO cells. To investigate this possibility, CHO cells transfected with mNdst1 were incubated with [35S]sulfate. The recombinant enzyme was purified from the conditioned medium and then analyzed for [35S]glycosaminoglycans by electrophoresis. The data in Fig. 6 show that [35S]glycosaminoglycans copurified with mNdst1. The bound material was resistant to chondroitinase ABC degradation and fully degraded by heparitinases. Thus, the presence of heparan sulfate in secreted mNdst1 suggested that the peptide RGWRGEKIGN had been selected based on its ability to bind heparan sulfate.
FIGURE 6.
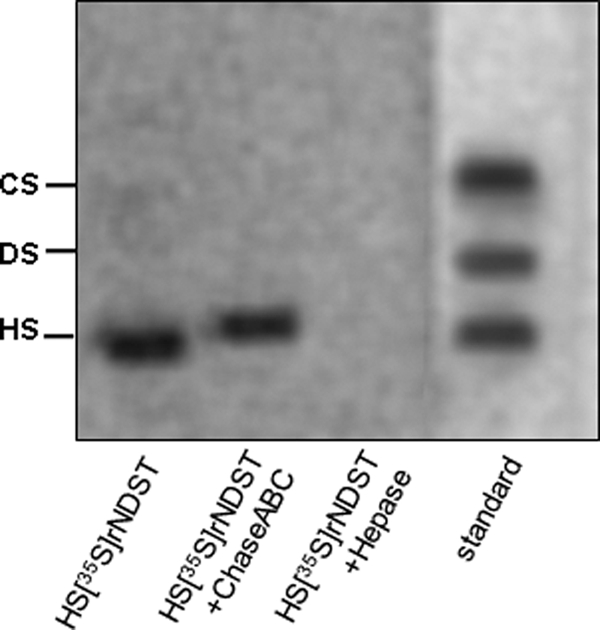
Heparan sulfate binds to recombinant Ndst1. Wild type CHO cells were transfected with mNdst1 and grown in medium containing [35S]sulfate. Recombinant enzyme was purified from the conditioned medium, and samples were analyzed for [35S]glycosaminoglycans by agarose gel electrophoresis before and after degradation with chondroitinase ABC (ChaseABC) and heparitinases I and II (Hepase). The positions of standard heparan sulfate (HS), dermatan sulfate (DS), and chondroitin sulfate (CS) are shown.
Molecular Docking
We next examined the docking of peptide NMQALSMPVT with mNdst1. Both heparan sulfate and heparin were capable of preventing the increase of fluorescence induced by the peptide (Fig. 7A). Furthermore, incubation of mNdst1 containing [35S]heparan sulfate with NMQALSMPVT displaced the bound chain (Fig. 7B). In contrast, the addition of the peptide RGWRGEKIGN was only partially effective (Fig. 7B). These findings suggested that the peptide NMQALSMPVT interacted with the binding site for the acceptor substrate.
FIGURE 7.

Peptide NMQALSMPVT blocks binding of heparan sulfate to mNdst1. A, emission spectra were obtained for 20 μm mNdst1 mixed with 50 μm heparan sulfate (□), 400 μm NMQALSMPVT (♦), 400 μm NMQALSMPVT, and 50 μm heparan sulfate (▿) or 400 μm NMQALSMPVT and 200 μm heparan sulfate (■). B, mNdst1 (2 μg) produced in wild type cells radiolabeled with [35S]sulfate was mixed with the indicated concentrations of the peptides. The beads were sedimented by centrifugation, washed, and analyzed by agarose gel electrophoresis. Peptide NMQALSMPVT displaced bound [35S]heparan sulfate above 50 μm, whereas peptide RGWRGEKIGN was only partially effective.
