

Abstract
The large apolar tunnel traversing the mini-hemoglobin from Cerebratulus lacteus (CerHb) has been examined by x-ray crystallography, ligand binding kinetics, and molecular dynamic simulations. The addition of 10 atm of xenon causes loss of diffraction in wild-type (wt) CerHbO2 crystals, but Leu-86(G12)Ala CerHbO2, which has an increased tunnel volume, stably accommodates two discrete xenon atoms: one adjacent to Leu-86(G12) and another near Ala-55(E18). Molecular dynamics simulations of ligand migration in wt CerHb show a low energy pathway through the apolar tunnel when Leu or Ala, but not Phe or Trp, is present at the 86(G12) position. The addition of 10–15 atm of xenon to solutions of wt CerHbCO and L86A CerHbCO causes 2–3-fold increases in the fraction of geminate ligand recombination, indicating that the bound xenon blocks CO escape. This idea was confirmed by L86F and L86W mutations, which cause even larger increases in the fraction of geminate CO rebinding, 2–5-fold decreases in the bimolecular rate constants for ligand entry, and large increases in the computed energy barriers for ligand movement through the apolar tunnel. Both the addition of xenon to the L86A mutant and oxidation of wt CerHb heme iron cause the appearance of an out Gln-44(E7) conformer, in which the amide side chain points out toward the solvent and appears to lower the barrier for ligand escape through the E7 gate. However, the observed kinetics suggest little entry and escape (≤25%) through the E7 pathway, presumably because the in Gln-44(E7) conformer is thermodynamically favored.
Keywords: Hemoglobin Myoglobin, Molecular Dynamics, Oxygen Binding, Oxygen Diffusion, Protein Structure, Cerebratulus lacteus Mini-hemoglobin
Introduction
Although molecular dynamics (MD)7 simulations suggest multiple interior pathways for O2 entry into and exit from globins, most experiments with mammalian myoglobins (Mbs) and hemoglobins (Hbs) suggest a well defined single pathway involving a short channel between the heme propionates and the heme iron atom that is gated by the distal E7 histidine (1). To search for and define an interior ligand migration trajectory, we chose to use the neuronal mini-hemoglobin from Cerebratulus lacteus as a model globin system to examine its long apolar tunnel that leads from the distal portion of the heme pocket to an exit point between the C-terminal regions of the E and H helices of the tertiary fold, a pathway that is roughly 180° opposite the E7 channel and appears to be a major route for ligand entry (2). This model globin provides a well defined system to examine both experimentally and theoretically the effects of xenon binding, mutagenesis, and conformational heterogeneity on the competition between movement through the E7 gate versus an internal apolar pathway.
Nerve tissue Hbs occur in both vertebrates and invertebrates (3). Among these, the nerve Hb from the nemertean worm C. lacteus (CerHb) is the smallest functional globin known, being composed of 109 amino acids instead of the ∼140–160 residues typical of most monomeric globins (4, 5). Analysis of the three-dimensional structure of CerHb has shown a markedly edited 3-over-3-globin fold with deletion of the N-terminal A-helix, extension of the GH region, and reduction of the C-terminal H-helix (6). Both sequence and fold comparisons indicate that CerHb is equally distant from all known globins, suggesting a specific mini-Hb family within the Hb superfamily. The affinity of CerHb for O2 is very similar to that of mammalian Mbs, and its function appears to be O2 storage and then release to axons and brain tissue during periods of burrowing under anoxic conditions (5). The evolution of a storage function in a highly altered globin structure has made CerHb an excellent model for testing biophysical mechanisms involved in the regulation of O2 affinity and reaction rates (2, 7–10).
Several distinct structural features make CerHb of interest for understanding structure-function relationships. First, the distal portion of the heme pocket contains Tyr-11(B10) and Gln-44(E7), which stabilize heme-bound O2 by hydrogen bonding in many invertebrate globins with high O2 affinity (11). However, unlike most of the globins containing this Tyr-Gln motif, CerHb also contains a polar Thr-48 residue at the E11 position, which in most globins is Val, Leu, Ile, or Phe. In CerHb, the non-bonded electrons of the Thr-48(E11) Oγ atom “pull” the proton on the Tyr-11(B10) hydroxyl group away from heme-bound O2, causing marked 1000- to 100-fold increases in the rate and equilibrium constants for O2 dissociation, respectively (6, 9). The net result is a P50 for O2 binding of ∼0.6 torr (Kd∼ 1 μm) for CerHb, which is similar to that of mammalian Mbs (10).
The second unusual structural feature of CerHb is a long apolar tunnel that connects the heme pocket to solvent through an exit pore between the C-terminal ends of the E and H helices. This tunnel is correlated with an unusually large association rate constant for O2 binding to CerHb (k′O2 = 230 μm−1s−1) when compared with those of other invertebrate globins containing the Tyr-Gln active site motif, which normally show bimolecular association rate constants on the order of 1–5 μm−1s−1 (11). The rate constant for O2 binding to CerHb is 10 times larger than that for sperm whale Mb and is similar to those for His(E7)Gly8 mutants of Mbs and Hbs and to unhindered pentacoordinate model hemes (12–14). The cause of these high rates appears to be a wide, ∼10 Å long channel that traverses the interior of the globin matrix, allowing relatively unhindered access to the heme pocket.
The CerHb tunnel has roughly the shape of an hourglass, is located between the E- and H-helices, is directed from the solvent to the heme pocket, and terminates at the active site as an aperture, which is circumscribed by the side chains of Val-7(B6), Phe-10(B9), and Thr-48(E11). This channel is lined by small hydrophobic residues and has a diameter that varies from 6.9 to 5.5 Å at the narrowest segment, which is close to residue Leu-86(G12) (6). Diatomic ligands are thought to enter and exit the tunnel through an opening between the E and H helix, which appears to have evolved by the complete loss of the N-terminal A-helix that is found in almost all other globins. Obstructing the tunnel entrance by increasing the size of Ala-55(E18) to Phe or Trp causes an ∼4-fold decrease of both k′O2 and kO2 and a 5-fold increase in the fraction of geminate recombination due to restriction of both entry into and escape from the apolar channel (2). In contrast, mutagenesis of Gln-44(E7) to either smaller or larger amino acids has little effect on internal rebinding after photolysis or bimolecular binding from solvent, suggesting strongly that the E7 gate for ligand entry/exit found in mammalian Hbs and Mbs (13, 14) is not a significant pathway in CerHb (2).
Apolar cavities and tunnel systems that could support ligand diffusion to and from the heme have been observed in selected globin families, although relative to CerHb, some of these channels are structurally and topologically distinct within the overall tertiary fold (15–17). However, in most cases there has been little experimental proof that these tunnels are used by diatomic ligands. Cavities large enough to accommodate xenon atoms have been found in most globins, including sperm whale Mb and Scapharca inaequivalvis HbI; however, in both of the latter cases, xenon binding has little effect on either the fraction of geminate recombination or on overall rates of ligand binding or release (13, 18, 19), implying that these internal spaces are not part of the route for ligand entry and escape.
In this work we have focused our attention on providing both direct experimental and theoretical evidence for ligand access to the heme pocket through the tunnel in CerHb and estimates of what fraction of the ligands use this route versus the E7 gate pathway, which dominates in mammalian Mbs and Hbs that contain a distal histidine. We have shown that xenon atoms can be accommodated within the CerHb tunnel by x-ray crystallography, examined the effects of xenon binding on CO geminate recombination, measured the effects of changing the width of the channel with Leu-86(G12) to Ala, Phe, and Trp mutations, and complemented these experiments with molecular dynamics simulations of how Leu-86 mutations modulate the tunnel shape, size, and energy barriers to ligand migration. Our results demonstrate that there is a strong correlation between tunnel size and observed rates of ligand binding to CerHb. More importantly, CerHb is the first example of a globin in which xenon binding markedly increases geminate recombination and slows bimolecular ligand entry.
