

Abstract
Although diffuse brain damage has been suggested to be the predominant predictor of neurological morbidity following closed head injury in infants and children, the presence of contusions also predicts long-term neurobehavioral dysfunction. Contusive brain trauma in the 17-day-old rat resulted in neurodegeneration and caspase activation in the cortex at 1 day, and in the thalamus at 3 days post-injury, and to a greater extent following a deeper impact. Cortical tissue loss in the 4-mm impact group was significantly greater than that in the 3-mm impact group (p < 0.05), and exhibited a time-dependent increase over the first 3 weeks post-injury. Traumatic axonal injury was observed in the white matter tracts below the site of impact at 1 day, and in the corpus callosum at 3 days, to a greater extent following 4-mm impact. In contrast, cellular caspase-3 activation in these white matter tracts was only observed at 24 h post-injury and was not affected by impact depth. Similarly, neurodegeneration and caspase activation in the hippocampus was restricted to the dentate gyrus and occurred to a similar extent in both injured groups. Only the 4-mm impact group exhibited learning deficits in the first week (p < 0.0001) that was sustained until the third week post-injury (p < 0.0001), while deficits in the 3-mm impact group were seen only at 3 weeks post-injury (p < 0.02). These observations demonstrate that increasing severity of injury in immature animals does not uniformly increase the extent of cellular damage, and that the progression of tissue damage and behavioral deficits varies as a function of injury severity.
Key words: apoptosis, caspases, children, cognition, contusion, infants, neurodegeneration, traumatic axonal injury, traumatic brain injury
Introduction
Pediatric traumatic brain injury (TBI) remains one of the leading causes of chronic disability and death (Langlois et al., 2003), with children less than 4 years of age exhibiting worse chronic outcomes than older children (Anderson et al., 2005; Levin et al., 1992). The phenotypes of long-term disability frequently observed following pediatric TBI include cognitive impairment (learning and memory), attention deficits, and emotional and psychosocial problems (Anderson et al., 2005; Catroppa et al., 2007, 2008; Ewing-Cobbs et al., 1997). A clear relationship between initial (acute) Glasgow Coma Scale (GCS) score and long-term neurobehavioral outcome exists, with a lower GCS score predictive of greater behavioral deficits in the chronic post-traumatic period (Anderson et al., 2005; Ewing-Cobbs et al., 2006; Yeates et al., 1995). The advent of sophisticated imaging modalities has led to a better understanding of the relationship between behavioral outcomes and the severity and type of intracranial pathology. Following diffuse brain injury in children (in an absence of contusive lesions), more extensive traumatic axonal injury (TAI) was associated with worse long-term outcome (Tong et al., 2004). While diffuse brain injury is a hallmark injury phenotype of TBI in infants and children, recent studies have highlighted the possibility that focal cortical lesions can also be associated and correlated with worsened cognitive and psychosocial outcomes (Levin et al., 2004; Wilde et al., 2005). The volume of focal lesions in the frontal cortex following closed head injury in children was predictive of deficits of memory performance (Di Stefano et al., 2000). These observations underscore the importance of understanding both the injury phenotype-specific (focal versus diffuse) and injury severity-dependent mechanisms of neuronal damage and associated behavioral deficits following pediatric TBI, with the ultimate goal of developing appropriate treatment strategies.
Models of contusive brain injury have been developed in immature rats (Adelson et al., 1998; Bayly et al., 2006; Bittigau et al., 1999; Grundl et al., 1994; Hickey et al., 2007; Huh et al., 2007; Jenkins et al., 2002; Ozdemir et al., 2005; Prins et al., 2005), immature mice (Pullela et al., 2006; Tong et al., 2002), and piglets (Duhaime et al., 2000). Traumatic injury resulted in a contusion, neuronal cell loss, and tissue atrophy in the injured hemisphere, which were associated with chronic cognitive deficits (Huh et al., 2007; Pullela et al., 2006). Furthermore, contusive brain injury can also have features of diffuse brain injury, such as TAI (Huh and Raghupathi, 2007), and apoptosis and neurodegeneration distant from the contusion area (Bayly et al., 2006; Bittigau et al., 1999). While the effects of increasing severity of contusive brain damage have been evaluated in adult rodents and were associated with graded deficits in behavioral function, the cellular pathological changes were more complex (Conti et al., 1998; Dixon et al., 1991; Fox et al., 1998; Goodman et al., 1994; Hannay et al., 1999; Hicks et al., 1997; Raghupathi et al., 2002; Saatman et al., 2006; Scheff et al., 1997; Sutton et al., 1993). Thus, moderate brain trauma, but not mild injury, induced axonal cytoskeletal changes and delayed apoptosis (Saatman et al., 1998; Raghupathi et al., 2002). These observations are indicative of the necessity to evaluate the effects of graded contusive brain trauma in immature rodents.
