

Abstract
Electron microscopy and fiber diffraction studies of reconstituted F-actin-tropomyosin filaments reveal the azimuthal position of end-to-end linked tropomyosin molecules on the surface of actin. However, the longitudinal z-position of tropomyosin along F-actin is still uncertain. Without this information, atomic models of F-actin-tropomyosin filaments, free of constraints imposed by troponin or other actin-binding proteins, cannot be formulated, and thus optimal interfacial contacts between actin and tropomyosin remain unknown. Here, a computational search assessing electrostatic interactions for multiple azimuthal locations, z-positions, and pseudo-rotations of tropomyosin on F-actin was performed. The information gleaned was used to localize tropomyosin on F-actin, yielding an atomic model characterized by protein-protein contacts that primarily involve clusters of basic amino acids on actin subdomains 1 and 3 juxtaposed against acidic residues on the successive quasi-repeating units of tropomyosin. A virtually identical model generated by docking F-actin and tropomyosin atomic structures into electron microscopy reconstructions of F-actin-tropomyosin validated the above solution. Here, the z-position of tropomyosin alongside F-actin was defined by matching the seven broad and narrow motifs that typify tropomyosin's twisting superhelical coiled-coil to the wide and tapering tropomyosin densities seen in surface views of F-actin-tropomyosin reconstructions. The functional implications of the F-actin-tropomyosin models determined in this work are discussed.
Introduction
Tropomyosin, a 40-nm-long coiled-coil, polymerizes end-to-end to form continuous superhelical strands that associate symmetrically alongside each of the helical chains of actin-based thin filaments (1–3). Tropomyosin itself is a modular protein consisting of seven tandem quasi-repeating units designed to bind to seven successive 55 Å long actin monomers over a 385 Å path of the thin filament (1–3). The azimuthal location of tropomyosin on the surface of actin governs the access and hence the interactions and activity of numerous actin-binding proteins in muscle and nonmuscle thin filaments (4–12). Relocation of tropomyosin brought about by other binding partners can further influence thin filament behavior (6–12). In the case of striated muscle thin filaments at low Ca2+, the troponin complex, lying adjacent to elongated tropomyosin, functions to constrain tropomyosin above myosin target sites on actin (6–10). This steric blocking by tropomyosin limits myosin binding and brings about muscle relaxation because myosin crossbridge cycling on actin and consequently contraction are inhibited. In contrast, Ca2+ binding to troponin after muscle activation leads to a ∼25 Å azimuthal movement of tropomyosin away from the blocking site, which partially relieves the structural restriction imposed by tropomyosin (6–10). In fact, the equilibrium position occupied by tropomyosin on actin in the absence of troponin or other constraints is, on average, very close to the position brought about by Ca2+-saturated troponin (13). In this C-state position, the myosin-binding site on actin is still partly closed. Now free of the constraints imposed by troponin, the tropomyosin strand oscillates on thin filaments by ∼10 Å, as if it were vibrating azimuthally over a locally flat energy landscape on actin. Such azimuthal displacements uncover the entire myosin-binding site and thus allow crossbridge cycling and contraction (10,14). The extent to which the initial weak binding of myosin plays a role in promoting movement of tropomyosin from the closed to the fully open position on actin is not completely understood. Moreover, it remains to be determined whether Ca2+ activation itself leads to a cooperative opening of the myosin-binding site (15) or, alternatively, occurs stepwise between discrete conformational states (closed and open) (10,16,17).
Tropomyosin's global shape and flexibility play key roles in the assembly, maintenance, and regulatory switching of thin filaments. Electron microscopy (EM) images and molecular-dynamics (MD) simulations of single tropomyosin molecules yield conformations of tropomyosin that closely resemble each other. Overall, isolated tropomyosin has a semirigid curved configuration with a design that is well matched to its superhelical shape on F-actin (18). Tropomyosin bends smoothly but anisotropically about its distinctive helically curved conformation, without signs of unfolding, chain separation, localized kinks, or joints (18). Taken together, these results are in accord with the view (19) that tropomyosin binds to thin filaments because of a helical preshaping that matches it to the contours of F-actin, an entropic advantage that is not satisfied by induced-fitting mechanisms. The relatively high stiffness of the tropomyosin strand also suggests that local azimuthal movements of tropomyosin will be propagated longitudinally along actin filaments. Indeed, the dynamic persistence length of tropomyosin (423 nm) is roughly one-half the length of the thin filament (18). It follows that local perturbation in tropomyosin's position caused by troponin, myosin, or other factors will be transmitted distally, which is consistent with the prevailing view that the tropomyosin switching system is highly cooperative (20). Nevertheless, although single-molecule studies have been quite informative, a complete elucidation of tropomyosin-based regulatory mechanisms requires more than an in-depth description of tropomyosin behavior and structure in isolation. A complete representation of the atomic structure and mechanical properties of the tropomyosin molecule linked to its biological substrate, F-actin, is needed, and thus is investigated here.
Both EM reconstruction and fiber diffraction studies of reconstituted F-actin-tropomyosin have revealed the azimuthal position of the end-to-end linked polymeric tropomyosin strands on the surface of actin (13,14); however, the longitudinal (z)-positioning of tropomyosin molecules on actin subunits along the axial length of the filament has not yet been determined. Moreover, the surface of tropomyosin that has been proposed to contact F-actin has not been verified (2,18). Thus, the manner in which the quasi-repeats of tropomyosin interact with actin subunits is uncertain. Given these deficiencies, all-atom models of F-actin-tropomyosin describing intermolecular amino acid contacts cannot be generated. In this work, we overcome the limitations of previous studies and describe the construction of a complete molecular model of actin-tropomyosin based on computational chemistry and EM reconstruction.