EXPERIMENTAL PROCEDURES
Sample Preparation
Wild-type (wt) and mutant recombinant CerHbs were expressed and purified as described previously using a synthetic gene with codon usage optimized for expression in Escherichia coli (6, 10). Most recombinant CerHb samples were isolated in the reduced state and used directly. When necessary, the unstable mutants were pre-reduced with dithionite, quickly passed through a Sephadex G-25 column, and eluted with buffer equilibrated with 1 atm of CO. All reactions were measured in 0.1 m phosphate buffer, pH 7.0, 1.0 mm EDTA, 20 °C.
Measurement of Overall Rates of Ligand Association and Dissociation
CO association time courses were measured after complete laser photolysis of 50 μm CerHbCO samples containing various CO concentrations under pseudo first order conditions, and CO dissociation was measured by mixing CerHbCO with high concentrations of NO (2). Time courses for O2 association and dissociation were measured after complete laser photolysis of CerHbCO samples containing various mixtures of O2/CO (2), and association rate constants for NO binding to deoxy-CerHb were measured using a flow-flash multi-mixing apparatus and the 500-ns dye laser system (2).
Measurement of Geminate Recombination
Time courses for internal rebinding within CerHb were measured at 436 nm after excitation with a 9-ns excitation pulse from a Lumonics YAG-laser system using a Tektronix TDS3052 digitizing oscilloscope and Hamamatsu high speed PM tube with a 2-ns rise time. Experimental procedures and fitting routines were performed as previously described (2). For experiments with xenon, a 0.3-ml sample of CerHbCO was transferred to a stainless steel pressure cell with a gas volume of ∼4 ml. The cell was equipped with anti-reflection-coated sapphire windows and had a path length of 1 mm as previously described (19). Data were first collected with 1 atm CO over the sample, and then the required pressure of xenon was introduced through a three-way valve attached to the top of the cuvette. The sample was then equilibrated with the CO-xenon mixture for several minutes by shaking the cuvette to expose the solution to the gas space. The fraction of geminate recombination (Fgem) and the rate of geminate recombination (kgem) were determined as previously described (2).
Crystallization, Data Collection, and Refinement
The oxygenated derivative of the Leu-86(G12)Ala (L86A) CerHb mutant was crystallized by vapor diffusion techniques (protein concentration 27 mg/ml) under conditions matching those for the wt protein (20). Elongated prismatic crystals (about 0.05 × 0.05 × 0.2 mm3) grew within 1 week. The crystals were transferred to 2.8 m ammonium sulfate, 50 mm sodium acetate, pH 6.0, and 15% glycerol (v/v) (cryoprotectant solution) immediately before data collection at 100 K. L86A CerHbO2 crystals are isomorphous with those of the wt protein.
High resolution data (1.60 Å) on the L86A CerHb mutant crystals were collected at the European Synchrotron Radiation Facility (beam line ID14-1, Grenoble, France) (Table 1). To promote xenon diffusion within the protein matrix, selected L86A CerHbO2 crystals in their cryoprotectant solution were exposed to 10 bar xenon for 5 min in a high pressure chamber (Xcell, Oxford Cryo-system). The x-ray diffraction data sets for the xenon-bound oxygenated derivative of the L86A CerHb mutant were collected at 100 K using a MAR-Research 345 imaging plate detector coupled to a Rigaku RU-H3R rotating anode generator (copper Kα radiation; resolution 2.3 Å) (Table 1).
TABLE 1.
Data collection and refinement statistics for L86A CerHbO2 and wild-type aquomet CerHb
| L86A CerHbO2 (PDB 2xkg) | L86A CerHbO2 10 bar xenon (PDB 2xkh) | wt aquomet CerHb (PDB 2xki) | |
|---|---|---|---|
| Data collection parameters | |||
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions (Å) | a = 42.7 | a = 42.9 | a = 42.5 |
| b = 43.4 | b = 43.0 | b = 43.2 | |
| c = 59.4 | c = 59.5 | c = 60.2 | |
| Resolution (Å) | 34.9-1.60 | 35.0-2.3 | 35.1-1.37 |
| (1.69-1.60) | (2.34-2.30) | (1.37-1.30) | |
| Observations | 58,823 | 20,818 | 156,248 |
| Unique reflections | 15,045 | 5,031 | 27,887 |
| Completeness (%) | 99.7 (100)a | 97.5 (93.0) | 99.9 (100) |
| R-mergeb (%) | 4.0 (9.5) | 8.8 (40.0) | 5.1 (34.5) |
| I/σ(I) | 24.1 (12.7) | 8.6 (4.5) | 15.5 (3.3) |
| Multiplicity | 3.9 (4.0) | 3.5 (3.0) | 5.6 (3.9) |
| Refinement | |||
| R-factorc/R-free (%) | 15.4/18.6 | 19.4/26.6 | 14.4/17.7 |
| Protein atoms in the asymmetric unit | 826 | 817 | 831 |
| Dioxygen molecules | 1 | 1 | |
| Water molecules | 135 | 61 | 131 |
| Sulfate ions | 3 | 1 | 1 |
| Glycerol | 1 | ||
| Model quality | |||
| Overall B-factor (Å2) | 11 | 17 | 14 |
| r.m.s.d. from ideal values: | |||
| Bond lengths (Å) | 0.011 | 0.010 | 0.011 |
| Bond angles (°) | 1.172 | 1.073 | 1.244 |
| Ramachandran plotd: | |||
| Most favored regions (%) | 98.1 | 99.1 | 100 |
| Additional allowed regions (%) | 1.9 | 0.9 | 0 |
a Outer shell statistics are shown within parentheses.
b R-merge = ΣhΣi|Ihi − 〈Ih〉|/ΣhΣiIhi.
c R-factor = Σh‖Fobs| − |Fcalc‖/Σ|Fobs|, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively.
d Data were produced using the program PROCHECK (26).
High resolution data (1.3 Å) for the aquomet form of the wt CerHb protein crystals were collected at the European Synchrotron Radiation Facility (beam line ID14-1, Table 1). The aquomet form of CerHb was obtained by soaking the oxygenated wt protein crystals (20) with 10 mm K3Fe(CN)6. Crystals were then transferred to the cryoprotectant solution supplemented with the same amount of K3Fe(CN)6 before data collection at 100 K.
All diffraction data were processed using MOSFLM and SCALA programs (21, 22) and phased by molecular replacement methods with the program MOLREP (23), as implemented in the CCP4 program package (CCP4, 1994), using the wt CerHbO2 structure as the starting model (PDB accession code 1kr7) (6). Crystallographic refinement was performed using the program REFMAC (24), and the program COOT (25) was used for model building/inspection. Xenon atoms were initially refined at 100% occupancy together with the protein structure. At the end of refinement, inspection of difference Fourier maps indicated that the xenon occupancies were overestimated. To obtain better values, the fractional occupancies were systematically decreased until no residual difference signal was detected. The relevant data collection and refinement statistics are reported in Table 1. The programs Procheck (26) and Surfnet (27) were used to assess the stereochemical quality of the protein structures and to explore the protein matrix cavities. Atomic coordinates and structure factors for the L86A CerHbO2, xenon-bound L86A CerHbO2, and wt aquomet CerHb have been deposited with PDB accession codes 2xkg, 2xkh, and 2xki, respectively (28).
Computational Methods
MD simulations were performed starting from the crystal structure of wt CerHbO2 (PDB accession code 1kr7 (6)), and in some cases the out conformation of Gln-44(E7) observed in the structure of the wt aquomet CerHb derivative was used in the model. All systems were solvated with water molecules leaving 10 Å between the protein surface and the system limit. Histidine protonation was assigned to favor H-bond formation. All simulations were performed with the parmm99 force field (29) using Amber8 (30). The equilibration process was performed by slowly heating the system to a final temperature of 298 K. The oxygenated and deoxygenated heme model system charges were determined by using restrained electrostatic potential charges (31) and HF/6–31G(d) wave functions according to the Amber standard protocol. This set of partial charges has been successfully used in similar systems (32).