In previous studies, we demonstrated that a 3-mm depth of impact on the intact skull over the parietal cortex resulted in a hemorrhagic contusion that developed into a defined necrotic cavity, which was accompanied by evidence of TAI in the underlying white matter tracts (Huh and Raghupathi 2007; Huh et al., 2006). Brain-injured animals also exhibited a spatial learning deficit in the fourth week post-injury (Huh and Raghupathi, 2007). To further characterize this model, we sought to determine whether (1) learning deficits would occur in an earlier time frame after injury, (2) regions other than the contused cortex would demonstrate evidence of neurodegeneration and apoptosis (caspase activation), and (3) depth of impact would affect the extent of histological alterations and behavioral deficits. We report here that at the 3-mm depth of impact, spatial learning deficits were not visible until the third week post-injury, neurodegeneration and caspase activation were observed in the hippocampus and thalamus, TAI was accompanied by cellular caspase activation in multiple white matter tracts, and that increasing the depth of impact only affected the degree of cortical cavitation and spatial learning deficits. These observations underscore the complex nature of severity-dependent alterations in structure and function of the traumatically-injured developing brain, and may have implications for acute and chronic treatment paradigms.
Methods
Brain injury
All surgical and behavioral procedures were done in accordance with the rules and regulations of the Institutional Animal Care and Use Committee of Drexel University College of Medicine, and were in compliance with the Guide for the Care and Use of Animals. The numbers of animals used in the present study are described in Table 1. Brain injuries were induced using the electronically-driven controlled cortical impact (eCCI) device (Custom Design International, Richmond, VA), as previously described (DiLeonardi et al., 2009; Huh et al., 2008). Seventeen-day-old male and female Sprague-Dawley rat pups (Charles River Laboratories, Wilmington, MA; 32–40 g) were anesthetized with isoflurane (5%) using a nose cone, and once a loss of a tail-pinch reflex was observed, a midline incision was made to expose the skull. The periosteum was reflected and the animal was placed in a standard mouse restrainer (Braintree Scientific, Braintree, MA); the head was supported by a soft foam pad in order to make it level with the body. The restrainer was positioned under the eCCI device with the impactor (5-mm-diameter hemispheric metal tip) positioned over the left parietal cortex, midway between the lambda and bregma sutures at an angle of 18° from the vertical to account for the curvature of the skull, and the zero point was set at the surface of the skull. At 30–45 sec after removal of anesthesia, with the animals still non-responsive to a tail pinch, the rats were subjected to either a 3-mm or 4-mm impact (i.e., the indenter traveled a distance of either 3 mm or 4 mm from the zero point). Immediately following the impact, the animals were monitored for length of apnea, length of loss of the righting reflex, and the return of the pain reflex. Sham-injured animals were anesthetized with isoflurane (5%) via nose cone, their scalps were opened, they were placed in the restrainer, the nose cone was removed, and the impactor tip was zeroed on the skull. The total time from initiation of anesthesia to removal of the nose cone prior to the zeroing of the impactor tip on the surface of the skull was 4 min. Apnea latency times were recorded for injured animals from the time of impact until they returned to normal, spontaneous breathing. The loss of the righting reflex was measured by the length of time taken by the animal to stand on all four limbs after being placed on its side immediately following injury or removal from anesthesia. Once the animal regained normal breathing, the skull was evaluated for the presence of fracture, herniation, and hematoma. Following recovery from injury/anesthesia, the animals were re-anesthetized with isoflurane in order to suture the scalp, and the pups were returned to the dam. The animals were placed on a heating pad maintained at 37°C to maintain body temperature throughout the procedures and recovery. The pups were weaned from the dam when they were 21 days old.