It is well known that the association of tropomyosin on actin is largely electrostatic (2,21). We carried out a full computational exploration of the electrostatic interaction by sampling multiple test z-positions of tropomyosin on F-actin while optimizing the tropomyosin rotation and azimuthal position. This computational approach is similar to that described by Lorenz et al. (21) but is based on improved atomic structures for F-actin and tropomyosin (18,22) and more recently developed programs (23,24). It defines an optimal z-location of tropomyosin on F-actin and yields sensible protein-protein contacts. The positioning of tropomyosin on F-actin agrees completely with a solution that is determined separately after fitting the atomic structures of F-actin and tropomyosin into EM reconstructions of F-actin-tropomyosin. In fact, the F-actin-tropomyosin structure generated by this dual approach of in silico computational chemistry and EM reconstruction identifies amino acid contacts between actin and tropomyosin that are remarkably similar to those first inferred by Brown and Cohen (2).
The atomic model that we propose here can serve as a reference location for characterizing tropomyosin regulatory movements on thin filaments, and offers a means of determining the stiffness of tropomyosin when bound to F-actin. Moreover, the map of F-actin-tropomyosin provides a structural platform for assessing mutations that affect actin-tropomyosin behavior and developing tropomyosin-mimicking peptide drugs designed to modulate actin-myosin or other interactions.
Materials and Methods
EM, image reconstruction, and docking of atomic structures into reconstruction volumes
Thin filaments were prepared by mixing a twofold molar excess of tropomyosin (6 μM) with F-actin (20 μM) to optimize the binding of the proteins to each other in a solution containing 100 mM NaCl, 3 mM MgCl2, 1 mM NaN3, 0.2 mM EGTA, 1 mM dithiothreitol, 5 mM sodium phosphate/5 mM Pipes buffer (pH 7.0) at 25°C (25,26). The mixture was diluted 20-fold in the same buffer, applied to carbon-coated grids, and negatively stained with 1% uranyl acetate (25,26). Micrographs were recorded on a Philips CM120 electron microscope (FEI, Hillsboro, OR) at a magnification of ×60,000 under low-dose conditions (∼12 e−/Å) by digitization with a 2K×2K F224HD slow-scan CCD camera (set at 3.6 Å/pixel; TVIPS, Gauting, Germany). Helical (27) and real-space (28) reconstructions were performed with the use of standard methods as previously described (25,26), yielding maps with ∼25 Å resolution. The image processing was carried out on filaments from five different preparations of F-actin-tropomyosin. Individual data sets yielded reconstructions that were indistinguishable from each other, and therefore all data were averaged together. The statistical reliability of densities in reconstructions was assessed by a Student's t-test (29,30). To isolate densities derived from tropomyosin molecules from those of F-actin, coordinates of F-actin reconstructions (13) (lacking tropomyosin) were aligned to those of F-actin-tropomyosin and the volume of one was then subtracted from the other. The Fit in Map tool in Chimera (31) version 1.3 was used to dock atomic models into the EM reconstruction volumes.
Computational chemistry
Building an initial reference structure
We developed a computational procedure to model the axial alignment of tropomyosin on F-actin by assessing the total electrostatic interaction energy for different test configurations of the two structures. To that end, we first had to build an initial F-actin-tropomyosin reference structure. To start, the atomic model of αα-rabbit striated muscle tropomyosin with a curvature matching that of the actin helix (18) was placed over seven monomers on one side of a 21 subunit actin filament model generated from the Oda rabbit F-actin structure (22) (PDB ID: 2ZWH; 21 subunits were chosen to obtain a filament length greater than that traversed by tropomyosin). Note that placing two tropomyosin molecules on opposite sides of the filament (to mimic native filaments) and then determining the optimal position for both would have been redundant and increased computational time. The known polarity of tropomyosin on F-actin (2) (i.e., with the N-terminus oriented toward the pointed end of F-actin) was chosen. The azimuthal position of tropomyosin on F-actin was matched to that in the EM reconstruction of F-actin-tropomyosin with the use of Chimera fitting tools (31). The radial position of tropomyosin was fixed at a distance of 43 Å from its own coiled-coil center to the central axis of the F-actin filament to correspond to the measured distance in the EM reconstruction. Thus, the initial reference position of tropomyosin was partially constrained by the geometry of the EM reconstruction, even though the longitudinal z-position of tropomyosin along the F-actin was set at an arbitrary starting point.
Coarse-grained search
A coarse-grained search was performed in which the electrostatic interaction energy between the F-actin and tropomyosin interface was determined for a sample of test structures, each representing a different tropomyosin conformation on F-actin. The computed electrostatic interaction energy terms between tropomyosin and F-actin that were used as criteria to assess the optimal position and conformation of tropomyosin were measured with utilities in CHARMM version C33b2 (23,24), and other interaction terms were not used in the search. Water was represented implicitly by the generalized Born solvation model by implementing the CHARMM GBSW protocol (32,33) at a salt concentration of 150 mM. During the search, tropomyosin was repositioned over different z-positions along the seven adjacent actin monomers while being pseudo-rotated (defined below) about its own axis and shifted azimuthally using a grid array of 21 × 19 × 11 (thus yielding 4389 different conformations). The matrix set up for this grid search covered z-positions extending over ±27.5 Å in 2.75 Å increments, an azimuthal range of ±5° in 1.0° increments, all coupled with a pseudo-rotational search of ±90° in 10° increments. Thus, tropomyosin was tested locally around the default azimuthal (closed) position on filaments but fully in the z-direction, so that each tropomyosin repeat domain was assessed over the entire length of a corresponding actin monomer and with a full set of tropomyosin rotations. Note that attempts to vary the radial distance of tropomyosin from the filament central axis had little effect on the optimal tropomyosin z-positioning, and the radius was held at 43 Å. The structure of F-actin was kept unchanged during the whole search procedure. The search was carried out on a Lenovo Think Station D10 (Morrisville, NC) and took 2–3 weeks to complete.
Pseudo-rotation of tropomyosin
Rotating superhelical tropomyosin as a rigid body about its axis diminishes its contact with the surface of F-actin. Therefore, we generated different pseudo-rotation conformers by first straightening the tropomyosin coiled-coil, then rotating the straightened tropomyosin incrementally about its z-axis, and finally rewrapping and energy-minimizing the rotated molecule superhelically around F-actin to again precisely fit to the helical shape of F-actin, as described by Lorenz et al. (21). (In effect, the coil-coil core of each pseudo-rotated tropomyosin is superimposable on the initial one.)
