

Abstract
We developed a new (to our knowledge) protocol to generate giant unilamellar vesicles (GUVs) composed of mixtures of single lipopolysaccharide (LPS) species and Escherichia coli polar lipid extracts. Four different LPSs that differed in the size of the polar headgroup (i.e., LPS smooth > LPS-Ra > LPS-Rc > LPS-Rd) were selected to generate GUVs composed of different LPS/E. coli polar lipid mixtures. Our procedure consists of two main steps: 1), generation and purification of oligolamellar liposomes containing LPSs; and 2), electroformation of GUVs using the LPS-containing oligolamellar vesicles at physiological salt and pH conditions. Analysis of LPS incorporation into the membrane models (both oligolamellar vesicles and GUVs) shows that the final concentration of LPS is lower than that expected from the initial E. coli lipids/LPS mixture. In particular, our protocol allows incorporation of no more than 15 mol % for LPS-smooth and LPS-Ra, and up to 25 mol % for LPS-Rc and LPS-Rd (with respect to total lipids). We used the GUVs to evaluate the impact of different LPS species on the lateral structure of the host membrane (i.e., E. coli polar lipid extract). Rhodamine-DPPE-labeled GUVs show the presence of elongated micrometer-sized lipid domains for GUVs containing either LPS-Rc or LPS-Rd above 10 mol %. Laurdan GP images confirm this finding and show that this particular lateral scenario corresponds to the coexistence of fluid disordered and gel (LPS-enriched)-like micron-sized domains, in similarity to what is observed when LPS is replaced with lipid A. For LPSs containing the more bulky polar headgroup (i.e., LPS-smooth and LPS-Ra), an absence of micrometer-sized domains is observed for all LPS concentrations explored in the GUVs (up to ∼15 mol %). However, fluorescence correlation spectroscopy (using fluorescently labeled LPS) and Laurdan GP experiments in these microscopically homogeneous membranes suggests the presence of LPS clusters with dimensions below our microscope's resolution (∼380 nm radial). Our results indicate that LPSs can cluster into gel-like domains in these bacterial model membranes, and that the size of these domains depends on the chemical structure and concentration of the LPSs.
Introduction
Lipopolysaccharides (LPSs) are critical components of the outer membrane of Gram-negative bacteria (e.g., Escherichia coli and Salmonella enterica). LPS lipids are part of a specialized barrier against macromolecules (e.g., lysozyme and antimicrobial peptide), hydrophobic compounds (e.g., antibiotics and bile salts), and other chemical agents that causes stress to Gram-negative bacterial cells. The negative charge contributed by LPSs and their association with divalent cations help to maintain the structural integrity of the overall outer bacterial membrane (1,2). In addition to their importance in the structure of Gram-negative bacterial cell's membranes, LPSs are important toxic agents in normal animal immune systems; for example, LPSs act as pyrogenic endotoxins that generate massive inflammatory responses by activating macrophages and other cell types (1).
The outer membrane of Gram-negative bacteria is highly asymmetric and the outer leaflet contains a high percentage of LPSs (covering ∼75% of the Gram-negative surface) (1). LPSs represent a group of complex amphiphilic molecules composed of a lipophilic part called lipid A, which anchors LPS molecules to the membrane, and a poly- or oligosaccharide portion that may extend up to 10 nm outside of the bacterial membrane surface. The lipid A structure consists of a biphosphorylated β-(1→6)-linked glucosamine disaccharide substituted with fatty acid ester linkages at positions 3 and 3′, and amide linked at positions 2 and 2′. The total amount of acyl groups per lipid A varies from 4 to 6, and generally comprises C10–C16 acyl chains (hydroxylated or unhydroxylated fatty acids), although longer chains can occur depending on the bacteria class (2) (see Fig. 1).
Figure 1.
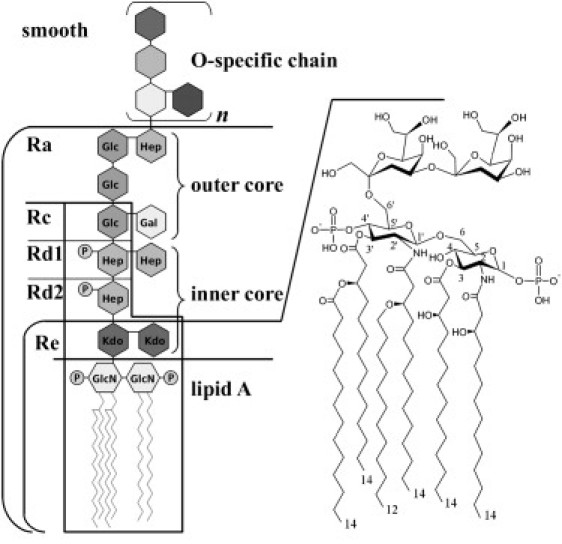
Molecular structure of LPS-Re of E. coli (left). Schematic representation of the different LPSs used in this work (right). Gal, galactopyranose; Glc, glucopyranose; GlcN, 2-amino-2-deoxyglucopyranose; Hep, L-glycero-α-D-manno-heptopyranose; Kdo, 3-deoxy-α-D-manno-oct-2-ulopyranosonic acid; P, phosphate. For more details, see Caroff and Karibian (2).
Gram-negative bacterial cells contain a diverse selection of LPSs that differ in the length of their polysaccharides. The polysaccharide core is divided into three parts: the inner core, the outer core, and an O-specific chain (known as O-antigen polysaccharide groups; Fig. 1). The complete LPS molecules (containing the inner and outer cores plus the O-specific chain) that are present in wild-type strains are generally called smooth LPSs. Typical LPS core structures (inner and outer) for enteric bacterial LPSs consist of eight to twelve, often branched, sugars (2), and the sugar at the reducing end is always α-3-deoxy-D-manno-oct-2-ulosonic acid (also known as keto-deoxyoctulosonate or Kdo) (2→6) linked to the glucosamine of lipid A. Other sugars present in the core include L-glycero-D-manno-heptose residues, such as glucose, galactose, and their derivatives (2).