To study the properties of the wt CerHb tunnel cavity system, the diffusion free energy profiles for O2 along the tunnels were calculated by performing constant velocity MSMD simulations using the Jarzynski inequality (33), which relates equilibrium free energy values with the irreversible work performed over the system and proceeds along a reaction coordinate from reactants to products. In the present study the reaction coordinate was chosen as the iron to O2 distance. Calculations were performed using a force constant of 200 kcal mol−1 Å−1 and a pulling velocity of 0.05 Å ps−1. To reconstruct the free energy profile of ligand migration along the tunnel, a set of MSMD runs was performed starting from equilibrated MD structures with the ligand (i) in the distal pocket, (ii) in the crystallographic xenon-binding sites, and (iii) outside the tunnel. Ten MSMD simulations were performed in each direction (forward/exit and backward/entry). In cases in which two overlapping profiles were obtained (from entry and exit sets), we confirmed that they matched.
The free energy profiles for the transition between the in and out conformations of Gln-44(E7) were obtained with Umbrella Sampling techniques (34). In this method the potential function is modified so that the unfavorable states are sampled sufficiently. The modification of the potential function can be written by adding a bias harmonic potential according to E′(r) = E(r) + k(r − ξ)2, where E(r) is the potential energy of the protein for a given configuration r (i.e. the force field equation), and k is the force constant. The harmonic potential is centered at some point along the reaction coordinate (ξ). The corresponding profile was computed using the Gln-44(E7) Nϵ2-iron distance as the selected reaction coordinate, and 12 different 2-ns runs were performed varying the center of the harmonic potential along the reaction coordinate.
In-silico L86A, L86F, and L86W mutant structures were generated using the Modeler program (35). The generated structures were equilibrated by slowly heating the system to a final temperature of 298 K. MD simulations for the in silico-generated mutants were performed for 10 ns using the specifications described above. From these simulations we obtained plots representing the interaction energy between the O2 molecule and the protein determined from classical molecular interaction potential calculations.
RESULTS
Crystal Structures of L86A CerHbO2 and Its Xenon Derivative
To examine the accessibility of the apolar tunnel, wt CerHbO2 crystals were equilibrated with high pressures of pure xenon gas. This gas has been used successfully to identify cavities in a variety of Mbs and Hbs (15, 16, 18, 36–40). Unfortunately, wt CerHbO2 crystals quickly lose diffraction power after exposure to xenon, even at low pressure and short times. This effect could be due to xenon binding at intermolecular crystal contacts, causing disruption of the crystal lattice, or to xenon binding within the apolar channel, expanding the protein and indirectly disrupting intermolecular contacts.
To solve this problem we crystallized the L86A CerHbO2 mutant, in which constriction near the center of the tunnel is relieved by removal of three side chain carbon atoms at the G12 helical position. When crystals of L86A CerHbO2 were exposed to high pressures of xenon, diffraction persisted after incubation with ∼10 atm of xenon, and a full diffraction data set was successfully collected at 2.3 Å resolution. The resulting structure was refined to a final R-factor of 19.4% and R-free of 26.6% and compared with the independently determined structure of the xenon-free L86A CerHbO2 mutant. For comparison, a full dataset was collected to 1.6 Å resolution for xenon-free L86A CerHbO2, and a model was refined to final R-factor and R-free values of 15.4 and 18.6%, respectively (Table 1).
The backbone structure of xenon-free L86A CerHbO2 is virtually identical to that of wt CerHbO2, with a r.m.s.d. of 0.15 Å calculated for all 109 Cα atom pairs. The only significant structural differences between the mutant and wt structures are small rotations of the side chains of Leu-98(H9) and Ile-102(H13). The net result of these changes and the Ala replacement is an increase in the tunnel inner diameter by ∼1.9 Å. This increase either promotes preferential xenon binding in the channel versus binding to external sites or facilitates internal xenon binding without requiring expansion of the protein structure and further alterations in intermolecular packing.
The heme cavity of xenon-free L86A CerHbO2 is identical to that of wt CerHbO2 (Table 2). Conservation of the heme distal-site structure in wt CerHbO2 and the xenon-free L86A mutant is supported by their similar kinetic parameters for diatomic ligand binding (Tables 3–5), which ensures that this mutant is a reliable model for studying ligand diffusion through the apolar matrix tunnel. The structure of the L86A mutant containing bound xenon is also very similar to that of wt CerHbO2, with a r.m.s.d of 0.19 Å for all 109 Cα atom pairs. Inspection of the residual difference electron density after the initial refinement of the xenon-bound mutant indicated the presence of two xenon atoms (Xe1 and Xe2) at refined occupancies of ∼ 65% and temperature factors of 36 and 22 Å2, respectively (Fig. 1). The Xe1 site is directly adjacent to Ala-86(G12) and surrounded by the heme ring B vinyl carbon atoms, the terminal side chains of Phe-10(B9) and Ile-52(E15). The Xe2 site is adjacent to the Xe1 site, surrounded by the heme ring B methyl group, Tyr-51(E14), Leu-98(H9), Ala-101(H12), and Ile-102(H13), and is near Ala-55(E18) and the exit to solvent. Together, the xenon atoms fill most of the tunnel located in the protein interior (transparent gray spaces in Fig. 1), help define the apolar channel, and are stabilized by favorable van der Waals contacts with Val-7(B6), Phe-10(B9), Thr-48(E11), Tyr-51(E14), Ile-52(E15), Ala-55(E18), Ala-86(G12), Leu-98(H9), Ala-101(H12), and Ile-102(H13).
TABLE 2.
Distances between polar atoms in the distal pockets of L86A CerHbO2, wild-type CerHbO2, and wild-type aquomet CerHb
The in and out conformation of the Gln-44(E7) side chain are indicated by the superscript “i” and “o” letters.
| wt CerHbO2 (PDB 1kr7)a | Xenon-free L86A CerHbO2 (PDB 2xkg) | Xenon-bound L86A CerHbO2 (PDB 2xkh) | wt aquomet CerHb (PDB 2xki) | |
|---|---|---|---|---|
| Fe—O2 (Å) | 1.94 | 1.97 | 1.90 | |
| Fe—water (Fe3+) (Å) | 2.14 | |||
| Fe-O1-O2 angle (°) | 103 | 110 | −105 | |
| Tyr-11(B10) OH—O2 (Å) | 2.57 | 2.47 | 2.46 | |
| Tyr-11(B10) OH—water (Fe3+) (Å) | 2.87 | |||
| Gln-44(E7) NE2—O1 (Å) | 2.60 | 2.96 | ||
| Gln-44(E7) NE2—O2 (Å) | 2.65 | |||
| Gln-44(E7) NE2—water (Fe3+) (Å) | 2.72i, 7.13o | |||
| Tyr-11(B10) OH—Thr-48(E11) OG1 (Å) | 2.59 | 2.62 | 2.75 | 2.68 |
| Tyr-11(B10) OH—Gln-44(E7) NE2 (Å) | 3.24 | 3.33 | 3.41i, 7.97o | 3.59 |
| Gln-44(E7) NE2—Thr-48(E11) OG1 (Å) | 3.53 | 3.34 | 3.48i, 7.50o | 3.34 |
| Gln-44(E7) NE2—solvent water (Å) | 2.72o | 2.83o | ||
| Solvent water—Ala-40(E3) O (Å) | 2.59o | 2.73o |
a Structure was reported in Pesce et al. (6).
TABLE 3.