Table 1.
Acute Physiological and Neurological Parameters After Contusive Trauma in the 17-Day-Old Rat
| Status | Survival time (days) | n | Apnea (sec) | Righting reflex (sec) | Fractures (%) | Hematoma (%) | Herniation (%) |
|---|---|---|---|---|---|---|---|
| Sham | 1 | 5 (3m, 2f) | NA | 130 ± 36 | NA | NA | NA |
| Sham | 3 | 5 (2m, 3f) | NA | 137 ± 50 | NA | NA | NA |
| Sham | 8 | 16 (7m, 9f) | NA | 153 ± 65 | NA | NA | NA |
| Sham | 18 | 16 (8m, 8f) | NA | 130 ± 52 | NA | NA | NA |
| 3-mm impact | 1 | 6 (3m, 3f) | 8 ± 2 | 127 ± 48 | 100 | 100 | 0 |
| 3-mm impact | 3 | 4 (2m, 2f) | 7 ± 2 | 67 ± 40 | 100 | 100 | 0 |
| 3-mm impact | 8 | 17 (9m, 8f) | 7 ± 1 | 119 ± 42 | 100 | 76 | 24 |
| 3-mm impact | 18 | 16 (8m, 8f) | 7 ± 2 | 85 ± 63 | 100 | 75 | 25 |
| 4-mm impact | 1 | 4 (2m, 2f) | 13 ± 2 | 221 ± 60 | 100 | 100 | 100 |
| 4-mm impact | 3 | 4 (2m, 2f) | 10 ± 2 | 167 ± 73 | 100 | 50 | 50 |
| 4-mm impact | 8 | 11 (5m, 6f) | 10 ± 3 | 248 ± 61 | 100 | 100 | 100 |
| 4-mm impact | 18 | 14 (8m, 6f) | 10 ± 4 | 146 ± 73 | 100 | 100 | 100 |
m, male; f, female.
Spatial learning
Learning in sham- and brain-injured animals was assessed using a 1-m-diameter Morris water maze (MWM) on days 4–8 or days 14–18 post-injury (separate groups of animals were used for each time point of assessment) as previously described (Huh et al., 2008). The evaluator was blinded to the injury status of the animals and the testing time point. At 24 h following the last training trial day (day 8 or 18), the platform was placed 2 cm above the surface of the water, a visual cue was adhered to the platform (visible platform trial), and the latency of the animal to reach the platform was measured. The swim speed (distance traveled/time) of the animal during the visible platform trial was recorded.
Tissue preparation for histology and immunohistochemistry
On days 1, 3, 8, and 18 post-surgery/injury, sham- and brain-injured animals were anesthetized (60 mg/kg sodium pentobarbital IP) and euthanized by trans-cardial perfusion with 10% phosphate-buffered formalin. The brains were post-fixed in the skull for 24 h, then outside the cranial cavity for an additional 24 h, cryoprotected in 30% sucrose, and frozen in liquid isopentane at −35°C. Coronal (40-μm-thick) sections were taken every 0.5 mm between the bregma and −4.3 mm posterior to the bregma. Each set contained 10 sections. One set of sections was mounted on gelatin-coated slides and stained with 2% cresyl violet and 0.2% cyanine R (Nissl-myelin) for assessment of gross histology. A second set was evaluated for neurodegeneration using Fluoro-Jade B histochemistry as previously described (Huh et al., 2008; Tong et al., 2002). Additional sets of sections were evaluated for caspase activation using a polyclonal antibody that recognizes the caspase-cleaved C-terminus fragment of actin (1:10,000, Fractin; Chemicon, Temecula, CA), and TAI (1:2000, polyclonal antibody recognizing the C-terminus of APP; Zymed, San Francisco, CA), as previously described (Huh et al., 2008). As negative controls, 1–2 sections from each animal were incubated with secondary antibody and the ABC reagent prior to exposure to diaminobenzidine (DAB).