Rough-chemotype LPSs are mutant LPS molecules and are named according to the size of the oligosaccharide domain. Ra, Rb, Rc, Rd, and Re correspond to the first, second, third, fourth, and fifth degrees of polysaccharide chain length in order of decreasing domain size (3) (see Fig. 1). One can obtain a variety of rough mutants that produce LPS lacking the sections of polysaccharide group (LPS-Ra to Re) by blocking steps in the LPS synthetic pathway. For example, minimal E. coli LPS (termed LPS-Re) consists of lipid A linking two Kdo residues. In general, rough mutants are more sensitive to hydrophobic agents such as antibiotics, detergents, and mutagens. They are also less virulent and more prone to detection by immune systems (4).
Most LPS-related studies to date have focused on two main aspects: 1), the structure/behavior of LPS-containing membranes (see below); and 2), interactions of LPS with relevant components of the mammalian immune system. LPSs act on the immune system when they are released from bacterial cells. Small amounts of LPS are released during cell division, but massive LPS release can occur when bacteria are killed by antibiotics or the immune system of the host organism (1). Activation of the immune system is mainly due to the interaction with macrophages; therefore, it is important to understand the interaction of LPS with macrophage membranes. To study this process (and also to test the use of LPS-containing liposomes as a potential vaccine), Dijkstra et al. (5) developed LPS-containing model membrane systems and successfully applied them to various LPS strains (5–7). Certain protocols for LPS incorporation in membranes used PC/PS/cholesterol lipid mixtures (which mimic eukaryotic membrane) as host membranes. Such LPS-containing liposomal formulations are generally less toxic than free LPSs (5,7). Giant unilamellar vesicle (GUV) models were further developed to study LPS partition in lipid mixtures that exhibit the coexistence of solid ordered/liquid disordered phases (i.e., DPPC/DOPC) (8).
Various studies have examined the structural and physical properties, including phase behavior and aggregation properties, of LPS-containing membranes (3,9–13). Different supramolecular assemblies (e.g., spherical (normal or inverted micelles), lamellar, tubular (normal or inverted HI and HII), and cubic assemblies) have been observed in samples of pure LPS above the critical micellar concentration (CMC). The structural characteristics of these assemblies depend on the molecular geometry of the monomeric LPS as well as environmental conditions such as hydration, temperature, and ion content (3). Accordingly, the phase behavior of pure LPS-containing membranes is also extremely dependent on the aforementioned factors. For example, it has been reported that the main phase transition temperature (Tm from ordered to disordered-like phases) for fully hydrated smooth E. coli LPS is 37°C. It has been shown that this Tm decreases by shortening the polysaccharide group. However, lipid A, which lacks the polysaccharide group, has a larger Tm of 45°C (9). It has also been reported that the transition temperatures dramatically increase in presence of divalent ions. For example, in a study using isolated LPS lipids from E. coli in the presence of Mg2+ no thermal transition was observed up to 75°C (14). Furthermore, thermal transitions associated with LPS were not observed up to ∼60°C (where proteins denaturate) in E. coli intact cells, suggesting that LPS can display gel-like organization in bacterial outer membranes at physiological temperatures (14). Furthermore, the existence of cardiolipin or LPS-rich membrane domains and their role in the action of antimicrobial peptides was recently reported (15).
In this work, we focused on investigating the physical properties of LPS/E. coli lipid-containing membranes. Although these model systems lack lipid asymmetry, they have previously been used as models mimicking bacterial membranes (mixtures containing LPS plus PE/PG or LPS plus E. coli lipid extracts (PE/PG/cardiolipin ∼81:17:2 molar)) (12,16,18). We developed a modified protocol to generate GUVs containing LPSs under physiological salt and pH conditions. We then sought to ascertain the efficiency of LPS incorporation into model membranes at high LPS/phospholipid (or E. coli lipids) ratios, since such information is generally lacking in the literature. Finally, we used GUVs to evaluate the impact of different LPS species on the lateral structure of these bacterial model membranes. By using multiphoton excitation fluorescence microscopy-based techniques, we were able to obtain spatially resolved information (i.e., lateral structure and dynamics) at the level of single vesicles as a function of the concentration of different LPS species.
Materials and Methods
Materials
E. coli polar lipid extract (hereafter referred to as E. coli lipids) was purchased from Avanti Polar Lipids (Alabaster, AL). LPS from E. coli O55:B5 (LPS-smooth), LPS E. coli O55:B5 fluorescein isothiocyanate conjugate (LPS-FITC), E. coli EH-100 (LPS-Ra mutant), E. coli J5 (LPS-Rc mutant), E. coli F-583 (LPS-Rd mutant), and lipid A monophosphoryl from E. coli F583 (Rd mutant) were obtained from Sigma-Aldrich (St. Louis, MO). We purchased 6-dodecanoyl-2-dimethylaminonaphthalene (Laurdan), Lissamine-rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (rhodamine-DHPE), and Alexa Fluor 488 hydrazide sodium salt from Molecular Probes (Eugene, OR).
Methods
Labeling LPS with Alexa Fluor 488 hydrazide
LPS from E. coli O55:B5 (smooth) was labeled with Alexa Fluor 488 hydrazide according to a protocol described by Luk et al. (19) with modifications (see section 1 in the Supporting Material).
Incorporation of LPS into oligolamellar vesicles
To incorporate LPS into the liposomal membrane, we used the dehydration-rehydration method proposed by Dijkstra et al. (5) with some modifications. The dehydration-rehydration method is more efficient for incorporating LPS into liposomal membranes (particularly those containing negative charges) compared with prolonged sonication methods (tested in this study) (5). The phospholipid vesicles were prepared from stocks of E. coli polar lipid extracts mixed with fluorescent probes (either rhodamine-DHPE or Laurdan) in chloroform/methanol 2:1 (vol). Briefly, organic stocks of fluorescently labeled E. coli lipids were deposited in borosilicate glass vials in various amounts (to obtain various LPS/E. coli lipid ratios), dried under an N2 stream, and exposed to low pressure in a desiccator for at least 2 h. The lipid films were hydrated in 0.4–0.5 mM LPS (LPS-smooth, LPS-Ra, LPS-Rc, or LPS-Rd) water solutions at 55°C for 20 min with continuous vortexing (Eppendorf Thermomixer Comfort R, Eppendorf–Netheler-Himz GmbH, Hamburg, Germany). The samples were further sonicated in a bath sonicator (Branson 1510; Branson Ultrasonic, Danbury, CT) at 55°C for 20 min, frozen in liquid nitrogen, and lyophilized for 5 h. The dried samples were resuspended in 150 mM NaCl to a final phospholipid concentrations of 2 mM, vortexed, and further sonicated in a bath sonicator (Branson 1510) at 55°C for 20 min. The LPS-containing liposomes were separated from nonincorporated LPS (both aggregates and monomers) by size exclusion chromatography (Sephacryl S-400, Promega, Glostrup, Denmark). We evaluated the separation of LPS-containing liposomes from LPS monomers and aggregates by using controls with fluorescently labeled liposomes and LPS (i.e., the liposomal fraction was detected in the column void volume and LPS was eluted later; see Fig. S1). We then obtained the liposomal fraction for further analysis and used it to prepare GUVs. The total phospholipid concentration and LPS concentration in these samples were determined by means of an inorganic phosphate assay (20) and a Kdo assay (21), respectively. Oligolamellar vesicles containing lipid A were prepared by mixing E. coli polar lipid extract with lipid A in chloroform/methanol 2:1 (vol), followed by removal of the organic solvent, hydration with 10 mM phosphate buffer (150 mM NaCl, pH 7.4), and sonication at 55°C as indicated above.