Rate constants for bimolecular and geminate recombination of CO with wild-type and L86A CerHb at pH 7, 20 °C
| CerHb | k′CO | kgem,CO | Fgem,CO | k′entry,CO |
|---|---|---|---|---|
| μm−1s−1 | μs−1 | μm−1s−1 | ||
| Leu-86(wt)a | 32 ± 5 | 80 ± 40 | 0.05 ± 0.03 | 640 ± 400 |
| Leu-86(wt) + xenon | 26 | 63 ± 10 | 0.29 ± 0.5 | 90 |
| Ala-86 | 30 | ≥150, 15 | 0.13 | 190 |
| Ala-86 + xenon | 27 | ≥150, 28 | 0.34 | 80 |
a The numbers for wt CerHb are based on the average of all determinations for the past 6 years (≥10 separate preparations, see Refs. 5, 8, and 9). The error in k′entry is very large because Fgem is close to 0.0, poorly defined, and has an error of ±60%. When Fgem is ≥0.1, the error in k′entry diminishes greatly to ∼±20%.
TABLE 4.
Rate and equilibrium constants for O2 and CO binding to position 86(G12) and 55(E18) mutants of CerHb at pH 7.0, 20 °C
| CerHb | k′O2 | kO2 | KO2 | k′CO | kCO | KCO | KCO/KO2 |
|---|---|---|---|---|---|---|---|
| μm−1s−1 | s−1 | μm−1 | μm−1s−1 | s−1 | μm−1 | ||
| Ala-55/Leu-86 (wt)a | 230 ± 17 | 190 ± 25 | 1.2 ± 0.2 | 32 ± 5 | 0.054 ± 0.005 | 590 ± 100 | 490 ± 110 |
| Ala-86 | 230 | 153 | 1.5 | 30 | 0.053 | 570 | 380 |
| Phe-86 | 120 | 71 | 1.7 | 24 | 0.024 | 710 | 600 |
| Trp-86 | 97 | 60 | 1.6 | 16 | 0.014 | 1100 | 700 |
| Trp-55 | 63 | 66 | 0.95 | 15 | 0.029 | 500 | 520 |
a The numbers for wt CerHb are based on the average of all determinations for the past 6 years (≥10 separate preparations).
TABLE 5.
Observed bimolecular rate constants for NO association and calculated rate parameters for ligand entry into 86(G12) and 55(E18) mutants of CerHb at pH 7.0, 20 °C
| CerHb | k′NO | kgem,CO | Fgem,CO | k′entry,CO |
|---|---|---|---|---|
| μm−1s−1 | μs−1 | μm−1s−1 | ||
| Ala-55/Leu-86(wt)a | 230 ± 30 | 80 ± 40 | 0.05 ± 0.03 | 640 ± 400 |
| Ala-86 | 200 | ≥150, 15 | 0.13 | 190 |
| Phe-86 | 110 | ≥150, 53 | 0.17 | 140 |
| Trp-86 | 67 | ∼80 | 0.52 | 31 |
| Trp-55 | 45 | 9.4 | 0.44 | 33 |
a The numbers for wt CerHb are based on the average of all determinations for the past 6 years (≥10 separate preparations, see Table 3).
FIGURE 1.

Effects of 10 atm of xenon on the structure of L86A CerHbO2 (PDB code 2xkh). A, side view looking through the heme propionates into the protein interior. B, back view looking between the E and H helices through the apolar tunnel. The empty space in the tunnel is highlighted in gray. The heme group, proximal His, and Gln-44(E7) are shown as sticks with O atoms in red, N atoms in blue, and C atoms in white. The xenon atoms are shown as green spheres at half their van der Waals radii to indicate partial occupancy and designated as Xe1 and Xe2. The Ala-86(G12) and Ala-55(E18) methyl side chains are shown as green sticks. C, electron density map of the amino acids and xenon atoms lining the tunnel using the same orientation as in panel B are shown. Amino acid side chains are shown as ball and stick models, and the xenon atoms are drawn as green spheres. The electron densities (2Fo − Fc map contoured at 1σ) are shown in magenta for the side chains and in blue for the xenon atoms.
Unexpectedly, xenon binding to the L86A mutant induces small but detectable changes in the position of the Gln-44(E7) side chain, which alter the hydrogen bonding interactions with bound O2 and the heme propionates (Fig. 2). The second bound O atom rotates outward toward the solvent interface and closer to the terminal O or N atom of Gln-44(E7) (Fig. 2A). The B factors for heme-bound O2 (BO(1) = 35 Å2, BO(2) = 38 Å2) are higher in the presence of xenon compared with wt CerHb (BO(1) = 23 Å2, BO(2) = 28 Å2). In contrast, the B factor averages for the two independent structures are essentially the same, 17 versus 15 Å2, respectively, for the xenon bound L86A versus wt CerHbO2. Thus, the difference in mobility and orientation of coordinated O2 appears to be real and coupled to the conformational heterogeneity of Gln-44(E7).
FIGURE 2.

Multiple conformations of Gln-44(E7) in various structures of CerHb. A, distal pocket, E-helix, multiple conformations of Gln-44(E7), and bound O2 of xenon-bound L86A CerHbO2 (2xkh, cyan). B, the electron density map (2Fo − Fc map contoured at 1σ) for xenon-bound L86A CerHbO2 is shown in magenta for the double conformation of Glu-44(E7), the heme group, and the oxygen molecule coordinated to the iron atom and for the Gln-44(E7) side chain refined in two (in and out) conformations. C, heme pocket, E-helix, in and out conformers of Gln-44(E7), and coordinated water of wt aquomet CerHb (2xki, green). D, electron density map (2Fo − Fc map contoured at 1σ) for wt aquomet CerHb are shown in magenta for the heme group, the heme-coordinated water molecule, and for the Gln-44(E7) side chain refined in two (in and out) conformations. In the upper panels A and C, the Gln-44(E7) side chain conformer from wt CerHbO2 (1kr7) is superimposed as white sticks. The position of bound O2 in wt CerHbO2 is also shown in panel A as white sticks. Residues Tyr-11(B10), Gln-44(E7), Lys-47(E10), and Thr-48(E11) are represented as sticks; heme-bound O2 and coordinated H2O are represented as red sticks; the heme group is displayed as sticks with the carbon atoms either light blue (panel A) or green (panel B), the iron atom in orange, and nitrogen and oxygen atoms in blue and red, respectively; external water molecules are displayed as red spheres; hydrogen bonds are drawn as dashed black lines.
The innermost Gln-44(E7) conformer (in) is close enough to stabilize the ligand by hydrogen bonding if the Nϵ atom is near the second bound O atom. The Tyr-11(B10) hydroxyl H atom is still hydrogen-bonded to the Thr-48(E11) hydroxyl O atom, and the non-bonded electrons of the Tyr-11(B10) hydroxyl O atom are still close enough to destabilize heme-bound O2. Thus, the O2 dissociation rate constant is still expected to be large in the xenon-bound mutant, as is observed for both xenon-free L86A and wt CerHbO2.
In the outermost conformation of Gln-44(E7) (out) in xenon-bound L86A CerHbO2, the amide side chain has moved toward the solvent and away from the iron atom by a small ∼56° rotation about the Cα-Cβ bond, has lost contact with the ligand, and instead is stabilized by a hydrogen bonding network, which links the Nϵ or Oϵ atom to the carbonyl O of Ala-40(E3) and the heme A propionate carboxyl groups through two well defined water molecules (Table 2; Fig. 2A).
Similar in and out conformations for Gln-44(E7) occur in the structure of wt aquomet CerHb (Fig. 2B). The terminal amide side chain atoms in the in conformation appear to be interacting with the coordinated water molecule, whereas in the out conformation the amide side chain has moved away from the coordinated ligand and again is fixed in position by a hydrogen bonding network involving the carbonyl of Ala-40(E3). In both the in and out conformers of aquomet CerHb, there is an electrostatic interaction between Lys-47(E10) and the heme-7-propionate. In the out conformer of both xenon-bound L86A CerHbO2 and wt aquomet CerHb, the Gln-44(E7) amide side chain still blocks access to the iron atom by filling the outer portion of the channel between the N-terminal region of the E-helix and the heme propionates. This position contrasts with the truly open position of His(E7) in sperm whale MbCO when it is protonated at low pH or when Phe-46(CD4) is mutated to Val (41, 42).