Quantification of cortical tissue loss
The extent of cortical tissue loss was quantified using Nissl-myelin-stained sections (n = 5–7 brain-injured animals/time point at 8 and 18 days post-injury), as previously described (Huh et al., 2007; Raghupathi and Huh, 2007). Sections were captured at 1 × magnification using an Eclipse E400 microscope (Nikon Corp., Tokyo, Japan), and digitized as TIFF images using Nikon ACT-1 software version 2.62. Using ImageJ software (Rasband, 2007), tissue loss in the injured cortex was calculated as a percentage of the cortical tissue volume in the contralateral hemisphere using the following formula utilizing the Cavalieri method:
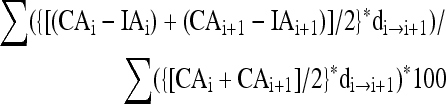 |
where IAi = area of the cortex in the injured hemisphere of section “I,” IAi+1 = area of the injured cortex in the next section from “i,” CAi and CAi+1 = areas of cortex in the contralateral hemisphere of section “i” and the next section “i+1,” and di→i+1 is the distance between sections “i” and the next section “i+1.”
Statistical analysis
All values are expressed as mean ± standard deviation (SD). Average latencies to locate the hidden platform in the MWM were compared using a one-way repeated-measures analysis of variance (ANOVA; injury status × training days). Times of apnea, pain reflex, and righting reflex recovery were compared between the 3-mm and 4-mm groups using a multiple ANOVA (MANOVA) as a function of time of euthanasia. Latencies to the visible platform, and swim speeds were compared using a two-way ANOVA (injury status × depth of impact) for each of the two time points evaluated, while percentage of tissue loss and volume of contralateral hemisphere values were compared using a two-way ANOVA (depth of impact × time of euthanasia). When necessary, post-hoc comparisons between groups were performed using the Neuman-Keuls test. Differences were considered significant at p < 0.05; in all analyses, gender was used as a covariate.
Results
Acute neurologic responses
Impact to the intact skull of the 17-day-old rat with a metal-tipped indenter did not result in acute or delayed mortality. On impact, linear skull fractures were observed in all injured animals, regardless of the distance traveled by the impactor (Table 1). However, in rats subjected to 4-mm impact, tissue extravasation (herniation) via the fractured skull was observed (Table 1). While herniation was noted only in a small subset of animals that received 3-mm impact, severe hematomas circumscribing the tip diameter were almost always observed in these animals (Table 1). Lateral impact resulted in a brief period of apnea ranging from 5–15 sec, regardless of the depth of injury. Interestingly, both sham- and brain-injured animals recovered righting reflexes in approximately similar times, suggesting that recovery from anesthesia overwhelmed any injury-induced effects. No differences in the acute neurological responses were observed between groups of animals that were euthanized at the different time points post-injury, indicative of the consistency of the injury paradigm. Acute neurological responses were not markedly different between male and female rat pups.
Pathological alterations in the cortex
Tissue tears in the cortex and subcortical white matter tracts with accompanying hemorrhage were observed below the site of impact at 24 h following a 3-mm-deep impact (Fig. 1A). By 8 days, the site of maximal injury contained small-diameter (non-neuronal) cells (Fig. 1B and G), and by 18 days the injured cortex and underlying white matter (Fig. 1C and H) exhibited substantial atrophy. Frank loss of Nissl-stained cortical and myelin-stained subcortical white matter tissue was observed at 24 h following 4-mm depth of impact (Fig. 1D). The injured cortex was surrounded by non-neuronal cells and the underlying white matter tract was substantially disrupted (Fig. 1E and I) at 8 days post-injury. By 18 days, a marked cavity was present in the injured hemisphere (Fig. 1F). Quantitative analysis of cortical tissue loss in the injured hemisphere revealed that the depth of the impact regulated the temporal pattern of tissue loss. Following an impact of 3 mm into the skull, the extent of the tissue loss was not significantly different between 8 and 18 days post-injury (Fig. 1J). In contrast, animals subjected to a 4-mm impact demonstrated a significant increase in the extent of tissue loss between the 8-day and 18-day time points (p < 0.0002; Fig. 1J). At the 8-day time point, there was no difference in the extent of tissue loss between the two impact depths; however, at 18 days post-injury, tissue loss was significantly greater in the 4-mm impact group than in the 3-mm impact group (p < 0.0002, Fig. 1J).
