

Abstract
Although it has been suggested that the C-terminal tail of the β1a subunit of the skeletal dihyropyridine receptor (DHPR) may contribute to voltage-activated Ca2+ release in skeletal muscle by interacting with the skeletal ryanodine receptor (RyR1), a direct functional interaction between the two proteins has not been demonstrated previously. Such an interaction is reported here. A peptide with the sequence of the C-terminal 35 residues of β1a bound to RyR1 in affinity chromatography. The full-length β1a subunit and the C-terminal peptide increased [3H]ryanodine binding and RyR1 channel activity with an AC50 of 450–600 pM under optimal conditions. The effect of the peptide was dependent on cytoplasmic Ca2+, ATP, and Mg2+ concentrations. There was no effect of the peptide when channel activity was very low as a result of Mg2+ inhibition or addition of 100 nM Ca2+ (without ATP). Maximum increases were seen with 1–10 μM Ca2+, in the absence of Mg2+ inhibition. A control peptide with the C-terminal 35 residues in a scrambled sequence did not bind to RyR1 or alter [3H]ryanodine binding or channel activity. This high-affinity in vitro functional interaction between the C-terminal 35 residues of the DHPR β1a subunit and RyR1 may support an in vivo function of β1a during voltage-activated Ca2+ release.
Introduction
Contraction in skeletal muscle fibers depends on the rapid release of Ca2+ ions from the internal sarcoplasmic reticulum (SR) Ca2+ store following a surface membrane action potential. This excitation-contraction (EC) coupling process is likely to depend on a physical interaction between the dihydropyridine receptor (DHPR) voltage sensor in the transverse (t)-tubule membrane and the ryanodine receptor (RyR1) Ca2+ release channel in the closely opposed SR membrane (1,2). Of the five subunits of the DHPR, the skeletal isoforms of the membrane-spanning α1S subunit and the cytoplasmic β1a subunit are essential for the skeletal muscle EC coupling mechanism (2–5), which is defined as EC coupling that does not depend on external Ca2+ entry. In the α1S subunit, a central segment of the intracellular II-III loop (amino acid residues 724–760) between the second and third transmembrane repeats forms a minimal essential region for transmitting the EC coupling signal from the voltage sensor in the α1S subunit to the RyR1 Ca2+ release complex in the SR membrane. Deletion/mutation of this region abolishes skeletal-type EC coupling.
The β1a subunit plays two roles in skeletal EC coupling. It has an essential role in targeting the DHPR to a precise position in the t-tubule membrane that will allow it to interact physically with the RyR (4). This targeting depends on the well-characterized binding of the guanylate kinase (GK) domain of β1a to the I-II loop of the α1S subunit. In addition to this essential role, several findings suggest that the β1a subunit may play a direct role in the physical EC coupling process. Beurg et al. (6) reported that deletion of the 35-residue C-terminal tail of β1a led to a significant reduction in depolarization-induced Ca2+ release from the SR through RyR1 in β1a-null mouse myotubes transfected with mutant β1a c-DNA. This C-terminal modification did not appear to affect targeting of the DHPR to RyR1 (6). Furthermore, Cheng et al. (7) demonstrated a direct interaction between the β1a subunit and RyR1 by affinity chromatography using RyR1 fragments, and by interrupting β1a binding by deleting or substituting the basic residues K3494–R3502 in RyR1. Finally, in a study by Schredelseker et al. (8), expression of the β1a subunit in β1a-null zebrafish restored targeting of the DHPR to the RyR and skeletal EC coupling, whereas expression of the cardiac/neuronal β2a or housefly βM subunits restored targeting of the DHPR to RyRs but not skeletal EC coupling. This result could be explained by a direct contribution of β1a to EC coupling. However, the results could equally well be explained by an allosteric influence of β1a on the precise geometry of the DHPR and RyR in the surface/SR junction that reduced the efficacy of EC coupling (8).
These studies indicate that there may be a direct interaction between β1a and RyR1 in vivo; however, the functional consequence of the interaction between the β1a subunit and the RyR1 protein has not previously been reported. In addition, although the C-terminal tail of β1a has been implicated in voltage-activated Ca2+ release, binding of the C-terminal tail to RyR1 has not previously been reported. Therefore, we examined interactions between the isolated RyR1 channel and the β1a subunit and its C-terminal tail. We explored the ability of the 35-residue C-tail of β1a to bind to RyR1 using affinity chromatography, and the ability of the full-length β1a subunit and its C-terminal tail peptide to interact with the gating mechanism of the RyR1 channel in lipid bilayer and [3H]ryanodine-binding experiments.
Materials and Methods
Expression and purification of the DHPR β1a subunit protein
The His-ubiquitin-β1a vector was generated by inserting the β1a cDNA (GenBank accession No. NM_031173) in-frame downstream of a poly-histidine-tagged ubiquitin sequence in the plasmid pHUE (9). The construct was checked by sequencing to exclude amplification error. The plasmid was transformed into Escherichia coli BL21 and the cells were grown at 37°C. Fusion protein expression was induced by addition of 0.1 mM isopropyl β-D-thiogalactoside to the culture medium. The cells were spun, resuspended in phosphate buffer A (300 mM NaCl, 8 M Urea, 5 mM imidazole, 12 mM BME, 10% glycerol, pH 8), and briefly sonicated. The suspension was centrifuged at 11,000 × g for 30 min. The supernatant was incubated with Ni-nitrilotriacetic acid agarose (Qiagen, Valencia, CA) for 1 h with gentle agitation. Ni-nitrilotriacetic acid agarose beads were packed into a Polyprep column (Bio-Rad), washed with buffer A plus 10 mM imidazole, and eluted with buffer A plus 250 mM imidazole. Urea was adjusted to 2.6 M by addition of buffer A without urea. Ubiquitin was cleaved by digestion with a His-tagged ubiquitin protease (9). Imidazole and ubiquitin protease were removed by dialysis against buffer A, and ubiquitin was removed by Ni-agarose chromatography. The β1a protein was purified by denaturing preparative electrophoresis in a Bio-Rad model 491 prep cell, and eluted with 25 mM Tris/HCl and 192 mM glycine, pH 8.3. The protein was precipitated with acetone to remove sodium dodecyl sulfate (SDS), resuspended in 6 M guanidine hydrochloride, dialyzed against phosphate buffer (300 mM NaCl, pH 8), and stored at −70°C.