Effects of Xenon Binding on Geminate CO Recombination
As shown in Fig. 3 and Table 3, Fgem for both wt and L86A CerHbCO increases almost linearly with increasing xenon pressure, from ∼0.1 at 0 atm to ∼0.35 at 15 atm (Table 3). Equilibration with 15 atm of N2 has no effect on Fgem for wt CerHbCO, ruling out a non-specific pressure effect. As observed previously for wt MbO2 (13), there is no effect of 15 atm of xenon on the fraction of geminate CO rebinding in Mb, which remains close to 0 (expected value is 0.05, Fig. 3B). The linear dependence of Fgem on xenon pressure indicates weak binding of the gas to CerHbCO in solution, with a P50 > 15 atm. The fractional occupancy estimated from the crystal structure at 10 atm of xenon was ∼0.65, indicating a somewhat higher affinity under the crystallization conditions. Assuming a lower estimate of 15 atm for the P50 of both xenon sites, the extent of geminate recombination at full occupancy of sites 1 and 2 is predicted to be ∼0.7 compared with ≤0.1 when the channel is completely open. Thus, xenon binding in the channel reduces the extent of CO escape at least 7-fold, strongly supporting the view that the apolar channel is the major pathway for ligand escape. It should be noted that the results in Fig. 3 are the first to show a large effect of xenon binding on the total fraction of geminate recombination in a globin.
FIGURE 3.

Effect of xenon on CO geminate recombination in wild-type and L86A CerHbCO at pH 7.0, 20 °C. The smooth solid black lines represent fits to single or double exponential functions with an offset. The gas pressure above each solution is marked next to each time course. There was no effect of 15 atm of xenon on geminate rebinding in wt sperm whale Mb (WT MbCO, panel B) nor was there an effect of 15 atm of N2 on geminate recombination in wt CerHbCO (panel A).
Effects of L86A, L86F, and L86W Mutations
We have already examined the effects of increasing the size of the amino acid at the Ala-55(E18) position, which is adjacent to the Xe2 site, and found that Leu, Phe, and Trp substitutions trap CO in the tunnel after laser photolysis, increasing Fgem to ∼0.50, and decreasing the bimolecular rate constant for ligand entry roughly 4–10-fold (Fig. 4). To examine the Xe1 site, we measured ligand binding to L86A, L86F, and L86W mutants (Figs. 4 and 5; Tables 4 and 5).
FIGURE 4.

Geminate recombination of CO in various CerHbCO mutants at pH 7.0, 20 °C. Geminate recombination time courses for 86A (L86A), 86F (L86F), 86W (L86W), 55W (A55W), and wt CerHbCO in the presence of 15 atm of xenon are represented by black lines. The smooth gray lines represent fits to single or double exponential functions with an offset.
FIGURE 5.

NO binding to position 86(G12) mutants of CerHb at pH 7.0, 20 °C. Time courses for NO binding after photolysis of 86A (L86A), 86F (L86F), 86W (L86W), and wt CerHbCO complexes at low CO concentration and high NO concentration are represented by black lines. The smooth gray lines represent fits to a single exponential function with an offset. The inset shows the linear dependence of kobs (s−1) on NO concentrations with a y intercept of 0.
The L86F and L86W CerHbCO mutants show progressive increases in geminate recombination, with Fgem for the L86W mutant being roughly the same as that for the A55W variant. However, for the 86(G12) mutants, the rates of geminate recombination remain very large, ≥100 μs−1 compared with that for the A55W mutant, which is 10-fold smaller, ∼10 μs−1 (Fig. 4). The Trp-86(G12) side chain keeps the photodissociated ligand closer to the iron atom, facilitating its rebinding and lowering the entropic barrier to bond reformation. In the A55W mutant, the entire tunnel is available to dissociated ligands because only the exit to solvent is blocked, and as a result, rebinding to the heme iron is slower (2).
The rates of O2 and CO binding to the 86 mutants are compared with those for wt and A55W CerHb in Table 4, and a summary of NO association and calculated bimolecular rates of ligand entry are presented in Fig. 5 and Table 5. Both the association and dissociation rate constants for O2 binding decrease 2–3-fold when Leu-86 is replaced with Trp, demonstrating that the indole ring limits both diatomic gas escape and entry into the active site but has little effect on the overall affinity. A similar ∼3-fold decrease in k′NO is observed. Because of the free-radical nature of NO, its reactivity with reduced iron is intrinsically very high, and virtually every NO molecule that enters the active site binds, causing ligand movement into the distal portion of the heme pocket to be the rate-limiting step. Thus, k′NO should equal the bimolecular rate of ligand entry, k′entry.
The lower reactivities of O2 and CO cause them to enter and leave the active site many times before bond formation occurs, and for a two-step scheme the overall association rate constant is given by Equation 1 (2),
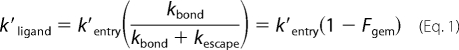 |
where kbond and kescape are the first order rates for internal iron-ligand bond formation and ligand escape, respectively. Thus, k′entry can be estimated from the overall association rate constant and the corresponding fraction of geminate recombination. Unfortunately, geminate rebinding of O2 to CerHb is very fast, and a significant amount occurs during the excitation pulse, making an accurate estimate of Fgem,O2 difficult (2). Estimates of k′entry based on bimolecular association rate constants and Fgem values for CO binding are given in Tables 3 and 5.
Equilibrating L86A CerHb with 15 atm of xenon causes an ≥2-fold decrease in the value of k′entry. Assuming that the two xenon sites are ≤50% occupied, this result suggests that filling the Xe1 and Xe2 pockets would decrease k′entry at > 4-fold. Similarly, the L86W mutation decreases the calculated k′entry value ∼6-fold compared with the computed value for L86A CerHb. The magnitude of this decrease is similar to that observed for the A55W mutation (Table 5).
Dynamics of O2 Entry/Exit for wt CerHb and the Leu-86(G12) Mutants
To analyze the resistance to ligand migration through the xenon sites, we computed the free energy profile for O2 movement through the apolar tunnel in wt CerHbO2 (PDB accession code 1kr7 (6)). The free energy profile and positions of the O2 molecule as it was moved along the tunnel are presented in Fig. 6. After entry into the channel, a small barrier was observed between 8 and 11 Å from the heme-iron atom, with two well-separated local minima (Fig. 6A). This barrier is the result of steric restriction of the tunnel diameter by the Leu-86(G12) side chain in wt CerHb. The O2 positions in Fig. 6B show that the ligand can pass consecutively through both xenon sites, which tentatively correlate with the observed free energy minima. Much larger free energy barriers were calculated on the two ends of the broad free energy well of the tunnel. A barrier of ∼5.5 kcal/mol was computed for O2 movement from the distal pocket into the Xe1 binding site, and then a ∼2 kcal/mol barrier was estimated for ligand movement out of the Xe2 site, past Ala-55(E18), and into solvent. The 5.5 kcal/mol barrier is due to steric restrictions of the side chains of Phe-10(B9), Tyr-11(B10), and Val-7(B6) (Fig. 6B).
FIGURE 6.

O2 migration in the wt CerHb channel. A, free energy profile for O2 migration. B, path followed by the O2 ligand during a selected MSMD simulation. The reaction coordinate, d, in panel A is the iron to O2 distance in Ångstroms (Ang).