Preparation of GUVs
GUVs were prepared from LPS-containing liposomes according to a recently reported protocol (22). Briefly, 0.1 mM solution of LPS-containing oligolamellar vesicles were deposited on the Pt electrodes (1 μl per electrode) of a custom-built electroformation chamber (23) and dried under vacuum for 10 min. This procedure was repeated three to four times before electroformation was performed. After final deposition of the oligolamellar vesicles on the Pt wires, 500 μL of 10 mM phosphate buffer (150 mM NaCl, pH 7.4) was added to the electroformation chamber and an alternate electric field was applied (Digimess Fg 100, Fürth, Germany) in three steps: 1), freq. 500 Hz, amp. 35 V/m for 5 min; 2), freq. 500 Hz, amp. 313 V/m for 20 min; and 3), freq. 500 Hz, amp. 870 V/m for 90 min. Electroformation was performed at 55°C. The GUVs were directly visualized in the GUV electroformation chamber (23). For the microscopy experiments, ∼25 vesicles per sample were analyzed.
Laurdan GP function and two-photon excitation Laurdan GP measurements
The Laurdan GP function is sensitive to membrane lateral packing (24–28). The spatially resolved GP information obtained from two-photon excitation microscopy allows one to infer the local membrane phase state at the level of single vesicles under equilibrium conditions (27). Information regarding the definition of the GP function and technical aspects of the two-photon excitation microscope experimental setup (including data analysis) is given in section 2 of the Supporting Material.
Fluorescence correlation spectroscopy of Alexa Fluor 488-labeled LPS
Fluorescence correlation spectroscopy (FCS) was performed on a custom-built, two-photon excitation fluorescence microscope as previously described (28). We obtained measurements using the SimFCS software package and globally fitted the fluorescence fluctuation data using the Globals for Spectroscopy software package (both software packages developed at the Laboratory for Fluorescence Dynamics, University of California, Irvine, CA (29)). For details regarding the FCS experiments, see section 3 in the Supporting Material.
Results
Our preparation procedure for GUVs containing LPS rendered a good yield of GUVs with an average diameter of 20 μm. The most laborious part of this protocol was the optimization of LPS incorporation into oligolamellar vesicles and further purification of these vesicles from the remaining LPS monomers and self-aggregates. Of importance, we observed that for values above 30 mol % of LPS with respect to total lipids in the initial LPS/E. coli lipids mixtures, the formation of GUVs from the purified lipid dispersions was completely impaired. Therefore, we decided to characterize samples containing an initial LPS concentration below 30 mol % with respect to total lipid.
Quantification of LPS concentration in oligolamellar vesicles containing LPS showed that not all of the LPS was incorporated into the oligolamellar vesicles, in agreement with previous observations (5). In Fig. 2, the final concentration of LPS incorporated into the oligolamellar vesicles is compared with the initial concentration of LPS used to prepare the different LPS/E. coli lipids mixtures. The results show that concentrations of no more than 15 mol % for LPS-smooth and LPS-Ra, and up to 25 mol % for LPS-Rc and LPS-Rd (with respect to total E. coli lipids), are present in the oligolamellar vesicles.
Figure 2.
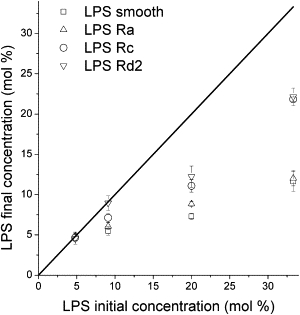
Summary of the LPS incorporation procedure for four studied LPS strains. The x axis represents the starting LPS concentration as a fraction of the total amount of lipids in the sample. The y axis shows the concentration of LPS in the oligolamellar vesicles as a fraction of the total amount of lipids in the samples after completion of the purification procedure. The solid black line indicates an ideal 100% incorporation efficiency. These vesicles were used to form GUVs. The efficiency of LPS incorporation increases as the length of the polysaccharide decreases. Error bars are standard deviations.
To ascertain whether the incorporation of LPS in the GUVs was the same as that in the oligolamellar vesicles, we performed LPS incorporation using fluorescently labeled LPS-FITC (corresponding to LPS-smooth) in membranes composed of E. coli lipids labeled with rhodamine-DHPE. We chose to use an LPS-smooth analog because this lipid (together with LPS-Ra) shows a lower incorporation efficiency in the host membrane (see Fig. 2). The rhodamine-DHPE/LPS-FITC fluorescence emission intensity ratio obtained in oligolamellar vesicle solution was comparable with that obtained in single GUV experiments (0.67 ± 0.02 and 0.72 ± 0.09, respectively), showing that the lipid composition in both model membranes is very similar. This result is in agreement with previous findings for GUVs composed of binary and ternary lipid mixtures, in which the lipid composition before and after GUVs electroformation was invariable (30,31). Another indication of LPS incorporation in the host membrane is shown in Fig. S2 (fluorescence images obtained with LPS-Alexa 488). To further validate our protocol, we explored other lipid compositions (at physiological conditions in the presence and absence of divalent cations, e.g., Ca+2 up to 2 mM) such as PE/PG-containing mixtures or single PC species such as POPC. In all cases, the formation of GUVs was successfully achieved at LPS concentrations similar to those observed for E. coli lipids (data not shown).