Peptide synthesis
The following peptides were synthesized by the Biomolecular Resource Facility of the John Curtin School of Medical Research (Australian National University, Canberra, Australia) with the use of an Applied Biosystems 430A peptide synthesizer, with purification to 98–100%, by high-performance liquid chromatography and mass spectroscopy. Additional HPLC purification steps were required with the hydrophobic b1a peptide to obtain the desired purity and prevent peptide aggregation:
β1a peptide: 490VQVLTSLRRNLSFWGGLEASPRGGDAVAQPQEHAM524
Biotinylated β1a C-tail peptide: Biotin-SNLQ-VQVLTSLRRNLSFWGGLEASPRGGDAVAQPQEHAM
βscrambled peptide: GEAVLAPLGFQSHPNGLQARLVWSDGSRQVTMREA
Biotinylated βscrambled peptide: Biotin-SNLQ-GEAVLAPLGFQSHPNGLQARLVWSDGSRQVTMREA
An additional four residues were added to the N-terminals of the β1a and βscrambled peptides as a linker to biotin. Stock peptide (1 mM in H2O) was frozen in 20 μl aliquots.
SR vesicle isolation, [3H]ryanodine binding, single-channel recording, and analysis
Rabbit skeletal SR vesicles were isolated and [3H]ryanodine binding was performed as described previously (10), except that for the [3H]ryanodine binding, the buffer contained 150 mM NaCl, 20 mM MOPS, 1 mM CaCl2 with BAPTA for the desired [Ca2+] (determined with a Ca2+ electrode); the filters were washed four times with 20 mM MOPS and 150 mM NaCl; and the SR vesicles were preincubated with peptides for 30 min on ice. In some cases, a mixture of 5 mg/ml γ-globulins and 5 mg/ml bovine serum albumin in 200 mM NaCl with 10% (w/v) polyethylene glycol was added after washing. The use of polyethylene glycol in early experiments did not alter [3H]ryanodine binding.
For single-channel experiments, SR vesicles were incorporated into lipid bilayers (11) via a cis (cytoplasmic) solution containing 230 mM CsCH3O3S, 20 mM CsCl, 1.0 mM CaCl2, 10 mM TES, and 500 mM mannitol (pH 7.4 with CsOH) and a trans (luminal) solution containing 30 mM CsCH3O3S, 20 mM CsCl, 1.0 mM CaCl2, and 10 mM TES (pH 7.4). After RyR incorporation was achieved, the cis solution was replaced with a similar solution with BAPTA added in amounts determined using a Ca2+ electrode, for the desired free [Ca2+]. After RyR1 was incorporated, 200 mM CsCH3O3S was added to the trans solution to achieve a symmetrical 250 mM Cs+. The bilayer potential, Vcis-Vtrans, was switched between −40 mV and +40 mV every 30 s. Channel activity under each condition was analyzed over 90–180 s using the program Channel 2 (developed by P. W. Gage and M. Smith, John Curtin School of Medical Research). The threshold levels for channel opening were set to exclude baseline noise at ∼20% of the maximum single-channel conductance and open probability (PO), mean open time (TO), and mean closed time (TC) measured. The peptide concentration dependence of relative PO (RPO) was fitted with a standard Hill equation (12):
where RPOmax and RPOmin are the relative PO of the minimally and maximally activated channels, respectively; AC50 is the β1a peptide or β1a subunit concentration for half-maximal activation; Ha is the Hill coefficient; and X is the concentration of β1a peptide or subunit. The least-squares method was used to fit the equation to the data.
RyR1 purification
RyR1 was solubilized and purified as described previously (13). Sucrose gradient fractions containing purified RyR1 (identified by SDS-PAGE) were concentrated using a Vivaspin 20 tube (Sartorius Stedim Biotech, Bohemia, NY), frozen in 15 μl aliquots, and stored at −70°C.
Steptavidin agarose and affinity chromatography
Streptavidin-agarose was prepared according to the manufacturer's specifications (Thermoscientific, Rockford, IL) and diluted to 50% in wash buffer (150 mM NaCl, 20 mM MOPS, 1 mM BAPTA, and ∼1 mM CaCl2, added in amounts determined by means of a Ca2+ electrode) to obtain 10 μM Ca2+. The streptavidin-agarose was preincubated at 4°C overnight with SR vesicles or purified RyR1 to saturate nonspecific binding and then incubated/rotated with biotinylated peptide for 3 h at room temperature. The mixture was centrifuged (×5) to remove unbound peptide. SR vesicles and purified SR were incubated with streptavadin-agarose-bound biotinylated βscrambled or β1a peptide or with streptavadin-agarose alone at 4°C for 48–72 h. Unbound protein was removed by five centrifugations (500 × g, 5 min) at 4°C. The washed pellet (streptavadin-agarose complex) was added to sample buffer (250 mM Tris HCL (pH 6.8), 40% glycerol, 10% SDS, 20% mercaptoethanol, and 0.1% bromophenol blue) and boiled for 5 min. The supernatant was run on SDS-PAGE, transferred, and immunodetected (10,11). The membrane was cut in half to allow separate detection of RyR1 with anti-RyR1 antibody and biotinylated peptide with StrepTactin.
Statistics
Average data are given as the mean ± SE. Statistical significance was evaluated by means of a paired or unpaired Student's t-test or analysis of variance as appropriate. The numbers of observations (N) are given in the figure legends. Additional high-performance liquid chromatography purification steps were required with the hydrophobic β1a peptides to obtain the desired purity and to prevent peptide aggregation. For the data shown in Figs. 2–4, one concentration of β1a subunit or β1a peptide was tested in each experiment. To reduce the effects of variability in control parameters (POC, TOC, TCC), and to evaluate parameters after β1a subunit (POB, TOB, TCB) or β1a peptide (POP, TOP, TCP) addition, data were expressed as the difference between log10XB (or log10XP) and log10XC for each channel (e.g., logPOB − logPOC). The difference from control was assessed with a paired t-test applied to log10POC and log10POF. The difference between each concentration was assessed by analysis of variance on log10PXB-log10PXC with the post hoc multidimensional Mahalanobis test.
Figure 2.