Molecular docking of both peptides was performed using the crystal structure of the sulfotransferase domain (35), assuming that the minimum energy represents the best model for the peptide-binding site. Both linear and cyclic RGWRGEKIGN peptides did not present reasonable ΔG docking energies, consistent with the studies described above showing that it most likely interacted with the heparan sulfate substrate rather than the enzyme. In contrast, NMQALSMPVT had a preferred docking site. A cavity formed between the phosphate-sulfate binding loop and helix α6 defines the PAP(S)-binding site, with a cleft perpendicular to the PAP(S)-binding cavity, that can accommodate a hexasaccharide chain. Helix α6 and the random coil between β2 and helix α2 constitute the cleft near the 5′-phosphate of PAP and thus may be part of the substrate-binding site. The optimal docking site for the peptide NMQALSMPVT was in this domain, suggesting that its inhibitory action was due to shielding of the heparan sulfate entry site and interactions with crucial residues at the active site (Table 1). In this model, the serine and valine residues from the peptide interact with the side chains of Lys833 and Tyr837 from the random coil formed after an anti-parallel β-sheet (β6, β7, and β8) from Ndst1 (Fig. 8A). These protein residues are within hydrogen bonding distance to two oxygen atoms of the 5′-phosphate and the oxygen atom of the 3′-phosphate, respectively (47). The valine residue from peptide NMQALSMPVT form a 0.24-Å hydrogen bond with the Lys833 residue, which is found in position to form a hydrogen bond with the 5′-sulfuryl oxygen from PAPS or serve as a proton donor, neutralizing the negative charge of the sulfate group (47). The carbonyl group of the asparagine residue in peptide NMQALSMPVT forms a hydrogen bridge with Lys614, a residue with confirmed catalytic importance (Fig. 8B) (7). The theoretical Kd obtained for the binding of the peptide NMQALSMPVT is 0.82 μm at 25 °C based on the free energy (ΔG = 2.303RT × logKd).
TABLE 1.
Predicted interactions of the NST-1-binding site residues with the peptide NMQALSMPVT
| NST domain | Peptide contact | Distance | Angle |
|---|---|---|---|
| Å | ° | ||
| Ser860-HN | Thr-OC | 1.075 | 122.1 |
| Lys614-NH | Asn-OG | 1.062 | 142.9 |
| Lys833-NH | Val-CH | 0.241 | 149.5 |
| Thr618-HH | Met-O | 1.086 | 132.5 |
| Tyr837-HH | Ser-O | 2.163 | 106.8 |
FIGURE 8.

Representation of the complex of NMQALSMPVT with the sulfotransferase domain (Nst1) of Ndst1. A, view of the minimum energy conformation from docking, showing the ligand completely engulfed inside the protein with its side chains pointing into the large hydrated cavity. Dark blue, NMQALSMPVT peptide; light blue, Gln613; green, Lys614; yellow, Arg835; red, PAPS). B, stereo view of the catalytic domain from the complex between Nst1 and the linear peptide, NMQALSMPVT. Lys614 on the 5′-phosphate-sulfate binding loop is shown in green, and the asparagine residue from the NMQALSMPVT peptide is shown in light blue.
Possible binding and/or inhibition of other heparan sulfate sulfotransferases was analyzed by structural comparison between sulfotransferases, as well as blind docking with the peptides. Because the transferase reaction requires insulation from bulk water environment (48), solvent accessibility (49) and pocket size (50) were also calculated. The docking energies for both Hs3st1 and uronyl 2-O-sulfotransferase (Protein Data Bank codes 1T8T and 3F5F, respectively) with the peptide NMQALSMPVT were infinite, indicating no binding (supplemental Table S1). The major difference found between Ndst1 and other glycosaminoglycan sulfotransferases was the solvent relative accessibility for the PAPS molecule. In addition, conserved cleft volume and the proposed reaction mechanism were observed for the phosphate-sulfate binding motif after structural alignment of these sulfotransferases with NST (supplemental Fig. S1).
DISCUSSION
The present study demonstrates the success of phage display methodology for selecting specific peptides that inhibit the sulfotransferase activity of recombinant N-deacetylase-N-sulfotransferase-1. The inhibitory effect was confirmed using cyclic peptides displayed on the bacteriophage capsid or recombinant peptides expressed as a GST chimeras or as linear synthetic peptides. The data indicate that RGWRGEKIGN inhibits sulfotransferase activity in a noncompetitive manner, most likely by binding the potential substrate (heparan sulfate or heparin), whereas NMQALSMPVT targets the active site of mNdst1, acting as a competitive inhibitor with respect to heparan sulfate. The binding site identified by molecular modeling is similar to that in other Ndst isoforms, and NMQALSMPVT also inhibited hNdst3, consistent with this prediction. However, blind docking of NMQALSMPVT and RGWRGEKIGN peptides against both O-sulfotransferases did not suggest any interaction. This may be due to structural differences between the Ndst and O-sulfotransferase catalytic sites, where conserved residues and the PAPS-binding site present distinct solvent accessibilities, despite their similar pocket volumes at the active site. The selected peptides presented no inhibitory effect on Hs3st1, thereby confirming the specificity of the selected peptides for Ndst. Moreover, the selected peptides specifically inhibit the sulfotransferase activity of full-length Ndst, with no effect on the N-deacetylase activity. These results together confirm the specificity of the selected peptides for Ndst sulfotransferase.