To complement the kinetic results in Tables 4 and 5, we constructed the L86A, L86F, and L86W mutants in silico, performed 10-ns MD simulations, analyzed the tunnel structure for each of them, and displayed the results in Fig. 7. The L86A mutant displays a continuous tunnel with two secondary sites corresponding to the Xe1 and Xe2 positions. In contrast, the L86F and L86W mutants display a marked increase of the barrier between the Xe1 and Xe2 free energy wells shown in Fig. 6A and create spatial barriers that divide the tunnel into separate cavities as shown in Figs. 7, B and C. Blockage of the tunnel by these aromatic side chains is consistent with the 2–5-fold decreases in the rates of ligand binding and release and the increase in Fgem,CO observed for these mutants (Fig. 5 and Tables 4 and 5). However, the kinetic results suggest that blockage is not complete, and visual inspection of the MD simulation in time shows large movements of the Phe-86 and, to a lesser extent, the Trp-86 side chains. These fluctuations probably allow ligands to move through the restricted tunnel, even on the nanosecond time scale used in the simulations.
FIGURE 7.

Iso-contour energy plots associated with the ligand migration pathway in L86A, L86F, and L86W mutants of CerHb (panels A, B, and C, respectively). The iso-contour energy plots were obtained for the average structure of 10-ns MD simulations.
Molecular Dynamics of the Alternate Conformations of Gln-44(E7)
The observation of multiple Gln-44(E7) conformers in the xenon-bound L86A CerHbO2 and wt aquomet CerHb structures suggests that the out states may partially open the E7 gate as is observed for Phe-46(CD4)Val or low pH forms of sperm whale MbCO (41, 42). MD simulations of the free energy barriers for O2 movement were performed using the wt aquomet CerHb in and out conformers. The in Gln-44(E7) conformation matches that observed in wt CerHbO2 (6). In simulations of the out conformer, the amide side chain of Gln-44(E7) is stabilized by a strong interaction between its Oϵ atom and the Nζ atom of Lys-47(E10), which occurs by an ∼90° rotation of the Gln-44(E7) side chain about the Cδ-Cγ bond relative to its position for the in conformer in the wt aquomet CerHb crystal structure (Fig. 8A). However, the amide side chain still blocks the outer portion of the E7 channel.
FIGURE 8.

MD simulations of the in to out Gln-44(E7) transition in wt CerHbO2. A, hydrogen-bonding interactions involving the simulated Gln-44(E7) out conformer and its neighboring residues are shown. B, free energy profile for the in to out Gln-44(E7) side-chain conformational transition. Results for the Fe2+ and Fe3+ cases are depicted using black and red lines, respectively. The reaction coordinate, d, in panel B is the iron to NE2 Gln(E7) distance in Ångstroms (Ang).
To estimate populations and interconversion of the in and the out Gln-44(E7) conformations in wt CerHb, we performed free energy calculations for the transition in both redox states using Umbrella sampling schemes and the distance from the heme-iron atom to the Nϵ atom of Gln-44(E7) as the reaction coordinate. The in form is most stable in the ferrous CerHbO2 state (black line, Fig. 8B), as is observed in the x-ray crystal structure (6). In the ferric state, the in and out conformations have similar free energy wells, explaining why both conformations are observed in the crystal structure of aquomet CerHb.
When free energy profile calculations for O2 migration from the heme-iron to the solvent were performed using the out Gln-44(E7) conformation, further movement of the Gln-44(E7) side chain away from the heme-iron and out of the E7 channel occurred (Fig. 9B). The Nϵ and Oϵ atoms of the amide side chain are in positions similar to those seen for Nϵ and Cϵ atoms of His-64(E7) in the open conformation of sperm whale MbCO at low pH or in the F46V MbCO mutant (41, 42). When Gln-44(E7) is in this truly open conformation in CerHbCO, the calculations suggest only a small, ∼1 kcal/mol, free energy barrier to ligand entry and exit through the open E7 channel (Fig. 9). These results show that the overall barrier to ligand movement through the E7 gate is governed primarily by the free energy difference between the in and completely open conformation. In the case of wt CerHbO2 or CerHbCO, the in conformation of Gln-44(E7) is favored by at least a factor of 10, which probably explains why no out conformer is observed in the crystal structure (i.e. ≤10%). An additional barrier occurs for the rotation of the Gln-44(E7) side chain from the out conformer seen in the xenon-bound L86A CerHbO2 and wt aquomet CerHb structures to the open conformer required in the simulations for O2 exit through the E7 gate.
FIGURE 9.

O2 migration through the Gln-44(E7) gate in wt CerHb. A, free energy profile for O2 migration when Gln-44(E7) occupies a hypothetical completely open conformation, in which the amide side chain has rotated even further away from the heme-iron than in the out conformation shown in Fig. 8. B, path followed by the O2 ligand during a selected MSMD simulation for the hypothetical open conformer. The reaction coordinate, d, in panel A is the iron to O2 distance in Ångstroms (Ang).
DISCUSSION
The Size and Accessibility of the Apolar Channel
The crystal structure of L86A CerHbO2 equilibrated with ∼10 atm of xenon demonstrates that the central cavity between the E and H helices is readily accessible to apolar gases (Fig. 1). The exact positions of Xe1 and Xe2 are near the free energy wells calculated for wt CerHbO2 using dioxygen as a probe (Fig. 6). Xenon binding to both wt and L86A CerHbCO causes a significant increase of Fgem (Fig. 3), experimentally demonstrating that occupancy of the apolar cavity blocks ligand escape. Even though xenon binding to animal Hbs and Mbs is well documented, the results for CerHb are the first to show a large effect on the fraction of geminate recombination and that the xenon cavities are on the pathway for ligand entry and escape. In contrast, xenon binding to sperm whale Mb and S. inaequivalvis Hb causes little or no change in Fgem for either O2 or CO rebinding (13, 18, 19).
The extent of geminate recombination also increases significantly, to ∼50%, when the size of the Leu-86(G12) side chain is increased (Fig. 4). The L86F and L86W mutations cause marked decreases in both k′O2 and kO2 with little change in affinity as would be expected if the pathways for ligand entry and exit were blocked (Table 4). The magnitude of the L86W effect is similar to that observed when exit from the tunnel into the solvent is blocked by the A55W mutation (Fig. 4, Table 4, and Ref. 2). The tunnel mutations also cause large decreases (4–5-fold) of the rate constant for bimolecular NO binding, which is limited only by movement into the protein (Table 5). Similar decreases are obtained for the bimolecular rate constant for ligand entry calculated using k′CO, Fgem, and Equation 1 (Table 5).
Pathway for Ligand Binding
As described above, filling the apolar cavity with either xenon atoms or large aromatic amino acids at position 86(G12) markedly inhibits ligand entry and exit and increases both the rate and extent of geminate recombination. The simplest interpretation of these results is shown in Scheme 1 (2, 7). The apolar tunnel containing the xenon binding sites is assumed to be the major pathway for ligand entry and escape. Laser excitation photodissociates the iron-ligand bond, generating an initial intermediate with ligand remaining in the distal pocket (state B). From there the ligand either rebinds rapidly or moves into the apolar tunnel. Once inside the tunnel, the ligand can occupy multiple locations in the large space between the distal pocket and the solvent aperture. This large space slows return to the heme-iron and geminate recombination, allowing most of the ligands to escape into solvent.
SCHEME 1.