After we quantitatively determined LPS incorporation in the host E. coli lipid bilayer membranes, we explored the impact of the different LPS species on the lateral structure of these membranes using GUV/fluorescence microscopy-related techniques. Our results can be divided into two groups: 1), GUVs containing either LPS-smooth or LPS-Ra; and 2), GUVs containing LPS-Rc or LPS-Rd (as well as lipid A).
GUVs composed of E. coli lipid mixtures containing LPS-smooth and LPS-Ra
GUVs containing LPS-smooth or LPS-Ra show a homogeneous distribution of Laurdan or rhodamine-DHPE probes at all of the LPS concentrations explored (up to ∼15 mol %; Fig. 3, A and B). However, further analysis of the Laurdan emission signals using the Laurdan GP function (in both GUVs and oligolamellar vesicles) shows an increase in the GP function values as the concentration of LPS in the GUVs is increased (Fig. 4, A and B), in similarity to what was observed for GUVs containing LPS-Rc and LPS-Rd below 10 mol % (see below). To more closely evaluate this phenomenon, we performed FCS measurements using fluorescently labeled LPS. These experiments evaluate the diffusion coefficient (D) of fluorescently labeled LPS on the membranes as a function of LPS concentration. The diffusion coefficient shows a ∼5-fold decrease when the LPS (LPS-smooth or LPS-Ra) concentration reaches the maximum attainable in our model systems (∼15 mol % with respect to total lipids; Fig. 5). The behavior observed in these mixtures for the FCS and Laurdan GP data is attributable to the formation of LPS lipid clusters with sizes below the resolution of our microscope images.
Figure 3.
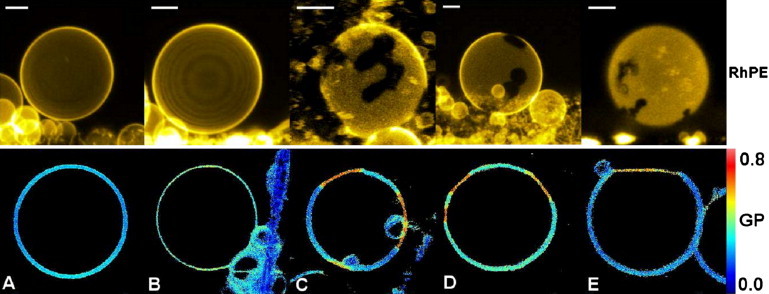
Representative images of GUVs (false color representation) composed of E. coli polar lipid extract and LPSs: (A) LPS-smooth, (B) LPS-Ra, (C) LPS-Rc (above 10 mol %), (D) LPS-Rd (above 10 mol %), and (E) lipid A (above 5 mol %). The upper row shows fluorescence images (false color representation) of GUVs labeled with rhodamine-DHPE. The bottom row displays Laurdan GP images of the GUVs. For LPS-smooth (A) and LPS-Ra (B), no visible phase separation can be seen in the GUVs at any of the explored concentrations (up to 15 mol %). For deep rough LPS strains (Rc (C) and Rd (D)), the coexistence of domains of high GP values (close to 0.6 indicating presence of a gel-like structure) is observed above 10 mol %. Scale bars are 10 μm.
Figure 4.
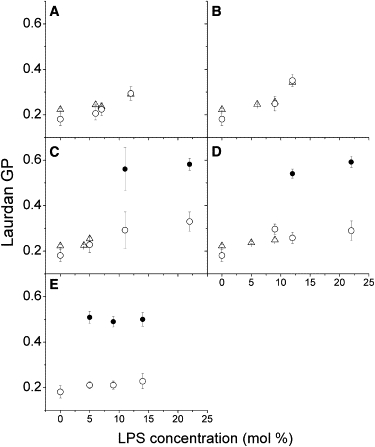
Laurdan GP values of LPSs containing membranes as a function of LPS concentration: (A) LPS-smooth, (B) LPS-Ra, (C) LPS-Rc, (D) LPS-Rd, and (E) lipid A. (○ and ●) Laurdan GP values calculated from GUVs. Double points at high LPS concentration for C–E represent visible phase separation GUVs. These Laurdan GP values were calculated separately for each domain, liquid-like (○) and gel-like (●). Each point represents an average of 15–25 separate vesicles; error bars are standard deviations. (Δ) Laurdan GP values from the oligolamellar vesicles used to form GUVs (measured in a fluorometer in the concentration range where domains are not observed in the GUVs).
Figure 5.
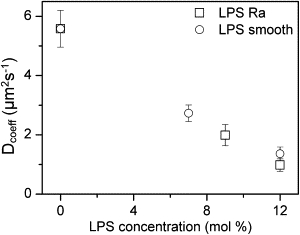
FCS experiments performed in GUVs containing LPS-smooth and LPS-Ra, respectively, doped with fluorescently labeled Alexa 488 LPS-smooth (0.001 mol % with respect to total lipids). The figure shows the corresponding LPS diffusion coefficient measured in GUVs (each point is an average of 15–20 separate vesicles; error bars are standard deviations). No microscopic phase separation was seen in these samples with either Laurdan GP or rhodamine-DHPE.
GUVs composed of E. coli lipid mixtures containing LPS-Rc and LPS-Rd
Compared with the results obtained in LPS-smooth (or LPS-Ra), we observed a different LPS concentration-dependent effect on the lateral structure of the LPS-Rc- and LPS-Rd-containing GUVs. Using rhodamine-DHPE (or Laurdan) as fluorescent markers, we observed the coexistence of distinct elongated micrometer-sized lipid domains only for LPS concentrations above 10 mol % (Fig. 3, C and D). Below a concentration of 10 mol % LPS, a homogeneous distribution of the probes was detected in our fluorescence images (data not shown).