The discovery that small cyclic peptides can be used to inhibit an enzyme involved in heparan sulfate biosynthesis raises the possibility that similar sequences might exist naturally in endogenous proteins. Screening various genome databases with the peptide NMQALSMPVT sequence only exhibited a significant resemblance to purine-binding proteins. These proteins possess a nucleotide-binding domain, which is known to be spatially similar to the PAPS-binding site in sulfotransferases (51, 52), but the relationship of these peptides sequences to sulfotransferase activity is not known.
Much interest exists in the discovery of antagonists of glycosaminoglycan function. Three classes of inhibitors have been described: (i) antagonists of heparan sulfate that block heparin-protein interactions (e.g. lactoferrin, protamine, single chain antibodies, and surfen) (17, 53); (ii) agents that block heparan sulfate metabolism (e.g. xylosides, rhodamine B, and genistein) (13, 14, 16); and (iii) agents that block specific enzymes in the pathway (54, 55). The peptides described in this report fall into two of these inhibitor subclasses. RGWRGEKIGN behaves as an antagonist by binding to heparan sulfate and preventing its use as a substrate. In that regard, RGWRGEKIGN behaves like the small molecule inhibitor surfen, which was recently shown to block uronyl 2-O-sulfotransferase from sulfating 2-O-desulfated heparin. Whether RGWRGEKIGN can block other protein interactions with heparin or heparan sulfate is unknown. This peptide may have similar properties to one described previously (CARSKNKDC) that binds to heparan sulfate and heparin (56). In contrast, NMQALSMPVT behaves like a true enzyme-based inhibitor, interacting with a distinct domain of the protein. Further modifications to the peptide sequence are underway to enhance its inhibitory activity, as a segue to in vivo studies.
Supplementary Material

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Fig. S1.
- Ndst
- N-deacetylase-N-sulfotransferase
- mNdst
- murine Ndst
- hNdst
- human Ndst
- PAPS
- adenosine 3′-phosphate,5′-phosphosulfate
- PAP
- 3′-phosphoadenosine,5′-phosphate
- MES
- 4-morpholineethanesulfonic acid
- Hs3st1
- glucosamine 3-O-sulfotransferase-1.
REFERENCES
- 1. Bishop J. R., Schuksz M., Esko J. D. (2007) Nature 446, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 2. Sampaio L. O., Tersariol I. L., Lopes C. C., Bouças R. I., Nascimento F. D., Rocha H. A., Nader H. B. (2006) Insights into Carbohydrate Structure and Biological Function, Research Signpost, Kerala, India [Google Scholar]
- 3. Esko J. D., Lindahl U. (2001) J. Clin. Invest. 108, 169–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grobe K., Ledin J., Ringvall M., Holmborn K., Forsberg E., Esko J. D., Kjellén L. (2002) Biochim. Biophys. Acta 1573, 209–215 [DOI] [PubMed] [Google Scholar]
- 5. Berninsone P., Hirschberg C. B. (1998) J. Biol. Chem. 273, 25556–25559 [DOI] [PubMed] [Google Scholar]
- 6. Wei Z., Swiedler S. J. (1999) J. Biol. Chem. 274, 1966–1970 [DOI] [PubMed] [Google Scholar]
- 7. Sueyoshi T., Kakuta Y., Pedersen L. C., Wall F. E., Pedersen L. G., Negishi M. (1998) FEBS Lett. 433, 211–214 [DOI] [PubMed] [Google Scholar]