FIG. 1.


Cortical tissue loss following contusive brain trauma in 17-day-old rats. Representative images of cresyl violet-cyanine R-stained tissue sections from animals subjected to 3-mm (A–C, G, and H), or 4-mm depth of impact (D–F, and I), demonstrating progressive loss of cortical tissue after impact. Note hemorrhage and tissue tears on day 1 (A and D) and day 8 (B and E), and pronounced cavitation on days 1 (D), 8 (E), and 18 (F) after 4-mm depth of impact; there was no overt cavitation on day 18 following 3-mm depth of impact (C). Boxes in panels B, C, and E represent areas illustrated at higher magnification in panels G, H, and I, respectively. Note the presence of non-neuronal cells in the gray-white matter interface at both 3-mm (G and H) and 4-mm depths of impact (I). The graph in J demonstrates the quantitative analysis of cortical tissue loss in the injured hemisphere at 8 and 18 days following either 3-mm or 4-mm depth of impact (*p < 0.0002 compared to 8 days after 4-mm depth of impact; #p < 0.0002 compared to the 3-mm group; scale bar in A–F = 1 mm, and for G–I = 100 μm).
The cortices of sham-injured animals did not contain any Fluoro-Jade B reactivity (Fig. 2A). At 1 day post-injury, Fluoro-Jade B-labeled neurons were observed in the cortex surrounding the site of maximal injury following either 3-mm (Fig 2B) or 4-mm depth of impact (Fig. 2C); Fluoro-Jade B reactivity was more extensive with increasing depth of impact. By 3 days post-injury, minimal Fluoro-Jade B staining was seen in the cortex in any brain-injured animal (data not shown). In regions where Fluoro-Jade B reactivity was observed, the presence of cells containing caspase-cleaved actin fragments was noted (Fig. 2E and F); fractin-labeled cells were absent in the cortices of sham-injured animals (Fig. 2D). Caspase activation was observed in both cell bodies and processes of neurons adjacent to the site of maximal injury at 1 day following 3-mm depth of impact (Fig. 2E). As observed for Fluoro-Jade B, increasing the impact depth to 4-mm resulted in fractin immunoreactivity in a greater area of the injured cortex (Fig. 2F). Fractin labeling was observed in both morphologically-intact cell bodies and neurites (inset in Fig. 2E), and in fragmented processes (inset in Fig. 2F). By 3 days, few if any cells exhibited fractin immunoreactivity. These results reveal that neurodegeneration and cellular caspase activation in the cortex below the impact site were maximally observed at 24 h post-injury. No overt differences in the extent of trauma-induced pathological alterations in the cortex were observed between male and female rat pups.
FIG. 2.

Neurodegeneration and caspase activation in the cortex at 24 h following contusive brain trauma in 17-day-old rats. Photomicrographs represent Fluoro-Jade B reactivity in the cortex adjacent to the site of maximal lesion following 3-mm (B) or 4-mm depth of impact (C). Similar regions in the cortices of sham-injured animals did not contain either Fluoro-Jade B-positive (A) or fractin-labeled neurons (D). In sections adjacent to those used for Fluoro-Jade B staining, fractin immunoreactivity was observed in cell bodies and neurites at both 3-mm (E) and 4-mm depth of impact (F).The inset in panel E demonstrates an example of a fractin-positive neuron that does not show neurite beading; these were occasionally seen within and around the site of maximal lesion. The inset in panel F illustrates a beaded neurite containing fractin. Scale bar in panel D represents 50 μm for A–F, and 25 μm for the insets in panels E and F.