The micrometer-sized lipid domains observed for LPS-Rc and LPS-Rd above 10 mol % very much resemble those shown in Fig. 3 E for lipid A (lacking the inner and outer core sugars; see Fig. 1). However, the presence of domain coexistence in GUVs containing lipid A is observed at lower lipid A molar fractions (i.e., 5 mol %). Analysis of Laurdan images using the Laurdan GP function provided additional information about the local packing of these distinct membrane regions. By taking into account the measured GP value for these elongated domains (∼0.55; see Fig. 4, C–E) and the GP of the surroundings (slightly below 0.2), we conclude that this scenario corresponds to a gel/liquid-disordered-like phase coexistence, where the elongated gel-like domains are enriched in LPS (or lipid A). This conclusion is supported by a comparison of the GP differences between the coexisting domains in our LPS-containing samples with those previously reported for phospholipid samples displaying gel/liquid-disordered phase coexistence (GP values for gel and fluid phases, i.e., >0.5 and <0.2, respectively) (32,33).
Although micrometer-sized domains are not observed below 10 mol % of LPS-Rc and LPS-Rd, the GP values measured in the GUVs increase to some extent relative to the GP measured in absence of LPS (Fig. 4, C and D). This behavior is also observed in the oligolamellar vesicles used to prepare the GUVs. These observations indicate an overall increase in membrane packing in the presence of these two LPSs before the occurrence of micrometer-sized lipid domains. In similarity to the results obtained for LPS-smooth or LPS-Ra, this observation can be explained by assuming LPS clustering in the membranes below the resolution of our microscope. Notice that this effect is not observed for GUVs containing lipid A, where the GP values for the fluid and gel-like phases are constant and independent of the lipid A concentration in the mixture (Fig. 4 E).
Discussion
In this work, we successfully extended the formation of GUVs under physiological conditions (22) (with some modifications; see Materials and Methods) to different compositionally complex LPS/E. coli polar lipid mixtures. This new (to our knowledge) protocol offers an interesting alternative for these lipid mixtures because it enables the preparation of appropriate freestanding bilayer model systems (i.e., GUVs) and thus paves the way for new studies using fluorescence microscopy-related techniques. Additionally, we carefully characterized our model system to obtain quantitative information on the amount of LPS incorporated into the model bilayer membrane (E. coli lipid extract in this case). The LPS molar fraction in the final model membrane is an important parameter not only in terms of this study but also because it has been reported that incorporation of LPS into bilayers models is generally not straightforward (5). A major consequence of this problem is that the LPS concentration in the GUV membrane can be overestimated, particularly if the initial concentration of LPS is assumed to be unchanged during the whole formation process. In fact, for all LPSs explored in our work, we found lower LPS concentrations in the final membrane preparations with respect to that anticipated from the initial E. coli lipid/LPS mixtures. This observation is in line with previous findings by Dijkstra et al. (5), who reported a similar situation for the incorporation of Salmonella minnesota wild-type LPS in PS/PC/cholesterol mixtures. By systematically exploring the incorporation of different LPSs at different concentrations, we were able to attain concentrations of no more than 15 mol % for LPS-smooth and LPS-Ra, and up to 25 mol % for LPS-Rc and LPS-Rd (with respect to total lipids) in our bilayer systems. These concentration ranges are broader than those observed in particular LPSs in previous studies on small unilamellar or multilamellar vesicles, which reported a maximum incorporation of ∼2 mol % (5,6).
The efficiency of LPS incorporation is also relevant in the context of GUV electroformation. We noticed that when we used initial concentrations above 30 mol % of LPS with respect to total lipids, the electroformation of GUVs was impaired. This situation may be connected to a decrease in the tendency to form lamellar structures at higher LPS concentrations for each particular LPS/E.coli lipid mixture. In fact, the formation of nonlamellar phases (cubic phases) was previously reported for high concentrations of a deep rough LPS mutant in mixtures of DPPE/DPPG (12). In our mixtures, the potential existence of nonlamellar structures can also be sustained with the decrease of LPS incorporation as a function of LPS polar headgroup size (particularly at high LPS concentrations; see Fig. 1 to compare the different LPS structures). This tendency can be rationalized by considering the influence of the critical packing parameter on the final membrane structure. For example, different supramolecular assemblies, including spherical (normal or inverted micelles), tubular (normal or inverted HI and HII), and cubic assemblies, have been observed in samples of pure LPS above their CMC. The characteristics of these assemblies largely depend on the molecular geometry of monomeric LPS and environmental conditions such as hydration, temperature, and ion content (3,34).
In a previous study, Henning et al. (8) formed GUVs containing DOPC/DPPC/LPS (LPS-FITC from E. coli 0111:B4; Sigma Chemical) mixtures in a 1:1:1 mol ratio using an electroformation-based method. They compared the effect of Triton X-100 on DOPC/DPPC/LPS membrane and the impact of LPS on the lipid lateral organization with the DOPC/DPPC/cholesterol 1:1:1 mol mixture (8). From a methodological standpoint, we notice two main differences with our method: 1), in their study the deposition of the lipid mixture on the Pt wires was made directly from CHCl3 (in our case, we used a suspension of oligolamellar vesicles containing a known concentration of LPS); and 2), they performed the electroformation of GUVs under nonphysiological conditions (1 mM Tris buffer, pH 8.0) by applying an alternate field with a frequency of 10 Hz, whereas we used 500 Hz (23). Unfortunately, Henning et al. (8) did not report the final amount of LPS in the DOPC/DPPC/LPS membrane (they assumed that all of the LPS used in the initial mixture was incorporated into the GUVS), which hampers a final comparison with our method. In any case, we believe that our protocol offers an improved way to generate GUVs containing LPS, for three main reasons: 1), it is difficult (if not impossible) to achieve proper solubilization of LPS (particularly the more-complex species, i.e., smooth LPS) in CHCl3; 2), our method provides quantitative information about the incorporation of LPS into the final membrane model; and 3), physiological conditions are attained.
Impact of different LPS on the lateral structure of E. coli lipid bilayers
The successful preparation of GUVs containing LPS allowed us to evaluate the spatial distribution of particular fluorescent parameters (e.g., Laurdan GP) at the level of single vesicles. This experimental approach, which is very useful for studying the lateral structure of membranes of diverse composition (27), was applied to evaluate the effect of LPS in GUVs composed of E. coli lipids/LPSs at different LPS compositions.