- 8. Aikawa J., Esko J. D. (1999) J. Biol. Chem. 274, 2690–2695 [DOI] [PubMed] [Google Scholar]
- 9. Aikawa J., Grobe K., Tsujimoto M., Esko J. D. (2001) J. Biol. Chem. 276, 5876–5882 [DOI] [PubMed] [Google Scholar]
- 10. Zhang L., Lawrence R., Frazier B. A., Esko J. D. (2006) Methods in Enzymology 416, 205–221 [DOI] [PubMed] [Google Scholar]
- 11. Bülow H. E., Hobert O. (2006) Annu. Rev. Cell Dev. Biol. 22, 375–407 [DOI] [PubMed] [Google Scholar]
- 12. Gorsi B., Stringer S. E. (2007) Trends Cell Biol. 17, 173–177 [DOI] [PubMed] [Google Scholar]
- 13. Jakóbkiewicz-Banecka J., Piotrowska E., Narajczyk M., Barańska S., Wegrzyn G. (2009) J. Biomed. Sci. 16, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roberts A. L., Thomas B. J., Wilkinson A. S., Fletcher J. M., Byers S. (2006) Pediatr. Res. 60, 309–314 [DOI] [PubMed] [Google Scholar]
- 15. Brown J. R., Crawford B. E., Esko J. D. (2007) Crit. Rev. Biochem. Mol. Biol. 42, 481–515 [DOI] [PubMed] [Google Scholar]
- 16. Fritz T. A., Esko J. D. (2001) Methods Mol. Biol. 171, 317–323 [DOI] [PubMed] [Google Scholar]
- 17. Schuksz M., Fuster M. M., Brown J. R., Crawford B. E., Ditto D. P., Lawrence R., Glass C. A., Wang L., Tor Y., Esko J. D. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 13075–13080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith G. P. (1985) Science 228, 1315–1317 [DOI] [PubMed] [Google Scholar]
- 19. Burritt J. B., Bond C. W., Doss K. W., Jesaitis A. J. (1996) Anal. Biochem. 238, 1–13 [DOI] [PubMed] [Google Scholar]
- 20. Scott J. K., Smith G. P. (1990) Science 249, 386–390 [DOI] [PubMed] [Google Scholar]
- 21. Cwirla S. E., Peters E. A., Barrett R. W., Dower W. J. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 6378–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wrighton N. C., Farrell F. X., Chang R., Kashyap A. K., Barbone F. P., Mulcahy L. S., Johnson D. L., Barrett R. W., Jolliffe L. K., Dower W. J. (1996) Science 273, 458–464 [DOI] [PubMed] [Google Scholar]
- 23. Koivunen E., Wang B., Ruoslahti E. (1994) J. Cell Biol. 124, 373–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pasqualini R., Koivunen E., Ruoslahti E. (1995) J. Cell Biol. 130, 1189–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peletskaya E. N., Glinsky G., Deutscher S. L., Quinn T. P. (1996) Mol. Divers. 2, 13–18 [DOI] [PubMed] [Google Scholar]
- 26. Barry M. A., Dower W. J., Johnston S. A. (1996) Nat. Med. 2, 299–305 [DOI] [PubMed] [Google Scholar]
- 27. Pasqualini R., Ruoslahti E. (1996) Nature 380, 364–366 [DOI] [PubMed] [Google Scholar]
- 28. Ruoslahti E., Rajotte D. (2000) Annu. Rev. Immunol. 18, 813–827 [DOI] [PubMed] [Google Scholar]
- 29. Esko J. D., Stewart T. E., Taylor W. H. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 3197–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nader H. B., Dietrich C. P., Buonassisi V., Colburn P. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 3565–3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nader H. B., Porcionatto M. A., Tersariol I. L., Pinhal M. A., Oliveira F. W., Moraes C. T., Dietrich C. P. (1990) J. Biol. Chem. 265, 16807–16813 [PubMed] [Google Scholar]
- 32. Inoue Y., Nagasawa K. (1976) Carbohydr. Res. 46, 87–95 [DOI] [PubMed] [Google Scholar]