When xenon atoms or the indole ring of Trp-86(G12) block the initial portion of the tunnel, ligands are trapped in or very near to the distal pocket and rapidly rebind to the heme iron. As a result, large values of kgem and Fgem are observed (Fig. 4, Tables 3 and 5). The ligand molecules that move past the bound xenon atoms or the Trp-86(G12) side chain rapidly escape because return to the distal heme pocket from the exterior portion of the tunnel is restricted. When the tunnel exit is blocked by the A55W mutation, Fgem also increases. However, in this case, the rate of internal rebinding decreases markedly because the trapped ligand molecules can access the entire apolar tunnel, making return to the heme iron and net escape much slower (Fig. 4 and Table 5). Scheme 1 is also supported strongly by the calculations shown in Fig. 6. Entry into the tunnel has a free energy barrier of ∼5.5 kcal/mol due to steric restrictions by Phe-10(B9), Tyr-11(B10), Thr-48(E11), and Val-7(B6) (Scheme 1). The steric barrier between the two xenon sites (near Leu-86) is small (≤1 kcal/mol), and the exit to solvent is near Ala-55, with a barrier of ∼2 kcal/mol.
This mechanism also explains the results for the L86A mutation. Even though the steric barrier is small in wt CerHbCO, the Leu-86(G12) side chain does separate the two xenon binding sites (Fig. 6). As a result, ligands are partially sequestered close to the distal pocket, causing the small amount of observed geminate rebinding to be fast in wt CerHbCO. At the same time, the Leu-86(G12) restriction decreases Fgem by inhibiting ligand return to the distal pocket from the more exterior portions of the tunnel. When Ala replaces Leu at this position, geminate recombination is biphasic. The observed time course shows a small rapid phase with Fgem = 0.05 and kgem ≥ 100 μs−1, values that are similar to those for wt CerHbCO, and a second, slow phase with Fgem ≈ 0.05 and kgem ≈ 15 μs−1. The latter slower phase represents ligand return from the exterior portion of the tunnel, presumably the Xe2 site, at a rate similar to that seen for geminate recombination in the A55W mutant, whereas the fast phase represents rapid ligand rebinding from the Xe1 site as is observed in wt CerHb.
Conformational Flexibility of Gln-44 and the Role of the E7 Gate
In the crystal structure of wt CerHbO2, the amide side chain of Gln-44(E7) appears to be fixed in position by multiple hydrogen bonding interactions with the bound ligand and the hydroxyl groups of Tyr-11(B10) and Thr-48(E11). However, when the structure of A55W CerHbO2 was examined in detail (2), two conformations for the Gln-44(E7) side chain were observed that differed primarily by rotation about the Cα-Cβ bond. In the in conformation the amide Nϵ atom donates a hydrogen bond to the bound ligand, whereas in the out orientation the amide side chain interacts with the carbonyl of Ala-40(E3) and the heme propionates through well defined crystallographic water molecules. Two similar Gln-44(E7) conformations occur in the xenon-bound L86A CerHbO2 and wt aquomet CerHb structures (Fig. 2). These structures suggest that the Gln-44(E7) side chain is not rigidly held in place but may fluctuate between positions in which the amide side chain interacts electrostatically with either internal polar side chains and bound ligand or with Lys-47(E10), the carbonyl O atom of Ala-40(E3), the heme propionates, and water molecules at the solvent interface.
Simulation of the interconversion between the in and out conformers in the wt CerHbO2 and aquomet CerHb structures suggests a free energy barrier of 2.5 to 3.5 kcal/mol (Fig. 8). The out conformer appears to be ≥2 kcal/mol less stable than the in conformation in CerHbO2. MD simulations of the out conformer of wt CerHbO2 leads to a modified orientation in which the amide Oϵ atom of Gln-44(E7) directly accepts a proton from the side chain of Lys-47(E10), which in turn interacts electrostatically with the heme A propionate. However, even in this out conformation, the Gln-44(E7) side chain still blocks the access to the heme-iron through the E7 gate.
A simulation of ligand migration through the E7 gate when Gln-44(E7) is initially in the out conformer is shown in Fig. 9A. To allow ligand escape from the E7 channel, the Gln-44(E7) side chain must rotate further by ∼180° about the Cβ-Cγ bond, which moves the amide atoms away from the heme plane and up against Phe-25(CD1) and Phe-27(CD3), pushing the benzyl rings away from the heme group. When the Gln-44(E7) residue is in this completely open conformation, the free energy barrier to ligand movement through the E7 gate is very small compared with the free energy difference and barrier between the in and out conformers in the reduced state (Figs. 9A versus 8B). The latter differences plus the free energy required to rotate the Gln-44(E7) side chain into the completely open conformation determine the kinetic barrier to ligand escape through the E7 gate, which must be ≥4 kcal/mol and is probably much higher than the ∼6 kcal/mol barrier estimated for ligand movement into the apolar channel from the distal pocket shown in Fig. 6A.
When Gln-44(E7) is replaced with small amino acid side chains, there are no changes in the rates of O2, CO, and NO binding or in the fractions of either O2 or CO geminate recombination (2). The Q44F and Q44W mutations do cause 10-fold decreases in the rates of ligand association, but the rates of dissociation stay the same or increase 2-fold. As a result, these mutations cause marked 10-fold decreases in ligand affinity. The decreases in the association rate constants appear to be due to direct steric hindrance of internal iron-ligand bond formation by the large aromatic side chains and not to a change in the rate of ligand entry and escape. This conclusion is supported by the lack of an increase in the fraction of CO geminate recombination when the size of the amino acid at position 44(E7) is increased progressively from Ala to Trp, and Fgem remains at 0.05–0.1 for all the Gln-44(E7) mutants (Fig. 3 in Ref. 2).
In contrast, the L86W and A55W mutations and xenon binding within the apolar channel cause large increases in Fgem,CO after laser photolysis (Figs. 3, 4, and 5). These effects are unambiguous, indicate that the apolar tunnel is the major pathway for ligand movement into the protein, and demonstrate that the side chains at positions 86(G12) and 55(E18) line the major pathway for ligand entry and exit.
However, the Trp tunnel mutations do not completely inhibit ligand binding. The observed bimolecular rate constants for ligand entry into the L86W and A55W mutants (k′NO and k′entry values, Table 4) are 30–60 μm−1s−1, which although ∼4-fold less than the wt CerHb values, are similar to the values for both wt and His(E7)Gln mutants of sperm whale Mb (13) and the α and β subunits of human HbA (14). Thus, ∼25% of the ligand molecules could be entering the active site of CerHb through the E7 gate as shown in Fig. 9B, and the motions of the Gln-44(E7) side chain shown in Figs. 2, 8, and 9 are very similar to those seen for opening of the His(E7) gate in mammalian Mbs and Hbs (41, 42).
The 4–5-fold higher rates of ligand entry observed for native CerHb are due to the evolution of an unhindered apolar tunnel, which results from the loss of the A-helix and replacement of large amino acid side chains with small ones between the E- and H-helices. The lower net barrier for entry into the active site through this tunnel is due to elimination of the need for large conformational changes to open the E7 gate and to favorable partitioning of apolar diatomic gases into the large pre-existing tunnel, which effectively pre-concentrates the ligand for movement into the heme pocket. Similar unhindered, continuous apolar channels are not found in mammalian Mbs and Hbs, which accounts for why the E7 channel appears to be the dominant pathway even though the barrier to complete opening of the distal histidine or glutamine gate is significant.
This work was supported, in whole or in part, by National Institutes of Health Grants GM035649 and HL047020 (to J. S. O.). This work was also supported by grants from the Italian Ministry of University and Scientific Research (FIRB Project “Biologia Strutturale,” RBLA03B3KC) and from the University of Milano (to M. B.), Universidad de Buenos Aires Grants 08-X625 (to M. A. M.) and X076 (to D. A. E.), Agencia de Promocion Cientifica y Tecnologica Grants 07-1650 (to M. A. M.) and 06-25667 (to D. A. E.), Consejo Nacional de Investigaciones Científicas y Técnicas Grant PIP 5218 and a Guggenheim Foundation fellowship (to D. A. E.), and Robert A. Welch Foundation Grant C-0612 (to J. S. O.).
The atomic coordinates and structure factors (codes 2xkg, 2xkh, and 2xki) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Mutations are defined by the single letter code for the native amino acid, its sequence position, and the single letter code for the mutant amino acid, e.g. Leu-86(G12)→Ala is represented as L86A.