Pathological alterations in the thalamus
In contrast to the cortex, Fluoro-Jade B reactivity in the thalamus did not begin to appear until at least 3 days post-injury (Fig. 3). There was no evidence of Fluoro-Jade B-labeled neurons in any thalamic nuclei in sham-injured animals (data not shown), or at 1 day following either 3-mm (data not shown), or 4-mm depth of impact (Fig. 3A). By 3 days following 3-mm depth of impact, Fluoro-Jade B-positive neurons were observed primarily in the lateral geniculate nucleus at 3 (Fig 3B) and 8 days (data not shown). Increasing the depth of impact to 4 mm resulted in Fluoro-Jade B-labeled degenerating neurons additional in the medial geniculate nucleus (Fig. 3C). Morphologically, the neuronal soma was shrunken and degenerating cell processes were readily apparent (Fig. 3D). Degenerating neurons were observed at 18 days post-injury, albeit to a lesser extent than at earlier time points (data not shown). Fractin immunoreactivity, indicative of caspase activation, was not observed in the thalamus of sham-injured animals (Fig. 3E), but was noted to a similar extent at 1 day following either 3-mm (data not shown) or 4-mm depth of impact (Fig. 3F), and appeared as intensely-stained cell bodies surrounded by diffusely-labeled neuropil, suggestive of oligodendrocytes. By 3 days post-injury, fractin immunoreactivity was observed in cell bodies of intact neurons (Fig. 3G), and as punctate profiles at 8 days post-injury (Fig. 3H). No evidence of caspase activation was observed at 18 days in either injured group (data not shown). As observed with Fluoro-Jade B reactivity, fractin immunoreactivity was restricted to the lateral geniculate nucleus following impact at 3-mm depth, and extended to the medial geniculate with increased depth of impact. In addition, at 8 days, the density of the punctate fractin profiles was greater in the 4-mm-impact injured animals (Fig. 3H). Gender did not appear to influence the spatial and temporal extent of TAI, neurodegeneration, and caspase activation in the thalamus.
FIG. 3.

Neurodegeneration and caspase activation in the thalamus following contusive brain trauma in 17-day-old rats. Photomicrographs represent Fluoro-Jade B reactivity in the thalamus at 1 day following 4-mm depth of impact (A), 3 days following either 3-mm (B) or 4-mm depth of impact (C and D). Note the presence of Fluoro-Jade B-labeled cells in medial geniculate neurons after 4-mm depth of impact (C), that were not observed at 3-mm (B). Note the shrunken morphology and beaded neurites indicative of neurodegeneration (D). Panels E–H represent fractin immunoreactivity in the thalamus in sham-injured group (E), and at 1 (F), 3 (G), and 8 days (H) following 4-mm depth of impact. Note the intense fractin staining in centrally-located cell somata surrounded by processes, suggestive of oligodendroglial cells (F). Note the neuronal morphology in panel G, and punctate labeling indicative of advanced degeneration at 8 days (H). Scale bar in panel H represents 250 μm for panels A–C, and 25 μm for panels D–H.
Pathological alterations in the hippocampus
Neurodegeneration and caspase activation in the hippocampus was restricted to the dentate gyrus in the hemisphere below the site of impact, and occurred to a similar extent following either 3- or 4-mm depth injury (Fig. 4) in both male and female rats. The dentate gyrus of sham-injured animals did not stain for either Fluoro-Jade-B (Fig. 4A) or caspase activation (Fig. 4D). At 1 day post-injury, Fluoro-Jade B reactivity was observed predominantly in the granule cell layer, and sporadically in the hilus (Fig. 4B); this was accompanied by evidence of caspase activation in cell bodies (granule cell layer) and processes (molecular layer, Fig. 4E). By 3 days post-injury, both Fluoro-Jade B reactivity (Fig. 4C), and fractin immmunoreactivity (Fig. 4F), were observed in a punctate labeling pattern in the molecular layer. At 8 and 18 days post-injury, Fluoro-Jade B reactivity and caspase activation were not observed (data not shown).
FIG. 4.

Neurodegeneration and caspase activation in the dentate gyrus following contusive brain trauma in 17-day-old rats. Representative photomicrographs depicting Fluoro-Jade B reactivity (A–C), and fractin immunoreactivity (D–F), in the dentate gyrus of sham-injured animals (A and D), and at 1 day (B and E) and 3 days (C and F) following injury. Note the absence of either Fluoro-Jade B- (A) or fractin-labeling (D) in sham-injured animals, and the appearance of punctate labeling in the molecular layer of the dentate gyrus for both Fluoro-Jade B (C) and fractin (F). Scale bar in panel F represents 50 μm for all panels.