- 33. Bame K. J., Reddy R. V., Esko J. D. (1991) J. Biol. Chem. 266, 12461–12468 [PubMed] [Google Scholar]
- 34. Bame K. J., Lidholt K., Lindahl U., Esko J. D. (1991) J. Biol. Chem. 266, 10287–10293 [PubMed] [Google Scholar]
- 35. Kakuta Y., Sueyoshi T., Negishi M., Pedersen L. C. (1999) J. Biol. Chem. 274, 10673–10676 [DOI] [PubMed] [Google Scholar]
- 36. Berman H. M., Bhat T. N., Bourne P. E., Feng Z., Gilliland G., Weissig H., Westbrook J. (2000) Nat. Struct. Biol. 7, (suppl.) 957–959 [DOI] [PubMed] [Google Scholar]
- 37. Schüttelkopf A. W., van Aalten D. M. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 38. Kuntz I. D., Blaney J. M., Oatley S. J., Langridge R., Ferrin T. E. (1982) J. Mol. Biol. 161, 269–288 [DOI] [PubMed] [Google Scholar]
- 39. Eldridge M. D., Murray C. W., Auton T. R., Paolini G. V., Mee R. P. (1997) J. Comput. Aided Mol. Des. 11, 425–445 [DOI] [PubMed] [Google Scholar]
- 40. Gasteiger J., Marsili M. (1981) Org. Magn. Resonance 15, 353–360 [Google Scholar]
- 41. Cornell W. D., Cieplak P., Bayly C. I., Gould I. R., Merz K. M., Jr., Ferguson D. M., Spellmeyer D. C., Fox T., Caldwell J. W., Kollman P. A. (1995) J. Am. Chem. Soc. 117, 5179–5197 [Google Scholar]
- 42. Hetényi C., van der Spoel D. (2002) Protein Sci. 11, 1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burster T., Marin-Esteban V., Boehm B. O., Dunn S., Rotzschke O., Falk K., Weber E., Verhelst S. H., Kalbacher H., Driessen C. (2007) Biochem. Pharmacol. 74, 1514–1523 [DOI] [PubMed] [Google Scholar]
- 44. Weber G. (1992) Protein Interactions, Chapman and Hall, New York [Google Scholar]
- 45. Capila I., Linhardt R. J. (2002) Angew Chem. Int. Ed Engl. 41, 391–412 [DOI] [PubMed] [Google Scholar]
- 46. Conrad H. E. (1998) Heparin-Binding Proteins, Academic Press, San Diego, CA [Google Scholar]
- 47. Gorokhov A., Perera L., Darden T. A., Negishi M., Pedersen L. C., Pedersen L. G. (2000) Biophys. J. 79, 2909–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koike R., Amemiya T., Ota M., Kidera A. (2008) J. Mol. Biol. 379, 397–401 [DOI] [PubMed] [Google Scholar]
- 49. Hubbard S. J., Thornton J. M. (1993) NACCESS, computer program, University College London, London [Google Scholar]
- 50. Laurie A. T., Jackson R. M. (2005) Bioinformatics 21, 1908–1916 [DOI] [PubMed] [Google Scholar]
- 51. Chapman E., Ding S., Schultz P. G., Wong C. H. (2002) J. Am. Chem. Soc. 124, 14524–14525 [DOI] [PubMed] [Google Scholar]
- 52. Verdugo D. E., Cancilla M. T., Ge X., Gray N. S., Chang Y. T., Schultz P. G., Negishi M., Leary J. A., Bertozzi C. R. (2001) J. Med. Chem. 44, 2683–2686 [DOI] [PubMed] [Google Scholar]
- 53. Jenniskens G. J., Koopman W. J., Willems P. H., Pecker I., Veerkamp J. H., van Kuppevelt T. H. (2003) FASEB J. 17, 878–880 [DOI] [PubMed] [Google Scholar]
- 54. Armstrong J. I., Portley A. R., Chang Y. T., Nierengarten D. M., Cook B. N., Bowman K. G., Bishop A., Gray N. S., Shokat K. M., Schultz P. G., Bertozzi C. R. (2000) Angew Chem. Int. Ed Engl. 39, 1303–1306 [DOI] [PubMed] [Google Scholar]
- 55. Armstrong J. I., Ge X., Verdugo D. E., Winans K. A., Leary J. A., Bertozzi C. R. (2001) Org. Lett. 3, 2657–2660 [DOI] [PubMed] [Google Scholar]
- 56. Järvinen T. A., Ruoslahti E. (2007) Am. J. Pathol. 171, 702–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.