Fine-grained search
The F-actin-tropomyosin conformation with the most favorable electrostatic interaction terms determined by the above coarse-grained search was used as a new reference structure to refine the optimal tropomyosin position on F-actin, using the same strategy as above. The matrix setup for this grid search covered z-positions over ±5 Å in 1 Å increments, an azimuthal range of ±3° in 1.0° increments, and a pseudo-rotational search of ±10° in 1° increments.
Results and Discussion
3D reconstruction of F-actin-tropomyosin
Thin filaments were reconstituted from F-actin and cardiac αα-tropomyosin under conditions known to saturate the filaments with tropomyosin (25,26). They were then negatively stained and EM was carried out (25,26). (The effect of troponin was not investigated.) Although EM images of the reconstituted thin filaments show characteristic double-helical arrays of actin monomers and tropomyosin strands (Fig. 1), a detailed picture of tropomyosin strands on filaments only becomes evident in helical or single-particle reconstructions (Fig. 2 a). As found previously (13,25), the longitudinally continuous tropomyosin strands seen in reconstructions are well defined and follow the path of the long-pitch actin helix along the junctions between the inner and outer domains on successive actin subunits. Tropomyosin approaches actin monomers most closely over the upper parts of actin subdomains 1 and 3, where the actin monomers show the greatest radial extension and appear to be the most bulbous. In contrast, a distinct separation between the surfaces of actin and tropomyosin is noted over subdomains 2 and 4 and the lower part of subdomains 1 and 3, where the surface of actin is the shallowest. Thus, tropomyosin appears to be attracted the most to one region on actin monomers while bridging over the rest, thereby connecting successive actin monomers along filaments. On average, the center of tropomyosin lies at a 43 Å radius from the center of the thin filament. This value is ∼2% greater than expected from measurements on reconstructions of F-actin-tropomyosin-troponin, where troponin may hold tropomyosin slightly closer to the surface of actin (14).
Figure 1.

Electron micrographs of negatively stained F-actin-tropomyosin. Note the obliquely oriented tropomyosin strands (arrows). Filaments are shown with their pointed ends facing up; polarity was determined with alignment tools as previously described (27). Scale bar: 50 nm.
Figure 2.
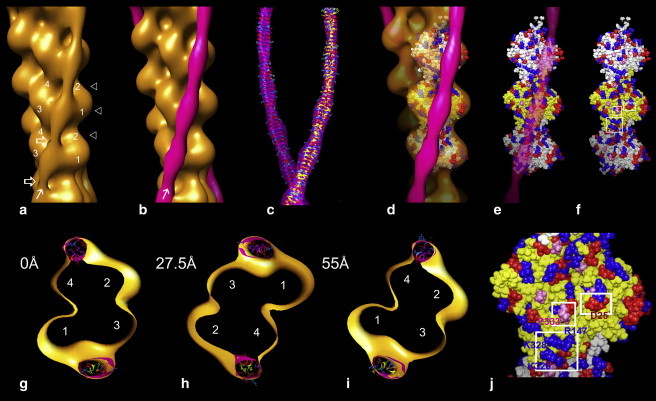
Fitting the atomic model of F-actin into the F-actin-tropomyosin reconstruction. (a) F-actin-tropomyosin reconstruction. Actin subdomains are marked on two actin subunits. An arrow indicates the tropomyosin strand (and points toward the pointed end of the filament), and the open white arrows point to locations where the tropomyosin strand coils appear to cross over each other. (b) Tropomyosin difference density (magenta) superposed on the F-actin-tropomyosin reconstruction. (c) Tropomyosin model fitted in tropomyosin difference density. (d) Three actin subunits from the Oda atomic model of F-actin (22) fitted into the reconstruction in b, with the actin part of reconstruction now made transparent. (e) Atomic model of actin as in d, with just the superposed tropomyosin difference density made transparent. (f) Atomic model of actin alone. Amino acid residues that appear likely to contact tropomyosin are shown enclosed in white rectangles on the middle actin subunit; the bottom rectangle includes basic residues Lys-326, Lys-328, Arg-147, and the two others include Pro-333, Asp-25, and Arg-28. Basic amino acids in the Oda model are painted blue, acidic amino acids are red, and proline is pink. The pointed end of actin is facing up in each panel. (j) Enlargement of the actin subunit containing the white rectangles shown in f; residues noted above are labeled. (g–i) Three transverse sections of the reconstruction in panel a (indicated by triangles in a). The sections are viewed from the barbed end upward toward the pointed end of the filament with each section separated from the previous one by 27.5 Å (i.e., traversing 55 Å and equaling the length of actin monomers on either side of the filament); actin subdomains are numbered in each section. Tropomyosin on each side of these filament sections is fitted with a coiled-coil tropomyosin model (18), thus filling the elliptically shaped densities. Sectioned tropomyosin difference densities are superposed.
The tropomyosin density seen in reconstructions of F-actin-tropomyosin is not uniform; rather, it appears to broaden and then taper over each successive actin monomer along filaments, suggestive of the wide and narrow faces of coiled-coil tropomyosin noted by McLachlan and Stewart (34) when viewing models of the coiled-coil face-on. Looking at the F-actin-tropomyosin reconstruction face-on, tropomyosin appears to be particularly wide over the bulbous part of actin subdomains 1 and 3 and narrower elsewhere. This attribute is statistically significant at >99.99% confidence levels and also becomes obvious in images of tropomyosin derived by aligning and then subtracting reconstructions of F-actin from F-actin-tropomyosin maps. These resulting tropomyosin difference densities clearly superpose on the corresponding density in the F-actin-tropomyosin reconstruction (Fig. 2, b and c). The widening and tapering observed is likely to be due to the different viewing angles recorded for the twisting tropomyosin coiled-coil on the actin surface. Inspecting a surface projection of an isolated double helix would in effect be comparable. In cross-section, the density attributed to tropomyosin is in fact slightly elliptical, and the ellipse appears to make a full rotation over each actin subunit (Fig. 2, g–i) as expected and as previously observed in corresponding reconstructions (35).