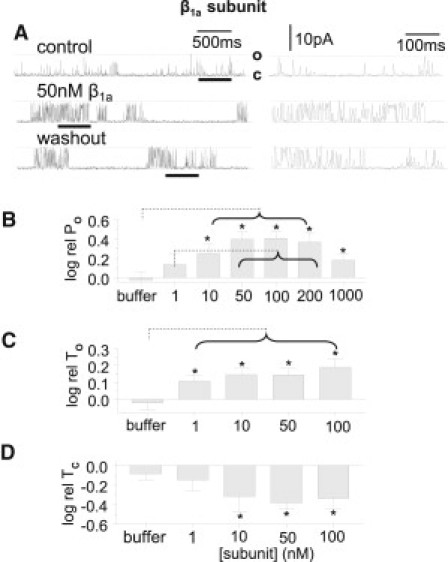
β1a subunit increases RyR1 channel activity with 10 μM cis Ca2+. (A) The β1a subunit (50 nM) was added to the cytoplasmic side of a single RyR1 channel in a lipid bilayer, recorded at +40 mV with channel opening upward from the closed (C) to the maximum open (O) level. Control activity was recorded before addition of β1a protein. The central record was obtained ∼2 min after addition of the β1a subunit, and the lower record was acquired several minutes after the cis chamber was perfused to remove the protein. The left panel shows 3-s records, and the right panel shows expansions of data underlined by the thick black line in the 3-s records to illustrate the changes in open duration. (B) Relative PO (log rel PO) is the average of differences between the log10 of PO in the presence of the β1a subunit (logPOB) and log10 of the control PO (logPOC) for each channel, with PO measured over 180 s. (C) The relative mean open time (log rel TO) is logTOB-logTOC. (D) The relative mean closed time (log rel TC) is logTCB-logTCC. N = 5–8 experiments for each bin in B–D. Asterisks indicate significant changes from control induced by the peptide. The broken lines indicate significant differences between each bin under the horizontal bracket and data at the far end of the line. In B, log rel PO with 10–200 nM subunit is significantly greater than PO with buffer; and log rel PO for 50–200 nM subunit is significantly greater than log rel PO with 1 nM subunit. In C, log rel TO with 1 nM to 100 nM subunit is significantly different from log rel TO with buffer.
Figure 3.
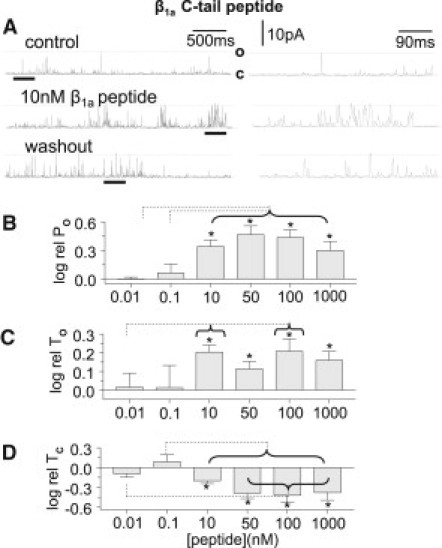
The 35-residue β1a peptide increases the activity of RyR1 channels with 10 μM cis Ca2+. (A) Addition of 10 nM of the peptide to the cytoplasmic side of a single RyR1 channel in a lipid bilayer, recorded at +40 mV with channel opening upward from the closed (C) to the maximum open (O) level. Control data were obtained before addition of the β1a peptide. The central record was obtained ∼3 min after addition of peptide, and the lower record was acquired several minutes after perfusion to remove peptide. The left panel shows 3-s records and the right panel shows expansions of data underlined by the thick black line in the 3-s records to illustrate the changes in open duration. (B) The relative PO (log rel PO) is the average of the difference between log10 of PO in the presence of β1a C-tail peptide (logPOP) and log10 of control PO (logPOC) for each channel, measured over 180 s. (C) The relative mean open time (log rel TO) is logTOP-logTOC. (D) The relative mean closed time (log rel TC) is logTCP-logTcC . N = 5–10 experiments for each bin in B–D. Asterisks indicate significant changes from control induced by the peptide. The broken lines indicate significant differences between each bin under the horizontal bracket and data at the far end of the line. In B, log rel PO for 10–1000 nM is significantly greater than PO with 0.01–0.1 nM peptide. In C, log rel TO for 10 and 100 nM is significantly greater than PO with 0.01 nM peptide. In D, log rel TC with 10–1000 nM peptide is significantly less than log rel TC with 0.1 nM peptide, and log rel TC with 50–1000 nM peptide is significantly less than log rel TC with 0.01 nM peptide.
Figure 4.
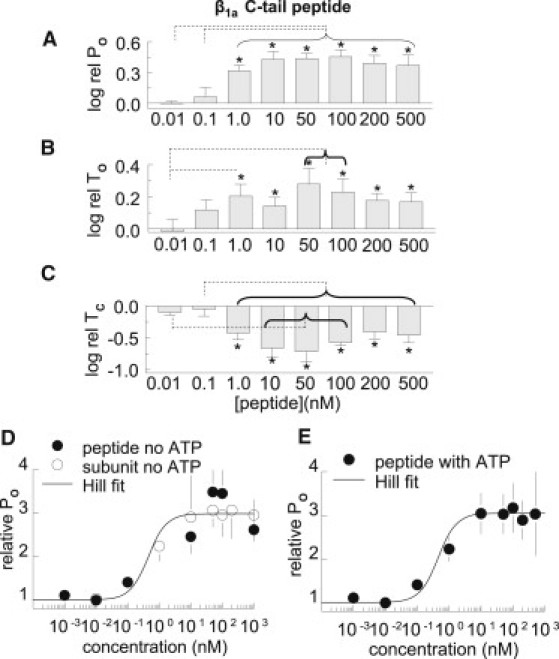
Actions of the 35-residue β1a peptide on RyR1 channels in the presence of 10 μM cis Ca2+. (A–C) Average values measured over 180 s before and after cytoplasmic addition of the β1a peptide. (A) The relative PO (log rel PO) is the average of the difference between log10 of PO in the presence of β1a C-tail peptide (logPOP) and log10 of control PO (logPOC) for each channel. (B) The relative mean open time (log rel TO) is logTOP-logTOC. (C) The relative mean closed time (log rel TC) is logTCP-logTcC. N = 4–6 experiments for each bin in A–C. Asterisks indicate significant changes from control induced by the peptide. The broken lines indicate a significant difference between each bin under the horizontal bracket and data at the far end of the line. In A, log rel PO with 1–500 nM peptide is significantly different from log rel PO with 0.01 and 0.1 nM peptide. In B, log rel TO with 0.01 nM peptide is significantly greater than log rel TO with 1, 50, and 100 nM peptide. In C, log rel TC with 1–500 nM peptide is significantly less than log rel TC with 0.1 nM peptide, and 10–100 nM is significantly less than 0.01 nM peptide. (D and E) Least-squares fit of a standard Hill equation to the average PO expressed as average POB/POC (open circles for β1a subunit) or POP/POC (solid circles for β1a peptide), calculated for individual channels in the absence (D) and presence (E) of ATP. The fit parameters are given in the text.