Pathological alterations in subcortical white matter tracts
Although impact to the intact skull over the parietal cortex of the 17-day-old rat resulted in a contusion in the cortex, evidence of TAI and cellular caspase activation were observed in the white matter tracts below the site of maximal cortical injury (Fig. 5). At 1 day post-injury following either 3-mm (Fig. 5A) or 4-mm impact, intra-axonal accumulation of amyloid precursor protein (APP) was observed, and was more extensive at 4-mm depth of impact (Fig. 5B). By 3 days post-injury, most of the APP labeling for both 3-mm (Fig. 5D) and 4-mm (Fig. 5E) depths of impact was present in terminal bulbs. Axonal injury was accompanied by cellular caspase activation, which was only observed at 1 day post-injury (Fig. 5C), and appeared to similar extents in both injured groups. Cellular fractin (Fig. 5F) and intra-axonal APP (inset in Fig. 5D) immunoreactivities were absent in white matter tracts of sham-injured animals.
FIG. 5.

Traumatic axonal injury and caspase activation in white matter tracts below the cortical lesion following contusive brain trauma in 17-day-old rats. Representative photomicrographs illustrate the accumulation of amyloid precursor protein (APP) in axons at 1 (A and B) and 3 days (D and E) following 3-mm (A and D) or 4-mm depth of impact (B and E). Note the presence of APP-positive terminal bulbs at 3 days post-injury (D and E). The inset in panel D represents a sham-injured animal with no visible APP immunoreactivity. Panel C illustrates cellular fractin immunoreactivity at 1 day post-injury following 3-mm depth of impact; note the intensely labeled cell soma surrounded by diffusely-labeled processes. (F) Fractin immunoreactivity was not observed in sham-injured brains. Scale bar in panel F represents 50 μm for panels A–F.
In addition to the white matter directly below the impact site, intra-axonal accumulation of APP and cellular fractin immunoreactivity was also observed in the corpus callosum (Fig. 6). Axonal swellings were observed following 3-mm depth of impact (Fig. 6A), and to a greater extent in the 4-mm-deep impact groups (Fig. 6B). By 3 days, APP accumulation was also observed in terminal bulbs in both groups (Fig. 6D and E). In contrast to the white matter below the site of impact, where the extent of axonal injury at 3 days was less than that observed at 24 h, TAI in the corpus callosum appeared to increase between 1 and 3 days post-injury. However, no evidence of axonal APP accumulation was observed at the 8- and 18-day time points (data not shown). Evidence of caspase-mediated proteolysis of actin was observed in cells of the corpus callosum (Fig. 6C) to similar extents in the 3- and 4-mm groups, and was observed only at the 24-h time point; no fractin-positive cells were observed in the corpus callosum of sham-injured animals (Fig. 6F). As observed for pathological alterations in other regions, gender did not appear to influence the extent of neuropathological damage in the white matter.
FIG. 6.

Traumatic axonal injury and caspase activation in the corpus callosum following contusive brain trauma in 17-day-old rats. Representative photomicrographs illustrate accumulation of amyloid precursor protein (APP) in axons at 1 (A and B) and 3 days (D and E) following 3-mm (A and D) or 4-mm depth of impact (B and E). Panel C illustrates cellular fractin immunoreactivity at 1 day post-injury following 3-mm depth of impact; note the intensely labeled cell soma surrounded by diffusely-labeled processes. (F) Fractin immunoreactivity was not observed in sham-injured brains. Scale bar in panel F represents 50 μm for all panels.