Fitting atomic models of F-actin (22) uniquely into the volume described by the actin component of the reconstruction shows that tropomyosin lies over a set of basic amino acids (Lys-326, Lys-328, and Arg-147) near the base of the actin monomers where subdomains 1 and 3 intersect (Fig. 2, d–f, and j). These basic amino acids are clustered next to the path of tropomyosin and appear poised to clasp onto tropomyosin. Tropomyosin is also found near a group of hydrophobic amino acids centered on actin residue Pro-333, which lies at a relatively high radius from the center of the thin filament, and together with acidic Asp-25 may form a similar molecular clasp (Fig. 2, d–f, and j). However, since the z-position of tropomyosin is not evident, contacts between tropomyosin and F-actin cannot be explicated on the basis of the above evidence alone.
Computational search to identify the optimal z-position of tropomyosin on F-actin
The likely optimal z-location of tropomyosin on the surface of F-actin was established based on scores from an in silico search of electrostatic interaction terms of a large number of possible F-actin-tropomyosin configurations. (As described in the Materials and Methods section, the coarse- and fine-grained grid searches covered ∼6000 different combinations of azimuthal shifts, longitudinal positions, and pseudo-rotations of tropomyosin on F-actin, with tropomyosin fixed at a 43 Å radius.) The search indicates that tropomyosin falls into a discrete electrostatic minimum (Fig. 3). This minimum defines an optimized conformation in which tropomyosin makes meaningful contacts with F-actin, just as tropomyosin appears to do in the EM reconstruction (Fig. 4). In the favored conformation, basic amino acids on the surface of F-actin clustered around residues 147, 326, and 328 (mentioned above) are centered on a corresponding patch of acidic residues on each of the tropomyosin quasi-repeating modules (Table 1, Fig. 4, and Supporting Material). These are the same amino acids that Brown et al. (36) and Li et al. (18) suggested form interfacial contacts with F-actin. Cross-sectional views of F-actin-tropomyosin through this region of electrostatic interaction show the two α-helices of the sliced tropomyosin coiled-coil transverse to the surface of actin and in line with the long axis of the corresponding elliptical section taken from EM data. Such an arrangement is likely to minimize the distance and hence maximize the contact between the side chains of oppositely charged amino acids (Fig. 5 g highlights this pattern at the level of tropomyosin quasi-repeat 5). In addition to these charge-charge interactions, negatively charged Asp-25 on actin flanks basic residues on one edge of each of the tropomyosin quasi-repeats (Fig. 5 d). The largest number of favorable electrostatic interactions is found to occur on quasi-repeating modules 4 and 5 of tropomyosin (Table 1). Indeed, these two modules are thought to contribute most strongly to actin binding (2,3).
Figure 3.
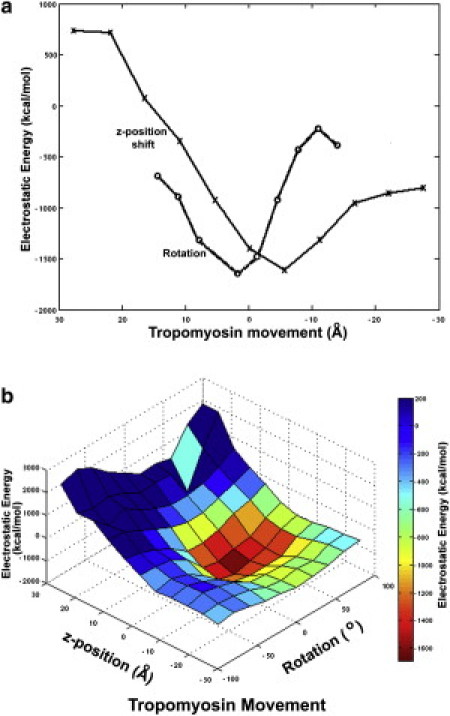
Evaluating electrostatic interactions between tropomyosin and F-actin. (a) Change in the electrostatic interaction energy values for tropomyosin repositioned on F-actin in the z-direction (plot marked by x) starting from the initial reference structure (0 point on the x axis of the plot), in this case holding the rotation and azimuth of tropomyosin fixed, and for tropomyosin rotated on F-actin (plot marked by o), in this case holding the z-position and azimuth constant. Positive values for the z-position change indicate longitudinal shifting of tropomyosin toward the pointed end of F-actin, and positive values for rotation indicate a turning of tropomyosin toward the open position on F-actin. Note the strong minima in each of the plots. The z-position and rotational shifts plotted in panel a represent 2D sections through the origin in the 3D plot in panel b. (b) A plot of the electrostatic energy landscape reflecting tropomyosin movement on F-actin as a function of both z-position and pseudo-rotation (0,0 reflects the z-position and rotation of tropomyosin on F-actin in the initial reference structure). Again note the energy minima.
Figure 4.