The Laurdan GP values measured in E. coli lipid membranes in the absence of LPS (Figs. 4 and 5) suggest that under our experimental conditions (i.e., room temperature, excess of water, and high ionic strength), the observed lamellar structures display a liquid-disordered phase (Lα), in agreement with previous observations for phospholipid membranes (33). This conclusion is supported by measurements of the diffusion coefficient of fluorescently labeled LPS at very low concentrations in E. coli lipid membranes (0.001 mol % with respect to total lipid, D = 5.58 ± 0.62 μm2 s−1), resembling those measured from various fluorescent probes in phospholipid membranes displaying a single liquid-disordered phase (35). When LPS concentration is increased in the membrane, the impact of LPS on the lateral structure of E. coli lipid membranes is apparent (Figs. 3–5). From our observations, we conclude that LPSs show a tendency to cluster into gel-like domains in the model mixtures explored, and that the size of these lipid domains depends on the chemical structure of the LPSs as well as their concentration. This conclusion is supported by the presence of micrometer-sized gel-like domains (for LPS-RC- and LPS-RD-containing membranes above 10 mol % of LPS), by the increase in the Laurdan GP values (below 10 mol % for LPS-Rc and LPS-Rd and up to 15 mol % for LPS-smooth and LPS-Ra), and by the fivefold decrease in the diffusion coefficient of fluorescently labeled LPS (up to 15 mol % for LPS-smooth- and LPS-Ra-containing membranes) under conditions in which lipid domains are not visible in the GUVs. To obtain further information about these submicroscopic domains, we employed our diffusion data to compute the domain size (and number of molecules) using the Saffman-Delbrück model (36). From our calculations (see section 4 of the Supporting Material), we found that that a fivefold reduction in the diffusion coefficient (in interfaces containing LPS-smooth or LPS-Ra) implies a clustering of ∼10,000 LPS molecules per leaflet (assuming that the domains span the bilayer). Taking into account the resolution of our microscope (∼380 nm in the radial direction), we estimate that a clustering of >1 × 106 LPS molecules per leaflet (assuming that the molecular area of LPS is ∼1.5 nm2 (37)) is needed to directly observe the domains under the microscope. This result is in line with our microscopy data. Additionally, using the same model, we estimate that the diffusion of micrometer-sized domains is ∼2 orders of magnitude lower than that of single LPS molecules (data not shown). The slow diffusion values for the LPS micrometer-sized domains are in line with the diffusion coefficient of lipid domains measured in GUVs composed of ternary mixtures containing cholesterol (38).
The observed dependence of domain size on the amount and nature of the LPSs in our experiments (reflected in the low concentration of lipid A with respect to LPS-Rc and LPS-Rd observed to form micrometer-sized domains, and the lack of this phenomenon in LPS-Ra and LPS-smooth species; see Figs. 1 and 3–5) could be interpreted as an interplay among different interactions occurring at the level of the LPS polar headgroup and those occurring among the hydrophobic part of LPS molecules. Because in our experiments the different LPS molecules all have the same hydrophobic part (i.e., all of our LPSs come from E. coli strains), it is reasonable to assume that interactions occurring at the level of the polar headgroup can regulate the dimension of these gel-like domains. We speculate that a change in the nature of the LPS polar headgroup can modulate supramolecular interactions through steric effects, electrostatic repulsions, or eventually changes in lipid miscibility. All of these effects may compromise the size of LPS-enriched domains in the plane of the membrane (see Fig. 6). In agreement with our results, LPS clustering was previously observed upon incorporation of LPS isolated from group B Neisseria meningitidis strain B125 in mixtures of egg PC/cholesterol at low LPS concentrations (39). Taking into account the aforementioned results, we can hypothesize that the clustering ability of LPS at low concentrations would also be observed in compositional environments that are representative of both mammalian and bacterial membranes. Because our protocol for LPS incorporation has been successfully extended to other lipid compositions (e.g., POPC) at LPS concentration ranges similar to those presented here, we believe that these model systems would be useful for testing this hypothesis.
Figure 6.
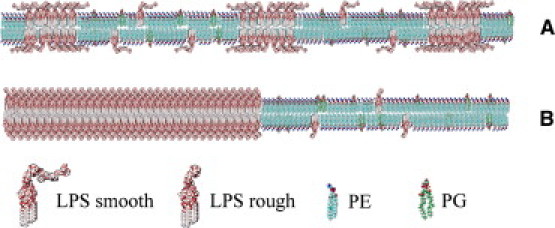
Sketch representing the different LPS-rich domain sizes observed in our experiments. LPS chemotypes (A: LPS-smooth and LPS-Ra; B: Rc and Rd2 (deep rough chemotypes of LPS)) are inserted into the lipid membrane (PE and PG, cardiolipin not shown). (A) The lateral organization of LPS-smooth (valid also for LPS-Ra) is represented for a low concentration of this lipid (the LPS domains in the sketch correspond to a diffusion twofold lower than that of the single LPS and are nanoscopic; see section 3 of the Supporting Material). (B) Formation of large membrane platforms (micrometer-sized) for the rough LPS chemotypes above 10 mol % LPS concentration.
Are LPS-enriched gel-like domains relevant in bacterial membranes?
Vanounou et al. (40) studied the phase behavior of intact bacterial cell membranes using the fluorescent probes Laurdan and DPH in regular cuvette GP and anisotropy fluorescence spectroscopy experiments. They interpreted their results in terms of the existence of two phases of different polarity and packing related to lipid-rich and protein-rich membranes, which is consistent with the presence of outer and inner membranes but is not related to gel-like enriched LPS domains. These observations are in contrast to our results, which were obtained with a similar approach (Laurdan GP function) and show LPS-enriched gel-like domains in model membranes.
Conclusions drawn from Laurdan GP experiments in cuvettes without further exploration of spatially resolved information (available from fluorescence microscopy experiments) can lead to a misinterpretation of the data. In other words, the presence of gel phase-like domains cannot be ruled out. As reported by Fidorra et al. (41), Laurdan GP experiments in cuvettes (where solutions of liposomes are used) using ceramide/POPC mixtures suggested the absence of gel/liquid-disordered phase coexistence, a situation that is in contrast to the data obtained from the same mixture using Laurdan GP imaging microscopy (since the membrane domains can be spatially resolved). As discussed in Fidorra et al. (41), because of the additive property of the GP function, the detection of solid-ordered (gel)/liquid-disordered phase coexistence by Laurdan GP measurements in cuvettes requires a particular quantity of gel-like phase areas in the membrane. This situation cannot be achieved with ceramide-containing mixtures because a maximum of ∼25 mol % of this lipid can be incorporated into the system, which is comparable to the maximum amount of LPS incorporated into our model mixtures.