Results
Binding of the β1a protein and its C-tail to RyR1
The full-length β1a subunit has been shown to bind to a fragment of RyR1 (7). We show here the 35-residue C-tail peptide binds to the full-length RyR1 (Fig. 1). Fig. 1 A shows that when the biotinylated β1a peptide was coupled to streptavidin-agarose, it bound both native RyR1 in SR vesicles and purified RyR1 (lanes 6 and 7, shaded gray). Thus, the tiny 35-residue C-tail peptide bound to and precipitated full-length RyR1. In control experiments, no RyR bound to streptavidin-agarose in the absence of biotinylated β1a peptide (lanes 10 and 11), and streptavidin-agarose coupled to biotinylated βscrambled peptide did not bind to RyR1 in SR vesicles or purified RyR1 (lanes 8 and 9). Positive controls are shown for the two peptides (lanes 2 and 3), for native RyR1 (lane 3) and purified RyR1 (lane 4). These results indicate that binding of the peptide to RyR1 is specific and requires the native C-tail sequence.
Figure 1.
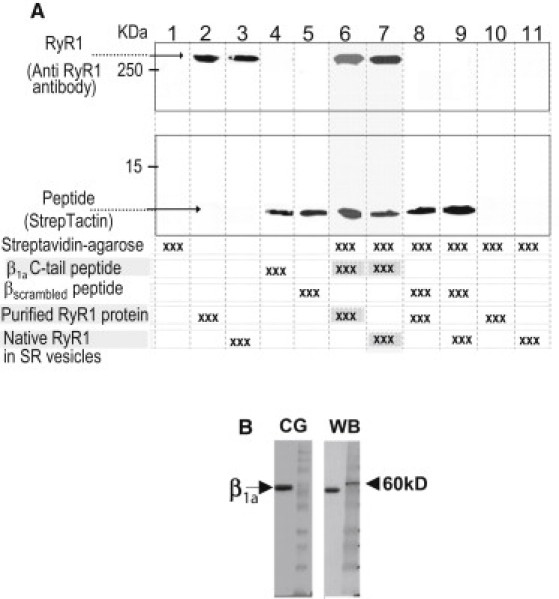
Binding of the 35-residue β1a C-terminal tail peptide to RyR1 and purification of the recombinant β1a subunit. (A) Western blots showing binding of the native RyR1 in SR vesicles (lane 7) and the purified RyR1 (lane 6) to the biotinylated 35-residue β1a C-terminal tail peptide coupled to streptavidin-agarose (highlighted). The peptide was detected with StrepTactin, and RyR1 was detected with anti-RyR1 antibody. The βscrambled peptide did not bind to either native (lane 9) or purified RyR1 (lane 8). Positive controls: purified RyR1 and SR vesicles containing native RyR1 alone in lanes 2 and 3, respectively; and biotinylated β1a and βscrambled peptides alone in lanes 4 and 5, respectively. Lane 1 contained streptavidin-agarose alone, lane 10 contained streptavidin-agarose incubated with purified RyR1, and lane 11 contained streptavidin-agarose incubated with SR vesicles. (B) A Coomassie blue-stained 10% SDS-PAGE gel (CG, left) and Western blot (WB, right) probed with anti-β1a antibody. The first lane shows purified β1a subunit and the second lane shows molecular mass markers; 5 μg and 2.5 μg of β1a subunit were loaded for the Coomassie gel and Western blot, respectively.
The β1a subunit increases RyR1 channel activity with 10 μM cis [Ca2+]
The β1a subunit (Fig. 1 B) increased the activity of native RyR1 channels in lipid bilayers when added to the cytoplasmic (cis) solution. Each channel was exposed to one concentration of β1a subunit for 15–20 min and then the β1a subunit was removed by perfusion. An increase in activity was apparent within 1 min of addition of only 10 nM of the protein, and this was maintained until the β1a was removed by perfusion, when activity fell toward control levels (Fig. 2 A). However, the effect of β1a was not always reversible within the lifetime of the bilayer (10–20 min after perfusion), with only 15 of 27 channels showing a decrease in activity after washout of ≥10 nM of β1a protein. This poor reversibility likely reflects the high-affinity binding of the β1a subunit to the RyR1 channel.
The effects of the β1a subunit on channel activity were similar at +40 and −40 mV. For example, there was an average 2.3 ± 0.4-fold increase in PO at +40 mV and 3.8 ± 1.0-fold at −40 mV with 100 nM β1a subunit (average POB/POC for individual experiments). Therefore, measurements at +40 and −40 mV were included in the average data. There was a significant increase in average relative open probability (PO for each channel with the subunit compared with its internal control before exposure to protein, i.e., logPOB-logPOC) with [subunit] ≥10 nM (Fig. 2 B). The increase in PO was due to significantly longer open times (Fig. 2 C) and shorter closed times (Fig. 2 D). The ∼5-fold reduction in closed times (Fig. 2 D) was greater than expected from the ∼3-fold increase in open times, indicating that the β1a subunit destabilized the closed state of the channel in addition to stabilizing the open state (see below). Buffer alone, added at the same volume as with 1 μM protein, did not alter channel activity (Fig. 2, B–D, first bar).
The β1a C-tail peptide increases RyR1 channel activity with 10 μM cis [Ca2+]
The 35-residue β1a peptide was as effective as the full-length protein at increasing RyR1 channel activity (Fig. 3 A), with concentrations as low as 100 pM in the cis solution causing an increase in activity in five of seven individual channels (see, e.g., Fig. 5 A below), although the average increase in open probability with 100 pM was not significant. Each channel was exposed to one concentration of β1a peptide for 15–20 min and then the peptide was removed by perfusion. As with the full-length protein, the effects of the peptide were not easily reversible, with a clear decrease in activity seen in only nine of 18 channels after washout of ≥10 nM peptide.
Figure 5.