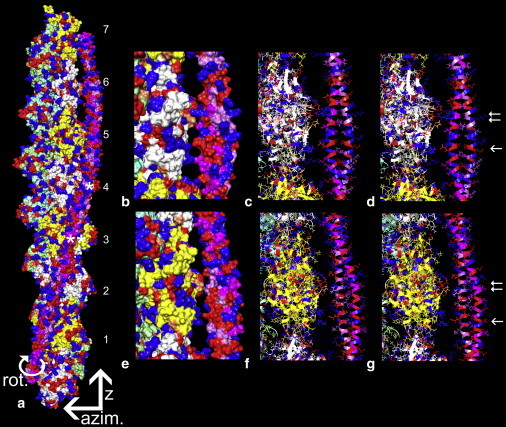
Optimal electrostatic interactions between F-actin and tropomyosin. (a) Surface view showing the location of tropomyosin on F-actin that optimizes electrostatic interactions between the two structures. The rotation, azimuthal, and z directions discussed in the text are indicated by arrows. The pointed end of F-actin is facing up (as the arrow for z indicates) and actin subunits are numbered as in Table 1, tropomyosin quasi-repeat 4 is marked with a single asterisk and quasi-repeat 5 with a double asterisk (note that, by convention, the numbering of actin subunits and tropomyosin repeats do not match). (b and e) Segments of the F-actin-tropomyosin model in panel a are enlarged and rotated to highlight potential interactions of tropomyosin quasi-repeats 4 (b) and quasi-repeat 5 (e) with actin subunits. (c and f) The same as b and e but in wire/ribbon format. (d and g) The corresponding region of F-actin as in c and f, but here displaying the alignment of F-actin and tropomyosin models that were fitted into the F-actin-tropomyosin reconstruction (note that the relative positions of interacting amino acids correspond closely to those in c and f). The single arrow in g points to Lys-326, Arg-147, and Lys-328 residues of actin subunit 3 approaching Glu-181 on tropomyosin quasi-repeat 5, and the single arrow in d points to Arg-147 and Lys-328 of actin subunit 4 approaching residue Glu-139 on tropomyosin repeat 4. The double arrows point to actin Pro-333 on actin subunits 3 and 4 approaching Val-170 and Ser-132 on tropomyosin repeats 5 and 4, as well as to actin Asp-25 and Arg-28 close to Arg-167 and Glu-163 on tropomyosin repeat 5, and Lys-128, Arg-125, and Asp-121 on repeat 4. Each of the other tropomyosin quasi-repeats shows a similar but not identical pattern of interactions with respective actin subunits (not displayed; see Table 1). Basic amino acid residues in actin and tropomyosin are painted blue, acidic amino acids are red, and proline is coral. An identical grid search was performed to determine the optimal electrostatic interaction for tropomyosin with the recently published F-actin structure proposed by the Namba Laboratory (41). The z-position determined for tropomyosin on this model was the same as that found for the Oda model (22).
Table 1.
List of tropomyosin and actin residues that closely contact each other
| Actin subunit | Tropomyosin quasi-repeat | Residue pairs in close contact |
Distance between residues (Å) |
|||
|---|---|---|---|---|---|---|
| MD simulation |
||||||
| Actin | Tropomyosin | Electrostatic search | Initial values (at 100 ps) | Average values (over 30 ns) | ||
| 1 | 7 | Arg-147 | Asp-258 | 6.0 | 4.2 | 4.3 |
| Lys-328 | Asp-258 | – | 6.6 | – | ||
| Asp-25 | Arg-244 | 1.8 | 2.3 | 3.3 | ||
| 2 | 6 | Lys-326 | Glu-223 | 3.1 | 2.7 | 4.5 |
| Arg-147 | Asp-219 | – | – | 6.6 | ||
| Lys-328 | Glu-223 | – | – | 3.3 | ||
| Lys-328 | Asp-219 | – | – | 3.7 | ||
| Pro-333 | Ala-209 | 6.8 | 6.3 | – | ||
| Asp-25 | Lys-205 | 3.1 | 3.0 | – | ||
| 3 | 5 | Lys-326 | Glu-181 | 5.6 | 3.0 | 6.5 |
| Arg-147 | Glu-181 | 4.3 | – | 5.4 | ||
| Lys-328 | Glu-181 | 5.0 | 3.4 | 4.2 | ||
| Lys-328 | Glu-184 | – | – | 4.3 | ||
| Pro-333 | Val-170 | 5.4 | 4.0 | 4.8 | ||
| Asp-25 | Arg-167 | 3.2 | 6.7 | 5.3 | ||
| Arg-28 | Glu-163 | 6.4 | 5.7 | – | ||
| 4 | 4 | Lys-326 | Glu-142 | – | – | 3.1 |
| Arg-147 | Glu-139 | 4.1 | 6.0 | 2.0 | ||
| Lys-328 | Glu-139 | 4.2 | 3.0 | 4.9 | ||
| Pro-333 | Ser-132 | 5.6 | 6.3 | 3.7 | ||
| Glu-334 | Lys-128 | 5.2 | 4.8 | 5.0 | ||
| Asp-25 | Lys-128 | 6.2 | 5.3 | – | ||
| Asp-25 | Arg-125 | 1.8 | 2.5 | 4.0 | ||
| Arg-28 | Asp-121 | 5.1 | 5.9 | – | ||
| 5 | 3 | Lys-326 | Glu-104 | 4.3 | 3.8 | 3.5 |
| Lys-328 | Glu-104 | – | – | 3.9 | ||
| Lys-328 | Asp-100 | – | – | 4.2 | ||
| Arg-147 | Glu-97 | 4.1 | 1.6 | 2.0 | ||
| Asp-25 | Arg-90 | – | – | 3.0 | ||
| Arg-28 | Glu-82 | – | 6.3 | – | ||
| 6 | 2 | Arg-147 | Glu-62 | 5.4 | 5.0 | 2.3 |
| Arg-147 | Asp-58 | – | – | 6.7 | ||
| Lys-328 | Asp-58 | – | – | 5.3 | ||
| Lys-328 | Glu-62 | – | – | 6.0 | ||
| Asp-25 | Lys-48 | – | – | 5.7 | ||
| Arg-28 | Asp-41 | 4.6 | 3.1 | 5.1 | ||
| 7 |
1 |
Lys-326 | Asp-20 | – | 4.3 | – |
| Arg-147 | Asp-20 | 6.2 | 4.7 | 6.3 | ||
| Arg-147 | Glu-16 | 6.6 | 3.7 | – | ||
| Lys-328 | Asp-20 | 6.9 | 3.7 | – | ||
| Asp-25 | Lys-6 | 4.6 | 1.8 | – | ||
| Asp-25 | Gln-9 | – | – | 4.6 | ||
| Residues approaching each other by ≤7.0 Å | n = 26 | n = 28 | n = 31 | |||
| Mean distances between residue pairs (± SD) | 4.8 ± 1.4 Å | 4.3 ± 1.5 Å | 4.4 ± 1.3 Å | |||
Distances were measured (31) between the carboxyl and amino ends of closely spaced actin and tropomyosin residues after the electrostatic grid search, at the beginning of a MD simulation at 100 ps, and for the these same distances averaged over a 30 ns MD run. Any distance ≤ 7 Å is noted. Distances between side chains of Pro-333 on actin that align close to hydrophobic residues on tropomyosin are also indicated. Note that the numbering of actin subunits is for one helical chain of F-actin starting, by convention, with the barbed end, whereas the corresponding numbering of the tropomyosin quasi-repeats, having an antiparallel sequence arrangement to F-actin, begins with the N-terminus of the molecule, at the pointed end of F-actin. Also note that MD reduced the tropomyosin radius from the thin filament central axis to 41.7 Å.