In line with our results, the possible existence of gel-like LPS-enriched domains in bacterial membranes has been addressed in previous publications; however, this remains a matter of debate (see Nikaido (14) and references therein). Nevertheless, there are some interesting arguments to support this hypothesis. For example, the lack of sterols in bacterial membranes (42) rules out the potential existence of liquid-ordered-like domains (to our knowledge, there are not any molecular species reported to exist in bacterial membranes that can generate liquid-ordered phase in model membranes), and instead favors the existence of gel-like domains promoted particularly by lipids with a high phase transition temperature, such as LPSs. The existence of this type of membrane organization (gel-like) in bacterial membranes would raise interesting questions about its function, especially as related to the barrier properties of the bacterial outer membrane (14). It has been proposed that a gel-like phase organization exists in skin stratum corneum membranes (43), which would be relevant for the barrier function of this tissue.
Conclusions
We have introduced a new (to our knowledge) protocol to incorporate LPS into GUVs composed of E. coli lipids. This method allows the incorporation of no more than 15 mol % for LPS-smooth and LPS-Ra, and up to 25 mol % for LPS-Rc and LPS-Rd (with respect to total lipids). By exploring the lateral structure of these membranes, we found that LPSs tend to form gel-like lipid domains with sizes depending on the chemical structure and concentration of the LPSs. This phenomenon may be relevant for the organization and modulation of the outer membrane of Gram-negative bacteria, and raises a question as to whether similar processes might be connected to LPS partition in biological membranes upon bacterial infection. We expect that the models developed here (GUVs) will be also useful for microscopically characterizing the interactions of particular host molecules and antimicrobial drugs with LPSs (experiments in progress). There is a continuous call for new drug candidates that can target both new and well-known microbial molecules. Such an approach would be particularly relevant for multiresistant Gram-negative bacteria (e.g., methicillin-resistant Staphylococcus aureus), which are becoming an increasing problem in hospitals worldwide.
Acknowledgments
We thank Prof. David Jameson for a critical reading of the manuscript, and the Danish Molecular Biomedical Imaging Center for the use of its multiphoton excitation facility.
This work was supported by grants from the Forskningsrådet for Natur og Univers, Forskningsrådet for Sundhed og Sygdom, and the Danish National Research Foundation (to MEMPHYS-Center for Biomembrane Physics).
Supporting Material
References
- 1.Alexander C., Rietschel E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001;7:167–202. [PubMed] [Google Scholar]
- 2.Caroff M., Karibian D. Structure of bacterial lipopolysaccharides. Carbohydr. Res. 2003;338:2431–2447. doi: 10.1016/j.carres.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson S.G. Bacterial lipopolysaccharides—themes and variations. Prog. Lipid Res. 1996;35:283–343. doi: 10.1016/s0163-7827(96)00004-5. [DOI] [PubMed] [Google Scholar]
- 4.Wang X., Quinn P.J. Lipopolysaccharide: biosynthetic pathway and structure modification. Prog. Lipid Res. 2010;49:97–107. doi: 10.1016/j.plipres.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Dijkstra J., Ryan J.L., Szoka F.C. A procedure for the efficient incorporation of wild-type lipopolysaccharide into liposomes for use in immunological studies. J. Immunol. Methods. 1988;114:197–205. doi: 10.1016/0022-1759(88)90174-3. [DOI] [PubMed] [Google Scholar]
- 6.Nakhla A.N., Banoub J.H., Keough K.M.W. Incorporation of the lipopolysaccharide and polysaccharide from Aeromonas salmonicida into liposomes. J. Liposome Res. 1996;6:141. [Google Scholar]
- 7.Wong J.P., Cherwonogrodzky J.W., Knodel M.H. Liposome potentiation of humoral immune response to lipopolysaccharide and O-polysaccharide antigens of Brucella abortus. Immunology. 1992;77:123–128. [PMC free article] [PubMed] [Google Scholar]
- 8.Henning M.F., Sanchez S., Bakás L. Visualization and analysis of lipopolysaccharide distribution in binary phospholipid bilayers. Biochem. Biophys. Res. Commun. 2009;383:22–26. doi: 10.1016/j.bbrc.2009.03.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandenburg K., Seydel U. Investigation into the fluidity of lipopolysaccharide and free lipid A membrane systems by Fourier-transform infrared spectroscopy and differential scanning calorimetry. Eur. J. Biochem. 1990;191:229–236. doi: 10.1111/j.1432-1033.1990.tb19114.x. [DOI] [PubMed] [Google Scholar]
- 10.Sasaki H., White S.H. Aggregation behavior of an ultra-pure lipopolysaccharide that stimulates TLR-4 receptors. Biophys. J. 2008;95:986–993. doi: 10.1529/biophysj.108.129197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santos N.C., Silva A.C., Saldanha C. Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. ChemBioChem. 2003;4:96–100. doi: 10.1002/cbic.200390020. [DOI] [PubMed] [Google Scholar]
- 12.Urbán E., Bóta A., Kocsis B. Non-bilayer formation in the DPPE-DPPG vesicle system induced by deep rough mutant of Salmonella minnesota R595 lipopolysaccharide. Colloids Surf. B Biointerfaces. 2006;48:106–111. doi: 10.1016/j.colsurfb.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 13.García-Verdugo I., Cañadas O., Casals C. Surfactant protein A forms extensive lattice-like structures on 1,2-dipalmitoylphosphatidylcholine/rough-lipopolysaccharide-mixed monolayers. Biophys. J. 2007;93:3529–3540. doi: 10.1529/biophysj.107.109793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Epand R.M., Epand R.F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta. 2009;1788:289–294. doi: 10.1016/j.bbamem.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 16.Kuzmenko A.I., Wu H., McCormack F.X. Pulmonary collectins selectively permeabilize model bacterial membranes containing rough lipopolysaccharide. Biochemistry. 2006;45:2679–2685. doi: 10.1021/bi0522652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reference deleted in proof.