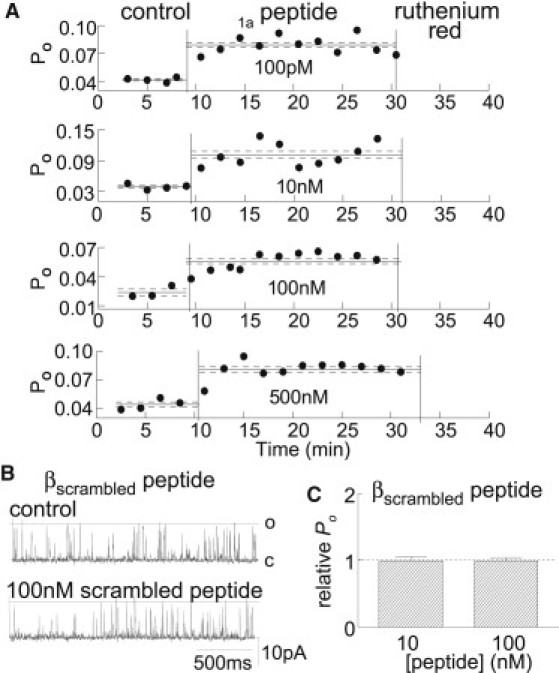
(A) Stability and specificity of the effects of the β1a peptide on RyR1 activity in the presence of ATP. Open probability (PO) is plotted against time for the entire experiment. Peptide concentrations (from the top down): 100 pM, 10 nM, 100 nM, and 500 nM. (B) The βscrambled peptide does not alter RyR1 channel activity. Channel opening upward at +40 mV from the closed (C) to the maximum open level (O) before (control) and after addition of 100 nM βscrambled peptide. (C) Average relative PO (POP/POC) after exposure to 10 and 100 nM βscrambled peptide (N = 7 experiments).
The changes in channel activity with the peptide were again similar at +40 and −40 mV, with an average 2.7 ± 0.6-fold increase in PO (average POP/POC for individual channels) at −40 mV and a 3.3 ± 1.1-fold increase at +40 mV with 100 nM peptide. Data obtained at +40 and −40 mV were combined in all averages. The average increase in open probability with peptide concentrations ≥10 nM (Fig. 3 B) could be attributed to a significant twofold increase in mean open time (Fig. 3 C), and a significant fourfold decrease in mean closed time (Fig. 3 D). These changes occurred over the same concentration range as those induced by the full-length protein and were similar in magnitude.
The β1a peptide induced a similar increase in RyR1 activity in the presence of 2 mM ATP (and 10 μM Ca2+) in the cytoplasmic (cis) solution. The changes in channel gating and the concentration dependence of the changes (Fig. 4, A–C) were again very similar to those described for the full-length β1a subunit. The decrease in the closed durations was greater than the increase in open times, again indicating an additional effect on the dwell time in the closed state. This resulted in an increase in the frequency of openings 3.8 ± 0.2-fold with 10 nM peptide and increases of ∼2.5–3.0-fold with other concentrations up to 500 nM.
The open probabilities of channels exposed to the full-length β1a subunit and the β1a peptide are plotted as the average POB/POC (for β1a subunit) or POP/POC (β1a peptide) in Fig. 4, D and E, respectively. The data were fitted with a standard Hill equation (see Materials and Methods). The best least-squares fit to the data was obtained with AC50 = 450 pM and Ha = 1.5, in the presence or absence of ATP. The Hill coefficient of 1.5 indicates that binding to two subunits of the RyR tetramer may be sufficient to increase RyR1 channel activity. It is possible that a binding pocket is formed at the interface between two subunits.
The stability of the β1a peptide effect on channel activity is illustrated in Fig. 5 A. The open probability was measured continuously during 20-min exposures to the β1a peptide with 1 mM cis ATP. Activity increased within 2–3 min of peptide addition and remained constant until the channel was blocked by ruthenium red (confirming that the channel was RyR1). Similar stability was observed with the full β1a subunit and peptide in the absence of ATP.
The specificity for the native sequence of the β1a C-terminal tail in increasing RyR1 activity was assessed by addition of 10 or 100 nM of the βscrambled peptide to the cis side of RyR1 channels. The βscrambled peptide had no effect on the channels (Fig. 5, B and C), indicating that an increase in activity required the specific native amino acid sequence in the C-terminal 35 residues of the β1a subunit.
The β1a C-tail peptide causes a Ca2+-dependent increase in [3H]ryanodine binding
[3H]Ryanodine binding was measured as an independent indicator of channel activity. A significant increase in [3H]ryanodine binding was observed when 50 nM and 1 μM of β1a peptide was added to SR vesicles in the presence of 10 μM Ca2+ (Fig. 6 A). This is indicative of an increase in RyR1 channel activity, since ryanodine binding increases as RyR1 activity increases (14). The binding with 1 μM peptide was significantly less than that achieved with 50 nM peptide, possibly due to a nonspecific effect of the high concentration of peptide. This was not explored further. The βscrambled peptide had no effect on [3H]ryanodine binding at either 50 nM or 1 μM (Fig. 6 A). The dependence of [3H]ryanodine binding on β1a peptide concentration is shown in Fig. 6 B. The curve through the data is the best least-squares fit of a Hill equation with AC50 = 600 pM and Ha = 1.1. These parameters are remarkably close to those obtained for the increase in open probability in single-channel experiments (above).
Figure 6.
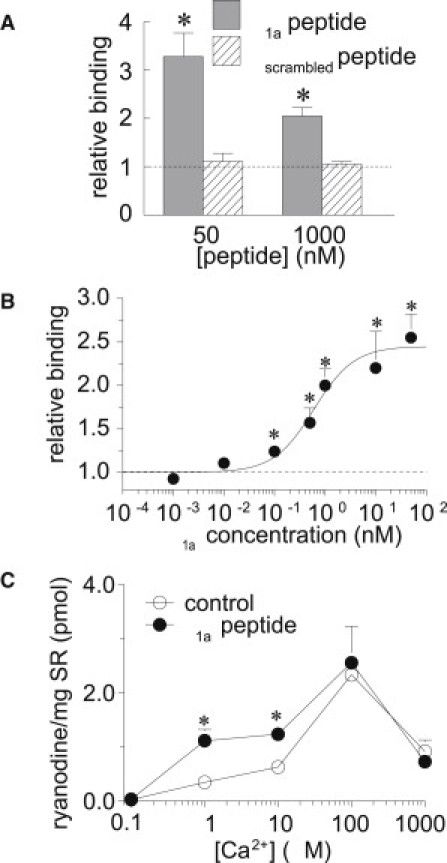
β1a peptide increases [3H]ryanodine binding in a Ca2+-dependent manner. (A) Average [3H]ryanodine binding with 50 or 1000 nM β1a peptide (solid bins) or βscrambled peptide (hatched bins) relative to binding with no peptide. (B) Average relative [3H]ryanodine binding as a function of β1a peptide concentration. Binding with 1, 10, and 50 nM peptide was significantly greater than with 1, 10, and 100 pM peptide, whereas binding with 500 pM was significantly greater than with 1 pM and significantly less than with 50 nM peptide. The line through the data is the best least-squares fit of a standard Hill equation with parameters given in the text. (C) Average [3H]ryanodine bound (pmol) per milligram of SR without (control), and with 50 nM β1a peptide at the indicated [Ca2+]. In A–C, asterisks indicate a significant increase in [3H]ryanodine binding with peptide compared with 3H]ryanodine binding in the absence of peptide (N = 6–18 for each bin).