Figure 5.
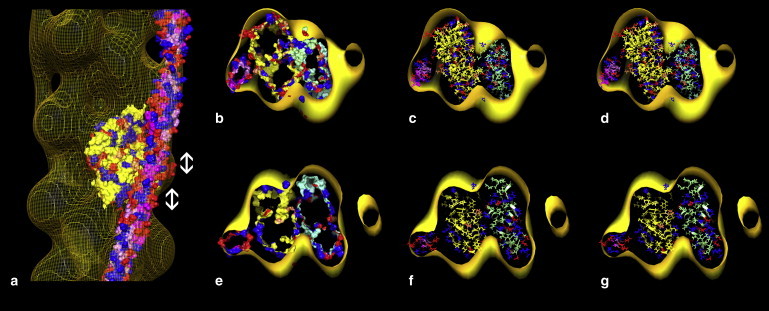
Relative location of tropomyosin and actin in the EM reconstruction of F-actin-tropomyosin. (a) Surface view of the reconstruction shown in yellow wire mesh to facilitate inspection of docked proteins. Here, both tropomyosin and F-actin models were fitted separately into the reconstruction using Chimera fitting tools (the atomic structure of just one of the docked actin subunits is displayed). (b–g) Cross-sections of actin viewed from the barbed end of F-actin (here, a was rotated by ∼180° before sectioning). Two cross-sections through regions of actin subunit 3 and tropomyosin repeat 5 are indicated by double-ended arrows in panel a and shown in b, c, e, and f. The outer boundaries of the docked actin and tropomyosin are shown as isosurfaces in b and e, and in more detail in wire/ribbon format in c and f. (b and c) Val-170, Arg-167, and Glu-163 on tropomyosin are seen oriented toward Pro-333, Asp-25, and Arg-28 on actin. (e and f) Glu-181 on tropomyosin is oriented toward Lys-326, Lys-328, and Arg-147 on actin. Note the perpendicular orientation of the tropomyosin elliptical axis with respect to the charged surface of actin. (This same arrangement is also seen over corresponding sections of the other tropomyosin quasi-repeats.) (d and g) The identical region of maps as in c and f, but here displaying the optimal alignment of F-actin and tropomyosin based on electrostatics, as in Fig. 4. Color-coding of amino acids is the same as in Fig. 4.
Pro-333 on actin (discussed by Brown et al. (36)) locates near hydrophobic residues on the surface of tropomyosin quasi-repeats 4 and 5 (Table 1, Figs. 4 and 5), likely contributing to binding strength. It is noteworthy that these nonpolar contacts between actin and tropomyosin became evident even though the search criterion relied solely on electrostatics (and not on hydrophobic interactions).
During the first 100 ps of an ongoing MD run in explicit water (NAMD version 2.6 (37)), the z-position of tropomyosin on actin remains constant, but the tropomyosin, no longer fixed at a 43 Å radius, moves incrementally closer to the surface of F-actin at a 41.7 Å radius. On average, the neighboring side chains mentioned above approach each other more closely by 0.5 Å (Table 1). This slightly closer distance between interacting side chains is maintained over a 30 ns MD run, as are the relative positions of amino acid residues on actin and tropomyosin (Table 1).
Validation of the computational chemistry
As a test of the accuracy of the top solution computed in the above grid search, the tropomyosin model, now with refined pseudo-rotation terms, was fitted into its corresponding volume in the EM reconstruction of F-actin-tropomyosin just as the Oda model (22) was in Fig. 1 above. Thus, tropomyosin (whose structure when not bound to F-actin is known from several crystal structures and further explicated here and elsewhere (18)) was docked to fit to the tropomyosin difference density shell (Fig. 2 c) as well as possible, and with minimal distortion of the corresponding atomic model, simply by gently bending the latter during pseudo-rotation into a superhelix with the same helical shape and periodicities as the actin filament. The fact that the slightly elliptical cross-section of the tropomyosin coiled-coil is exaggerated at one level in the EM map and somewhat diminished at another (Figs. 2, g–i) significantly restricts the range of possible z-positions and angular orientations of the fitting. The optimal docking arrangement is credible because of the good agreement with results of the electrostatic grid search and the MD results described below.
During the fitting, the tropomyosin model was provided the freedom to dock onto its optimal azimuthal and z-locations based on its match to the isosurface contours of the reconstruction. It is the wide and narrow interfaces on the superhelical tropomyosin model that lock the structure into corresponding features of the EM volume. In this way, tropomyosin fitted in a unique position on F-actin solely on the basis of its geometrical features (without any regard to electrostatic contacts; Fig. 5 a), just as F-actin fitted into the EM volumes described above (Figs. 2 and 5). In carrying out this procedure, we found that the tropomyosin model located to an identical position within the EM reconstruction volume at all thresholds tested, ranging from 3σ to 12.5σ above the mean density of the reconstruction, and regardless of whether the model was fitted as an atomic structure or as one with computationally reduced resolution (e.g., to 25 Å).