- 18.Takeuchi Y., Nikaido H. Persistence of segregated phospholipid domains in phospholipid-lipopolysaccharide mixed bilayers: studies with spin-labeled phospholipids. Biochemistry. 1981;20:523–529. doi: 10.1021/bi00506a013. [DOI] [PubMed] [Google Scholar]
- 19.Luk J.M., Kumar A., Staunton D. Biotinylated lipopolysaccharide binds to endotoxin receptor in endothelial and monocytic cells. Anal. Biochem. 1995;232:217–224. doi: 10.1006/abio.1995.0010. [DOI] [PubMed] [Google Scholar]
- 20.Bartlett G.R. Phosphorus assay in column chromatography. J. Biol. Chem. 1959;234:466–468. [PubMed] [Google Scholar]
- 21.Karkhanis Y.D., Zeltner J.Y., Carlo D.J. A new and improved microassay to determine 2-keto-3-deoxyoctonate in lipopolysaccharide of Gram-negative bacteria. Anal. Biochem. 1978;85:595–601. doi: 10.1016/0003-2697(78)90260-9. [DOI] [PubMed] [Google Scholar]
- 22.Méléard P., Bagatolli L.A., Pott T. Giant unilamellar vesicle electroformation from lipid mixtures to native membranes under physiological conditions. Methods Enzymol. 2009;465:161–176. doi: 10.1016/S0076-6879(09)65009-6. [DOI] [PubMed] [Google Scholar]
- 23.Ambroggio E.E., Bagatolli L.A. Membrane-active peptides: methods and results on structure and function. In: Castanho M., editor. Giant Unilamellar Vesicles, Fluorescence Microscopy and Lipid-Peptide Interactions. International University Line; La Jolla, CA: 2009. pp. 179–200. [Google Scholar]
- 24.Parasassi T., Krasnowska E.K., Gratton E. Laurdan and Prodan as polarity-sensitive fluorescent membrane probes. J. Fluoresc. 1998;8:365–373. [Google Scholar]
- 25.Parasassi T., De Stasio G., Gratton E. Phase fluctuation in phospholipid membranes revealed by Laurdan fluorescence. Biophys. J. 1990;57:1179–1186. doi: 10.1016/S0006-3495(90)82637-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parasassi T., De Stasio G., Gratton E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys. J. 1991;60:179–189. doi: 10.1016/S0006-3495(91)82041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagatolli L.A. To see or not to see: lateral organization of biological membranes and fluorescence microscopy. Biochim. Biophys. Acta. 2006;1758:1541–1556. doi: 10.1016/j.bbamem.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 28.Brewer J., de la Serna J.B., Bagatolli L.A. Multiphoton excitation fluorescence microscopy in planar membrane systems. Biochim. Biophys. Acta. 2010;1798:1301–1308. doi: 10.1016/j.bbamem.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 29.Beechem J.M., Gratton E. Fluorescence spectroscopy data analysis environment: a second generation global analysis program. Proc. SPIE Spectrosc. Biochem. 1988;909:70–81. [Google Scholar]
- 30.Fidorra M., Garcia A., Bagatolli L.A. Lipid domains in giant unilamellar vesicles and their correspondence with equilibrium thermodynamic phases: a quantitative fluorescence microscopy imaging approach. Biochim. Biophys. Acta. 2009;1788:2142–2149. doi: 10.1016/j.bbamem.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Juhasz J., Sharom F.J., Davis J.H. Quantitative characterization of coexisting phases in DOPC/DPPC/cholesterol mixtures: comparing confocal fluorescence microscopy and deuterium nuclear magnetic resonance. Biochim. Biophys. Acta. 2009;1788:2541–2552. doi: 10.1016/j.bbamem.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Bagatolli L.A., Gratton E. Two photon fluorescence microscopy of coexisting lipid domains in giant unilamellar vesicles of binary phospholipid mixtures. Biophys. J. 2000;78:290–305. doi: 10.1016/S0006-3495(00)76592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bagatolli L.A., Gratton E. A correlation between lipid domain shape and binary phospholipid mixture composition in free standing bilayers: a two-photon fluorescence microscopy study. Biophys. J. 2000;79:434–447. doi: 10.1016/S0006-3495(00)76305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seydel U., Labischinski H., Brandenburg K. Phase behavior, supramolecular structure, and molecular conformation of lipopolysaccharide. Immunobiology. 1993;187:191–211. doi: 10.1016/S0171-2985(11)80339-6. [DOI] [PubMed] [Google Scholar]
- 35.Machán R., Hof M. Lipid diffusion in planar membranes investigated by fluorescence correlation spectroscopy. Biochim. Biophys. Acta. 2010;1798:1377–1391. doi: 10.1016/j.bbamem.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 36.Saffman P.G., Delbrück M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiese A., Münstermann M., Seydel U. Molecular mechanisms of polymyxin B-membrane interactions: direct correlation between surface charge density and self-promoted transport. J. Membr. Biol. 1998;162:127–138. doi: 10.1007/s002329900350. [DOI] [PubMed] [Google Scholar]
- 38.Cicuta P., Keller S.L., Veatch S.L. Diffusion of liquid domains in lipid bilayer membranes. J. Phys. Chem. B. 2007;111:3328–3331. doi: 10.1021/jp0702088. [DOI] [PubMed] [Google Scholar]
- 39.Trubetskoy V.S., Koshkina N.V., Torchilin V.P. FITC-labeled lipopolysaccharide: use as a probe for liposomal membrane incorporation studies. FEBS Lett. 1990;269:79–82. doi: 10.1016/0014-5793(90)81123-6. [DOI] [PubMed] [Google Scholar]
- 40.Vanounou S., Pines D., Fishov I. Coexistence of domains with distinct order and polarity in fluid bacterial membranes. Photochem. Photobiol. 2002;76:1–11. doi: 10.1562/0031-8655(2002)076<0001:codwdo>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 41.Fidorra M., Duelund L., Bagatolli L.A. Absence of fluid-ordered/fluid-disordered phase coexistence in ceramide/POPC mixtures containing cholesterol. Biophys. J. 2006;90:4437–4451. doi: 10.1529/biophysj.105.077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volkman J.K. Sterols in microorganisms. Appl. Microbiol. Biotechnol. 2003;60:495–506. doi: 10.1007/s00253-002-1172-8. [DOI] [PubMed] [Google Scholar]
- 43.Plasencia I., Norlén L., Bagatolli L.A. Direct visualization of lipid domains in human skin stratum corneum's lipid membranes: effect of pH and temperature. Biophys. J. 2007;93:3142–3155. doi: 10.1529/biophysj.106.096164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.