The effect of the β1a peptide on [3H]ryanodine binding was dependent on [Ca2+] (Fig. 6 C). The usual increase in [3H]ryanodine binding, reflecting high-affinity Ca2+-activation of RyR1, was seen as [Ca2+] increased from 100 nM to 100 μM. A further increase in [Ca2+] to 1 mM was accompanied by a decline in ryanodine binding, reflecting lower-affinity Ca2+ inhibition (15,16). A significant increase in [3H]ryanodine binding with 50 nM β1a peptide was seen only with 1 and 10 μM Ca2+. There was no effect of the peptide with subactivating 100 nM Ca2+, maximally activating 100 μM Ca2+, or inhibiting 1 mM Ca2+. The lack of an effect with 100 μM Ca2+ cannot be attributed to saturation of channel opening, since PO in the maximally Ca2+-activated channel is well below 1.00, and significantly greater binding can be seen when additional activators are added (14). Ryanodine binding is clearly not saturated with 100 nM or 1 mM Ca2+.
The apparent Ca2+ dependence of β1a effects was further explored in single-channel experiments in which the peptide was added to a cytoplasmic solution containing 100 nM Ca2+. There was no change in activity during exposure to 10 nM β1a peptide or within 10 min after β1a peptide was increased to 100 nM (Fig. 7 A). There was no increase in average PO measured from 180 s of channel activity under either condition (Fig. 7 B). However in two of the five experiments, occasional longer channel openings could be seen (arrows in Fig. 7 A), perhaps reflecting a weak effect of the β1a peptide.
Figure 7.
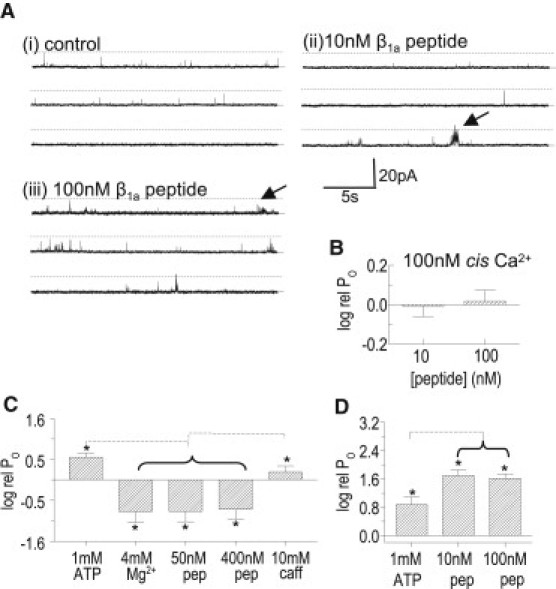
β1a peptide does not increase the activity of RyR1 channels that are inactive with 100 nM cytoplasmic (cis) Ca2+ or inhibited by Mg2+. (A) Three 30-s (90 s total) records from one channel at +40 mV: before (i) and 6–8 min after addition of 10 nM β1a peptide (ii) and 90 nM β1a peptide (iii). The arrows indicate occasional bursts of activity with peptide that did not significantly alter PO over the 90-s period. (B) Average relative PO (as defined for Figs. 2–4, for 10 and 100 nM β1a peptide added to a 100 nM Ca2+cis solution. (C) Average relative PO with 10 μM cis Ca2+ (control), and after sequential additions of 1 mM ATP, 4 mM Mg2+, 50 nM β1a peptide, 350 nM β1a peptide, and 10 mM caffeine (N = 7 experiments). (D) Average relative PO with 100 nM cis Ca2+ (control) and after addition of 1 mM ATP and then 10 nM and 90 nM β1a peptide. Asterisks indicate significant differences between control values and values with peptide (N = 4 observations from two experiments). The broken lines (C and D) indicate significant differences between average data under the horizontal bracket and data at the far end of the line. In C, log rel PO with 4mM Mg2+, 50 nM, and 400 nM peptide is significantly different from log rel PO with 1 mM ATP alone or with 10 mM caffeine. In D, log rel PO with 10 and 100 nM peptide is significantly different from log rel PO with ATP alone.
Under physiological conditions, low resting RyR1 activity is maintained by 1–4 mM [Mg2+] binding to the low-affinity inhibitory site, which has a similar sensitivity to Ca2+ and Mg2+ (16). Mg2+ inhibition is relieved by a signal from the DHPR during EC coupling (17,18). We examined the ability of the β1a peptide to activate RyR1 channels in the presence of 4 mM Mg2+ and 1 mM ATP. Addition of Mg2+ reduced channel activity to very low levels and there was no increase after 5–10 min incubation with 50 nM and then 400 nM β1a peptide (Fig. 7 C). Subsequent addition of 10 mM caffeine produced a significant increase in activity. In contrast to the lack of activation Mg2+-inhibited channels, the β1a peptide induced significant activation of channels with 100 nM cis Ca2+ when channel activity was enhanced by 1 mM cis ATP (but no Mg2+; Fig. 7 D). Consistent with the channel data, the β1a peptide was unable to increase Ca2+ release from SR vesicles (bathed in 1 mM ATP and 4 mM Mg2+ to load of Ca2+ (19); N = 30). Ca2+ release in these experiments was evoked by 1–10 mM caffeine. Together, these data indicate that RyR1 activity had a specific effect on the ability of the β1a peptide to activate the channel.
Discussion
The results presented here reveal for the first time, to our knowledge, a functional interaction between the β1a subunit of the skeletal DHPR and the skeletal RyR1 channel. The full-length β1a subunit was previously shown to bind to RyR1 fragments (7), and the 35-residue C-tail of β1a was shown to support EC coupling (6). Here, we show that the 35 C-terminal tail residues of β1a bind to the full-length RyR1. This result supports the possibility of a direct physical interaction between the C-terminal tail of the β1a subunit and RyR1 in vivo. In addition, we show for the first time, to our knowledge, that the full-length β1a subunit induces an increase in the activity of RyR1 when added to the cytoplasmic side of the channel, and that the C-terminal 35 residues are sufficient to support this increase in RyR1 channel activity. The functional interaction with the RyR1 channel was significant with only 500 pM to 1 nM of either β1a subunit or its C-terminal tail, and increases in individual RyR1 channel openings were seen with only 100 pM peptide. The binding and functional interaction between the β1a peptide and RyR1 was specific for the native sequence of the C-terminal 35 residues in the β1a subunit, as no binding or change in activity was seen with a peptide having a scrambled sequence of the same residues. Thus, there is a specific physical and very-high-affinity functional interaction between the C-terminal tail of the β1a subunit and the RyR1 channel.