Conspicuously, the relative axial position of the docked tropomyosin and F-actin models is virtually identical to that observed for the computational solution based solely on optimization of electrostatic interactions (Fig. 5 a), even though, in the case of the EM data fitting, we relied on completely different alignment criteria. Again, opposing charged groups and hydrophobic patches on the docked actin and tropomyosin models are as described above (Figs. 4, d and g, and 5, a–c, e, and f), and again the orientation of the wide and narrow faces of tropomyosin on actin is also the same as above, where oppositely charged groups approach each other at an optimally close distance (Fig. 4, b–g). This convergence of results despite the use of completely different alignment criteria cross-validates the approaches and is a powerful indicator of the correctness of the two solutions. The possibility that the distinctive shape of the tropomyosin density seen in reconstructions could be an artifact of the limited resolution of the F-actin-tropomyosin maps, or due to variable amounts of stain between tropomyosin and different actin subdomains, seems unlikely given the near identity of the solutions derived from the in silico work and the EM docking studies.
Conclusions
Our modeling of the interface between F-actin and tropomyosin strongly suggests that tropomyosin, when freed of constraints imposed by troponin, myosin, or other actin-binding proteins, localizes to a favored position on F-actin defined primarily by electrostatic interactions. We have indicated here that the modular structure of tropomyosin provides sufficient sequence specificity to ensure that such interactions occur over each successive actin subunit along the thin filament. Thus, our findings agree with the results of many previous biochemical and structural studies suggesting that tropomyosin is held onto F-actin primarily by electrostatic interactions (2,21,38). Our results are also consistent with other work (2,3) showing that the seven tropomyosin quasi-repeating modules make nonequivalent binding contacts with successive actin subunits along F-actin. As reviewed elsewhere (2,3), this binding variation may reflect a level of conformational specialization that allows tropomyosin to associate with both F-actin and other actin-binding partners while at the same time being able to form end-to-end links to build a tropomyosin polymer. The default favored position of tropomyosin on F-actin found here no doubt represents a conformation that in some cases blocks the binding of other actin-binding proteins. In striated muscle, this position coincides with or is close to the average position of tropomyosin on actin in the closed C-state. Without further information, how troponin, myosin S1, and other actin-binding proteins affect tropomyosin and thin filament structure cannot be easily predicted from the F-actin-tropomyosin model presented here. Although our computational search indicates that tropomyosin occupies an optimal z-position and rotation in the surrounds of the closed C-state position, no specific electrostatic minima were noted when tropomyosin was shifted computationally toward the open or blocked states.
Typically, electrostatic interactions confer specificity to protein-protein associations, and nonpolar interactions provide binding strength. Thus, the paucity of nonpolar interactions and the lack of stereospecific lock-and-key linkage between tropomyosin and actin result in a weak structural association. Preshaping of tropomyosin to contours of F-actin decreases the entropic expense of the association, but any loss of conformational entropy will also tend to weaken filament assembly. In fact, tropomyosin only binds to F-actin effectively as a narrow filament once it has polymerized end-to-end (19). Although polymer formation increases the collective binding strength of tropomyosin along F-actin, interaction of tropomyosin with individual actin subunits at a local level is still very weak (19). This dichotomy (strong overall binding but weak local interactions) allows tropomyosin to be readily perturbed by troponin, myosin, or other actin-binding proteins to initiate regulatory switching with a low energy cost. Cooperative signaling, propagating the movement of tropomyosin away from its favored associations, follows because tropomyosin is semirigid (18).
Recent structural studies of the overlap domain connecting the N- and C-terminal ends of adjacent tropomyosin molecules suggest that the chains of the tropomyosin coiled-coil splay open at the C-terminus of one molecule to envelop a more compact coiled-coil on the neighbor's N-terminus, thus forming a four-helix nexus (39,40). Consistent with this notion, our F-actin-tropomyosin model indicates that the ends of tropomyosin localize to a common site over actin subdomains on every seventh actin subunit along the actin filament. In this arrangement, tropomyosin is well positioned to form short four-helix connections and polymerize into extended strands. Future studies to fit the overlap structure into our model are planned.
Acknowledgments
This work was supported by grants from the National Institutes of Health to W.L. (HL86655 and HL36153), L.S.T. (HL38834 and HL63774), and R.C. (AR34711).
Supporting Material
References
- 1.Perry S.V. What is the role of tropomyosin in the regulation of muscle contraction? J. Muscle Res. Cell Motil. 2003;24:593–596. doi: 10.1023/b:jure.0000009811.95652.68. [DOI] [PubMed] [Google Scholar]
- 2.Brown J.H., Cohen C. Regulation of muscle contraction by tropomyosin and troponin: how structure illuminates function. Adv. Protein Chem. 2005;71:121–159. doi: 10.1016/S0065-3233(04)71004-9. [DOI] [PubMed] [Google Scholar]
- 3.Hitchcock-DeGregori S.E. Tropomyosin: function follows structure. Adv. Exp. Med. Biol. 2008;644:60–72. doi: 10.1007/978-0-387-85766-4_5. [DOI] [PubMed] [Google Scholar]
- 4.Gunning P.W., Schevzov G., Hardeman E.C. Tropomyosin isoforms: divining rods for actin cytoskeleton function. Trends Cell Biol. 2005;15:333–341. doi: 10.1016/j.tcb.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Gunning P.W., O'Neill G., Hardeman E. Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol. Rev. 2008;88:1–35. doi: 10.1152/physrev.00001.2007. [DOI] [PubMed] [Google Scholar]
- 6.Haselgrove J.C. X-ray evidence for a conformational change in actin-containing filaments of vertebrate striated muscle. Cold Spring Harb. Symp. Quant. Biol. 1972;37:341–352. [Google Scholar]