- MD
- molecular dynamics
- Hb
- hemoglobin
- Mb
- myoglobin
- CerHb
- C. lacteus hemoglobin
- CerHbO2
- oxygenated CerHb
- r.m.s.d
- root mean square deviation
- MSMD
- multiple steered molecular dynamics
- wt
- wild-type.
REFERENCES
- 1. Elber R. (2010) Curr. Opin. Struct. Biol. 20, 162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Salter M. D., Nienhaus K., Nienhaus G. U., Dewilde S., Moens L., Pesce A., Nardini M., Bolognesi M., Olson J. S. (2008) J. Biol. Chem. 283, 35689–35702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Geuens E., Dewilde S., Hoogewijs D., Pesce A., Nienhaus K., Nienhaus G. U., Olson J., Vanfleteren J., Bolognesi M., Moens L. (2004) IUBMB Life 56, 653–656 [DOI] [PubMed] [Google Scholar]
- 4. Bolognesi M., Bordo D., Rizzi M., Tarricone C., Ascenzi P. (1997) Prog. Biophys. Mol. Biol. 68, 29–68 [DOI] [PubMed] [Google Scholar]
- 5. Vandergon T. L., Riggs C. K., Gorr T. A., Colacino J. M., Riggs A. F. (1998) J. Biol. Chem. 273, 16998–17011 [DOI] [PubMed] [Google Scholar]
- 6. Pesce A., Nardini M., Dewilde S., Geuens E., Yamauchi K., Ascenzi P., Riggs A. F., Moens L., Bolognesi M. (2002) Structure 10, 725–735 [DOI] [PubMed] [Google Scholar]
- 7. Deng P., Nienhaus K., Palladino P., Olson J. S., Blouin G., Moens L., Dewilde S., Geuens E., Nienhaus G. U. (2007) Gene 398, 208–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hundahl C., Fago A., Dewilde S., Moens L., Hankeln T., Burmester T., Weber R. E. (2006) FEBS J. 273, 1323–1329 [DOI] [PubMed] [Google Scholar]
- 9. Martí M. A., Bikiel D. E., Crespo A., Nardini M., Bolognesi M., Estrin D. A. (2006) Proteins 62, 641–648 [DOI] [PubMed] [Google Scholar]
- 10. Pesce A., Nardini M., Ascenzi P., Geuens E., Dewilde S., Moens L., Bolognesi M., Riggs A. F., Hale A., Deng P., Nienhaus G. U., Olson J. S., Nienhaus K. (2004) J. Biol. Chem. 279, 33662–33672 [DOI] [PubMed] [Google Scholar]
- 11. Draghi F., Miele A. E., Travaglini-Allocatelli C., Vallone B., Brunori M., Gibson Q. H., Olson J. S. (2002) J. Biol. Chem. 277, 7509–7519 [DOI] [PubMed] [Google Scholar]
- 12. Mims M. P., Porras A. G., Olson J. S., Noble R. W., Peterson J. A. (1983) J. Biol. Chem. 258, 14219–14232 [PubMed] [Google Scholar]
- 13. Scott E. E., Gibson Q. H., Olson J. S. (2001) J. Biol. Chem. 276, 5177–5188 [DOI] [PubMed] [Google Scholar]
- 14. Birukou I., Schweers R. L., Olson J. S. (2010) J. Biol. Chem. 285, 8840–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Sanctis D., Dewilde S., Pesce A., Moens L., Ascenzi P., Hankeln T., Burmester T., Bolognesi M. (2004) Biochem. Biophys. Res. Commun. 316, 1217–1221 [DOI] [PubMed] [Google Scholar]
- 16. Milani M., Pesce A., Ouellet Y., Dewilde S., Friedman J., Ascenzi P., Guertin M., Bolognesi M. (2004) J. Biol. Chem. 279, 21520–21525 [DOI] [PubMed] [Google Scholar]
- 17. Raushel F. M., Thoden J. B., Holden H. M. (2003) Acc. Chem. Res. 36, 539–548 [DOI] [PubMed] [Google Scholar]
- 18. Knapp J. E., Pahl R., Cohen J., Nichols J. C., Schulten K., Gibson Q. H., Srajer V., Royer W. E., Jr. (2009) Structure 17, 1494–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scott E. E., Gibson Q. H. (1997) Biochemistry 36, 11909–11917 [DOI] [PubMed] [Google Scholar]
- 20. Pesce A., Nardini M., Dewilde S., Ascenzi P., Riggs A. F., Yamauchi K., Geuens E., Moens L., Bolognesi M. (2001) Acta Crystallogr. D Biol. Crystallogr. 57, 1897–1899 [DOI] [PubMed] [Google Scholar]
- 21. Evans P. R. (1993) Proceedings of the CCP4 Study Weekend on Data Collection and Processing, pp. 114–122, CLRC Daresbury Laboratory, Warrington, UK [Google Scholar]
- 22. Leslie A. G. M. (2003) MOSFLM User Guide, Mosflm Version 6.2.3. (Laboratory of Molecular Biology) Cambridge, UK [Google Scholar]
- 23. Vagin A. A., Teplyakov A. (1997) J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 24. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 25. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 27. Laskowski R. A. (1995) J. Mol. Graph. 13, 323–330 [DOI] [PubMed] [Google Scholar]
- 28. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J., Cieplak P., Kollman P. A. (2000) J. Comput. Chem. 21, 1049–1074 [Google Scholar]
- 30. Pearlman D. A., Case D. A., Caldwell J. W., Ross W. R., Cheatham T. E., III, DeBolt S., Ferguson D., Seibel G., Kollman P. A. (1995) Comp. Phys. Commun. 91, 1–41 [Google Scholar]
- 31. Bayly C. I., Cieplak P., Cornell W., Kollman P. A. (1993) J. Phys. Chem. 97, 10269–10280 [Google Scholar]
- 32. Marti M. A., Crespo A., Capece L., Boechi L., Bikiel D. E., Scherlis D. A., Estrin D. A. (2006) J. Inorg. Biochem. 100, 761–770 [DOI] [PubMed] [Google Scholar]
- 33. Jarzynski C. (1997) Phys. Rev. Lett. 78, 2690–2693 [Google Scholar]
- 34. Leach A. R. (2001) Molecular Modelling Principles and Applications, 2nd Ed., pp. 581–582, Pearson Education Limited, Edinburgh Gate, England [Google Scholar]
- 35. Martí-Renom M. A., Stuart A. C., Fiser A., Sánchez R., Melo F., Sali A. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 [DOI] [PubMed] [Google Scholar]
- 36. Moschetti T., Mueller U., Schulze J., Brunori M., Vallone B. (2009) Biophys. J. 97, 1700–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schoenborn B. P. (1965) Nature 208, 760–762 [DOI] [PubMed] [Google Scholar]
- 38. Schoenborn B. P., Watson H. C., Kendrew J. C. (1965) Nature 207, 28–30 [DOI] [PubMed] [Google Scholar]
- 39. Tilton R. F., Jr., Kuntz I. D., Jr., Petsko G. A. (1984) Biochemistry 23, 2849–2857 [DOI] [PubMed] [Google Scholar]
- 40. Savino C., Miele A. E., Draghi F., Johnson K. A., Sciara G., Brunori M., Vallone B. (2009) Biopolymers 91, 1097–1107 [DOI] [PubMed] [Google Scholar]
- 41. Lai H. H., Li T., Lyons D. S., Phillips G. N., Jr., Olson J. S., Gibson Q. H. (1995) Proteins 22, 322–339 [DOI] [PubMed] [Google Scholar]
- 42. Yang F., Phillips G. N., Jr. (1996) J. Mol. Biol. 256, 762–774 [DOI] [PubMed] [Google Scholar]