As noted in the Introduction, previous experiments in β1a-null mouse mytotubes transfected with various constructs of the C-terminal tail of the β1a subunit suggested that β1a may contribute directly to physical EC coupling in skeletal muscle (6,7). Experiments with the β1a-null zebrafish could also be interpreted in terms of a direct participation of the β1a subunit in the physical EC coupling process, although other interpretations were favored by the authors (8). The fact that the β1a subunit can bind to RyR1 fragments in affinity chromatography indicates that the effect of β1a on voltage-dependent Ca2+ release may be mediated by its binding to RyR1 (7). The impact of the C-terminal tail of β1a on EC coupling (6) further suggests that the C-terminal tail may be involved in β1a binding to RyR1. Our results provide the first direct evidence (to our knowledge) that the β1a C-terminal tail binds to the full-length RyR1, and that this results in a high-affinity (picomolar) functional interaction between the β1a subunit and the RyR1 proteins that may contribute to Ca2+ release during in vivo contraction.
However, the notion that the β1a subunit contributes directly to in vivo Ca2+ release is controversial (2). As mentioned above, Schredelseker et al. (8) questioned this view and argued that apparent effects on Ca2+ release could be due to a specific effect of the β1a on microtargeting of the DHPR to RyR1. In a study by Sheridan et al. (20), alanine substitution of specific residues in a hydrophobic heptad repeat (i.e., three hydrophobic residues each separated by seven residues) in the C-terminal 47 residues (β1a 478–524) of the β1a subunit reduced EC coupling. Curiously, however, alanine substitution of the hydrophobic residues that abolished voltage-dependent Ca2+ release in a mouse model did not alter Ca2+ release in a zebrafish model (21). It is worth noting that two of these three hydrophobic residues in the heptad repeat are outside the extreme 35-residue C-tail (residues 490–524) that was previously shown to influence EC coupling (6) and was shown here to be sufficient to reproduce the effects of the full-length β1a. The role of the heptad repeat remains to be resolved. It may be more important for the interaction between the β1a C-terminal tail and RyR1 in the mouse myotube than in the zebrafish because of small differences between the two tissues in either the structure of the T-tubule/SR junction or the RyR1 sequence. On the other hand, our preliminary data suggest that hydrophobic residues 409, 500, and 504 (in positions 7, 11, and 14 at the N-terminus of the 35-residue C-tail peptide) are required for the functional interaction between the peptide and RyR1. Of note, residues 11 and 14 are not part of the heptad repeat.
The action of the β1a C-terminal tail peptide on RyR1 channels was dependent on cytoplasmic Ca2+, ATP, and Mg2+ concentrations, such that the peptide was active when channel activity was high (in the presence of 10 μM cytoplasmic Ca2+ and/or 1 mM ATP) and was inactive when channel activity was low (inhibited by Mg2+). Thus, the peptide was most active under conditions achieved during voltage-activated Ca2+ release, i.e., 1 and 10 μM cytoplasmic Ca2+ and release of Mg2+ inhibition (17,18). It is not clear from our experiments whether the response to the β1a peptide is set by the level of channel activity or the actual concentrations Ca2+ and Mg2+. The data suggest that the level of activity may be the principal factor, because the peptide was inactive with 100 nM Ca2+ alone but active when ATP was added. Regardless of the mechanism, this result indicates that the functional interaction between β1a and RyR1 may become significant only after voltage-activated Ca2+ release has been initiated and Mg2+ inhibition has been relieved. In this case, the β1a → RyR1 pathway could act as a booster system in the Ca2+ release process.
The role of the C-tail of the β1a subunit in EC coupling and binding to RyR1 is also brought into question by the results of FRET experiments using a CFP-YFP tandem construct fused to β1a (22). First, β1a expressed without α1s was not colocalized with RyRs, suggesting that β1a cannot bind to RyR1 in the absence of α1S. However, this result could also be explained by other factors, including the geometry of the junction, which may be altered in the absence of α1s so that access to β1a is restricted. Second, the FRET efficiency of the CFP-YFP tandem construct fused to the β1a C-tail was similar when the construct was expressed in α1S-null or RyR1-null myotubes, indicating that the C-terminal tail does not bind to RyR1. Again, this could be explained by other factors, including the possibility that the binding site involves hydrophobic residues remote from the extreme C-terminus (as discussed above). Finally, fusion to the CFP-YFP tandem construct to the β1a C-terminus did not alter the percentage of myotubes that contracted in response to electrical stimulation, which could indicate that the C-tail is not essential for EC coupling. This may also be explained by the fact that the extremity of the C-tail is removed from the critical hydrophobic site for binding to RyR1. Additional questions arise because it has been shown that a biotin acceptor domain tag fused to the C-terminus of β1a is accessible to a 60-kD streptavidin probe, and EC coupling persists after streptavidin binds to the biotin acceptor (23). This suggests that the β1a C-terminus does not interact directly with RyR1 in the resting state (2). As with the other observations, this result could be explained by other factors. For example, because binding to RyR1 occurs at a site removed from the extreme β1a C-terminus, the conformation of the site is not influenced by streptavidin that is bound to an acceptor attached to the extreme C-terminus.
It remains curious that the β1a subunit does not copurify with RyR1 even though we see a high-affinity interaction between the proteins in vitro. This may be due in part to the similarly high-affinity binding of the β1a subunit to α1S I-II loop. It is possible that 1), β1a binding to RyR1 is destabilized by the detergents used for purification (since hydrophobic residues appear to be involved in the binding; see Discussion above); or 2), the effective β1a binding to RyR1 is only permitted with conformational changes that may occur within the α1S/β1a complex during EC coupling.
Conclusions
Our results show that there is a specific high-affinity activating interaction between the β1a subunit of the DHPR and RyR1. This effect of the full-length β1a subunit can be reproduced by a peptide corresponding to the 35-residue C-terminal tail of the β1a subunit, indicating that the action of the subunit is mediated by its C-terminal residues. We conclude that the high-affinity interaction between the C-terminal tail of β1a and RyR1 may contribute to an EC coupling signaling pathway. This conclusion is supported by previous results from mouse myotube experiments indicating that both the C-terminal tail of β1a and binding of the β1a subunit to RyR1 are required for proper skeletal-type EC coupling.