- 7.Huxley H.E. Structural changes in actin- and myosin-containing filaments during contraction. Cold Spring Harb. Symp. Quant. Biol. 1972;37:361–376. [Google Scholar]
- 8.Parry D.A.D., Squire J.M. Structural role of tropomyosin in muscle regulation: analysis of the x-ray diffraction patterns from relaxed and contracting muscles. J. Mol. Biol. 1973;75:33–55. doi: 10.1016/0022-2836(73)90527-5. [DOI] [PubMed] [Google Scholar]
- 9.Lehman W., Craig R., Vibert P. Ca2+-induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature. 1994;368:65–67. doi: 10.1038/368065a0. [DOI] [PubMed] [Google Scholar]
- 10.Vibert P., Craig R., Lehman W. Steric-model for activation of muscle thin filaments. J. Mol. Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 11.Hodgkinson J.L., EL-Mezgueldi M., Lehman W. 3-D image reconstruction of reconstituted smooth muscle thin filaments containing calponin: visualization of interactions between F-actin and calponin. J. Mol. Biol. 1997;273:150–159. doi: 10.1006/jmbi.1997.1307. [DOI] [PubMed] [Google Scholar]
- 12.Hodgkinson J.L., Marston S.B., Lehman W. Three-dimensional image reconstruction of reconstituted smooth muscle thin filaments: effects of caldesmon. Biophys. J. 1997;72:2398–2404. doi: 10.1016/S0006-3495(97)78885-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehman W., Galińska-Rakoczy A., Craig R. Structural basis for the activation of muscle contraction by troponin and tropomyosin. J. Mol. Biol. 2009;388:673–681. doi: 10.1016/j.jmb.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poole K.J., Lorenz M., Holmes K.C. A comparison of muscle thin filament models obtained from electron microscopy reconstructions and low-angle X-ray fibre diagrams from non-overlap muscle. J. Struct. Biol. 2006;155:273–284. doi: 10.1016/j.jsb.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y.-B., Irving M. The molecular basis of the steep force-calcium relation in heart muscle. J. Mol. Cell. Cardiol. 2010;48:859–865. doi: 10.1016/j.yjmcc.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKillop D.F.A., Geeves M.A. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tobacman L.S., Butters C.A. A new model of cooperative myosin-thin filament binding. J. Biol. Chem. 2000;275:27587–27593. doi: 10.1074/jbc.M003648200. [DOI] [PubMed] [Google Scholar]
- 18.Li X.E., Holmes K.C., Fischer S. The shape and flexibility of tropomyosin coiled coils: implications for actin filament assembly and regulation. J. Mol. Biol. 2010;395:327–339. doi: 10.1016/j.jmb.2009.10.060. [DOI] [PubMed] [Google Scholar]
- 19.Holmes K.C., Lehman W. Gestalt-binding of tropomyosin to actin filaments. J. Muscle Res. Cell Motil. 2008;29:213–219. doi: 10.1007/s10974-008-9157-6. [DOI] [PubMed] [Google Scholar]
- 20.Tobacman L.S. Thin filament-mediated regulation of cardiac contraction. Annu. Rev. Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 21.Lorenz M., Poole K.J.V., Holmes K.C. An atomic model of the unregulated thin filament obtained by X-ray fiber diffraction on oriented actin-tropomyosin gels. J. Mol. Biol. 1995;246:108–119. doi: 10.1006/jmbi.1994.0070. [DOI] [PubMed] [Google Scholar]
- 22.Oda T., Iwasa M., Narita A. The nature of the globular- to fibrous-actin transition. Nature. 2009;457:441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- 23.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 25.Lehman W., Hatch V., Craig R. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J. Mol. Biol. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- 26.Pirani A., Xu C., Lehman W. Single particle analysis of relaxed and activated muscle thin filaments. J. Mol. Biol. 2005;346:761–772. doi: 10.1016/j.jmb.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 27.Owen C.H., Morgan D.G., DeRosier D.J. Image analysis of helical objects: the Brandeis helical package. J. Struct. Biol. 1996;116:167–175. doi: 10.1006/jsbi.1996.0027. [DOI] [PubMed] [Google Scholar]
- 28.Egelman E.H. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- 29.Milligan R.A., Flicker P.F. Structural relationships of actin, myosin, and tropomyosin revealed by cryo-electron microscopy. J. Cell Biol. 1987;105:29–39. doi: 10.1083/jcb.105.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trachtenberg S., DeRosier D.J. Three-dimensional structure of the frozen-hydrated flagellar filament. The left-handed filament of of Salmonella typhimurium. J. Mol. Biol. 1987;195:581–601. doi: 10.1016/0022-2836(87)90184-7. [DOI] [PubMed] [Google Scholar]
- 31.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Im W., Feig M., Brooks C.L., 3rd An implicit membrane generalized born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Im W., Lee M.S., Brooks C.L., 3rd Generalized Born model with a simple smoothing function. J. Comput. Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 34.McLachlan A.D., Stewart M. The 14-fold periodicity in α-tropomyosin and the interaction with actin. J. Mol. Biol. 1976;103:271–298. doi: 10.1016/0022-2836(76)90313-2. [DOI] [PubMed] [Google Scholar]
- 35.Li X.E., Lehman W., Holmes K.C. Curvature variation along the tropomyosin molecule. J. Struct. Biol. 2010;170:307–312. doi: 10.1016/j.jsb.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown J.H., Zhou Z., Cohen C. Structure of the mid-region of tropomyosin: bending and binding sites for actin. Proc. Natl. Acad. Sci. USA. 2005;102:18878–18883. doi: 10.1073/pnas.0509269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eaton B.L., Kominz D.R., Eisenberg E. Correlation between the inhibition of the acto-heavy meromyosin ATPase and the binding of tropomyosin to F-actin: effects of Mg2+, KCl, troponin I, and troponin C. Biochemistry. 1975;14:2718–2725. doi: 10.1021/bi00683a025. [DOI] [PubMed] [Google Scholar]
- 39.Greenfield N.J., Kotlyanskaya L., Hitchcock-DeGregori S.E. Structure of the N terminus of a nonmuscle α-tropomyosin in complex with the C terminus: implications for actin binding. Biochemistry. 2009;48:1272–1283. doi: 10.1021/bi801861k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frye J., Klenchin V.A., Rayment I. Structure of the tropomyosin overlap complex from chicken smooth muscle: insight into the diversity of N-terminal recognition. Biochemistry. 2010;49:4908–4920. doi: 10.1021/bi100349a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujii T., Iwane A.H., Namba K. Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature. 2010;467:724–728. doi: 10.1038/nature09372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.