Acknowledgments
We thank S. Pace and J. Stivala for SR vesicle preparation and RyR solubilization, and M. Coggan for assistance with β1a subunit expression and purification.
This work was supported by the National Health and Medical Research Council (471462 to A.F.D., M.G.C., and P.G.B.).
References
- 1.Bannister R.A. Bridging the myoplasmic gap: recent developments in skeletal muscle excitation-contraction coupling. J. Muscle Res. Cell Motil. 2007;28:275–283. doi: 10.1007/s10974-007-9118-5. [DOI] [PubMed] [Google Scholar]
- 2.Beam K.G., Bannister R.A. Looking for answers to EC coupling's persistent questions. J. Gen. Physiol. 2010;136:7–12. doi: 10.1085/jgp.201010461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grabner M., Dirksen R.T., Beam K.G. The II-III loop of the skeletal muscle dihydropyridine receptor is responsible for the bi-directional coupling with the ryanodine receptor. J. Biol. Chem. 1999;274:21913–21919. doi: 10.1074/jbc.274.31.21913. [DOI] [PubMed] [Google Scholar]
- 4.Schredelseker J., Di Biase V., Grabner M. The β1a subunit is essential for the assembly of dihydropyridine-receptor arrays in skeletal muscle. Proc. Natl. Acad. Sci. USA. 2005;102:17219–17224. doi: 10.1073/pnas.0508710102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karunasekara Y., Dulhunty A.F., Casarotto M.G. The voltage-gated calcium-channel β subunit: more than just an accessory. Eur. Biophys. J. 2009;39:75–81. doi: 10.1007/s00249-009-0467-4. [DOI] [PubMed] [Google Scholar]
- 6.Beurg M., Ahern C.A., Coronado R. Involvement of the carboxy-terminus region of the dihydropyridine receptor β1a subunit in excitation-contraction coupling of skeletal muscle. Biophys. J. 1999;77:2953–2967. doi: 10.1016/S0006-3495(99)77128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng W., Altafaj X., Coronado R. Interaction between the dihydropyridine receptor Ca2+ channel β-subunit and ryanodine receptor type 1 strengthens excitation-contraction coupling. Proc. Natl. Acad. Sci. USA. 2005;102:19225–19230. doi: 10.1073/pnas.0504334102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schredelseker J., Dayal A., Grabner M. Proper restoration of excitation-contraction coupling in the dihydropyridine receptor β1-null zebrafish relaxed is an exclusive function of the β1a subunit. J. Biol. Chem. 2009;284:1242–1251. doi: 10.1074/jbc.M807767200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker R.T., Catanzariti A.M., Board P.G. Using deubiquitylating enzymes as research tools. Methods Enzymol. 2005;398:540–554. doi: 10.1016/S0076-6879(05)98044-0. [DOI] [PubMed] [Google Scholar]
- 10.Kimura T., Nakamori M., Sakoda S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005;14:2189–2200. doi: 10.1093/hmg/ddi223. [DOI] [PubMed] [Google Scholar]
- 11.Wei L., Gallant E.M., Beard N.A. Junctin and triadin each activate skeletal ryanodine receptors but junctin alone mediates functional interactions with calsequestrin. Int. J. Biochem. Cell Biol. 2009;41:2214–2224. doi: 10.1016/j.biocel.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laver D.R. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophys. J. 2007;92:3541–3555. doi: 10.1529/biophysj.106.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H.B., Xu L., Meissner G. Reconstitution of the skeletal muscle ryanodine receptor-Ca2+ release channel protein complex into proteoliposomes. J. Biol. Chem. 1994;269:13305–13312. [PubMed] [Google Scholar]
- 14.Kimura T., Pace S.M., Dulhunty A.F. A variably spliced region in the type 1 ryanodine receptor may participate in an inter-domain interaction. Biochem. J. 2007;401:317–324. doi: 10.1042/BJ20060686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meissner G., Darling E., Eveleth J. Kinetics of rapid Ca2+ release by sarcoplasmic reticulum. Effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry. 1986;25:236–244. doi: 10.1021/bi00349a033. [DOI] [PubMed] [Google Scholar]
- 16.Laver D.R., Baynes T.M., Dulhunty A.F. Magnesium inhibition of ryanodine-receptor calcium channels: evidence for two independent mechanisms. J. Membr. Biol. 1997;156:213–229. doi: 10.1007/s002329900202. [DOI] [PubMed] [Google Scholar]
- 17.Lamb G.D., Stephenson D.G. Effect of Mg2+ on the control of Ca2+ release in skeletal muscle fibres of the toad. J. Physiol. 1991;434:507–528. doi: 10.1113/jphysiol.1991.sp018483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamb G.D., Stephenson D.G. Effects of intracellular pH and [Mg2+] on excitation-contraction coupling in skeletal muscle fibres of the rat. J. Physiol. 1994;478:331–339. doi: 10.1113/jphysiol.1994.sp020253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jalilian C., Gallant E.M., Dulhunty A.F. Redox potential and the response of cardiac ryanodine receptors to CLIC-2, a member of the glutathione S-transferase structural family. Antioxid. Redox Signal. 2008;10:1675–1686. doi: 10.1089/ars.2007.1994. [DOI] [PubMed] [Google Scholar]
- 20.Sheridan D.C., Cheng W., Coronado R. Involvement of a heptad repeat in the carboxyl terminus of the dihydropyridine receptor β1a subunit in the mechanism of excitation-contraction coupling in skeletal muscle. Biophys. J. 2004;87:929–942. doi: 10.1529/biophysj.104.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dayal A., Schredelseker J., Grabner M. Skeletal muscle excitation-contraction coupling is independent of a conserved heptad repeat motif in the C-terminus of the DHPRβ (1a) subunit. Cell Calcium. 2010;47:500–506. doi: 10.1016/j.ceca.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papadopoulos S., Leuranguer V., Beam K.G. Mapping sites of potential proximity between the dihydropyridine receptor and RyR1 in muscle using a cyan fluorescent protein-yellow fluorescent protein tandem as a fluorescence resonance energy transfer probe. J. Biol. Chem. 2004;279:44046–44056. doi: 10.1074/jbc.M405317200. [DOI] [PubMed] [Google Scholar]
- 23.Lorenzon N.M., Haarmann C.S., Beam K.G. Metabolic biotinylation as a probe of supramolecular structure of the triad junction in skeletal muscle. J. Biol. Chem. 2004;279:44057–44064. doi: 10.1074/jbc.M405318200. [DOI] [PubMed] [Google Scholar]