Spatial learning deficits
As illustrated in Figure 7A, all animals learned the location of the platform over the 4 days of training during the first week post-injury, although the brain-injured animals took a longer time to do so than their uninjured littermates. One-way repeated-measures ANOVA revealed an injury effect (F(2,41) = 9.37, p < 0.0001), and a time effect (F(3,123) = 55.42, p < 0.0001). The absence of an interaction effect (injury × time) indicated that injury status did not affect the ability to learn the task. Post-hoc analysis for the injury effect revealed that animals receiving the 4-mm depth of impact were significantly different from both the sham group (p < 0.005), and animals receiving a 3-mm depth of impact (p < 0.005). Interestingly, injury from a 3-mm depth of impact did not result in a significant deficit in acquisition (learning) within the first week post-injury (Fig. 7A). In the third week post-injury (Fig. 7B), all brain-injured animals exhibited a learning deficit (injury effect, F(2,43) = 23.88, p < 0.0001), although all animals were able to locate the platform over the 4-day testing period (time effect, F(3,129) = 57.57, p < 0.0001). Post-hoc analyses indicated that injury at both the 3-mm and 4-mm depth of impact resulted in a significant learning deficit (p < 0.02 and p < 0.0005, respectively, compared to the sham-injured group). Moreover, impact of a 4-mm depth resulted in a greater deficit in learning compared to an impact of 3 mm (p < 0.001). Swim speeds of sham- and brain-injured animals ranged between 22 and 25 cm/sec, and were not significantly different between any groups at either of the two time points tested (data not shown), indicative of a lack of a perceptible motor deficit. Similarly, the absence of a significant difference in the latency to the visible platform (data not shown) between sham- and brain-injured animals at any time point post-injury suggests that a visual deficit did not contribute to the observed deficits in acquisition. In addition, statistical analyses indicated that gender (a co-variate) did not affect the outcomes.
FIG. 7.

Spatial learning following contusive brain trauma in 17-day-old rats. All animals were tested in the Morris water maze for their ability to learn the location of a submerged platform as described in the methods section. Data are presented as average latencies for four trials on each day, and error bars represent standard deviation of the mean values. (A) Days 4–7 following surgery/injury. Repeated-measures factorial analysis of variance (ANOVA) revealed an injury effect (p < 0.0001), and a time effect (p < 0.0001). Post-hoc Neuman-Keuls test for injury indicated that the 4-mm-injured group was significantly different from both the sham group (p < 0.005), and the 3-mm-injured group (p < 0.005), and that the 3-mm-injured group was no different from the sham-injured animals. (B) Days 14–17 following surgery/injury. Repeated-measures factorial ANOVA revealed an injury effect (p < 0.0001), and a time effect (p < 0.0001). Post-hoc Neuman-Keuls test for injury indicated that both the 3-mm- (p < 0.02) and 4-mm-injured groups (p < 0.005) were significantly different from the sham group, and the 4-mm-injured group was significantly more impaired than the 3-mm-injured group (p < 0.001).
Discussion
Contusive brain trauma in the 17-day-old rat resulted in acute and sustained deficits in spatial learning that was accompanied by neurodegeneration and caspase activation in the cortex, hippocampus, and thalamus, and axonal injury in various white matter tracts. Increasing the severity of the contusive trauma by increasing the depth of impact resulted in a greater extent of spatial learning deficits over the first 3 weeks post-injury, but learning deficits following the milder injury were only apparent in the third week post-trauma. Cortical tissue loss, neurodegeneration in the thalamus, and axonal injury in white matter tracts, were greater in the more severely-injured animals, although caspase activation in the white matter and hippocampus, and neurodegeneration in the hippocampus, appeared to be independent of impact depth. Moreover, at the lower injury severity (3-mm depth of impact), a time-dependent increase in cortical cavitation (tissue loss) was not observed. While these observations in large part confirmed the hypothesis that controlling the depth of impact at a constant velocity (5 m/sec) and dwell time (100 msec) would regulate injury severity, the differential responses in certain outcomes underscore the complex nature of contusive trauma to the developing brain.
Contusive traumatic injury to the immature rat resulted in a brief period of apnea, similar to that observed following diffuse trauma (Gurkoff et al., 2006; Huh et al., 2008). Despite the presence of skull fractures in all injured groups, and herniation of brain tissue in the 4-mm-injured group, neither apnea times nor loss of righting reflex appeared to be dependent on the severity of the impact. Furthermore, injured animals did not demonstrate an increase in loss of consciousness (measured as latency to right themselves after removal of anesthesia). In addition to an effect of the anesthetic used, this observation may indicate the relative resistance of the immature brain to acute neurological disturbance following trauma (Prins et al., 1996). The acute determination of injury severity following TBI in infants and young children poses practical difficulties, and in the extreme, may not be sufficient to predict chronic outcome. The verbal and motor scales of the GCS (Teasdale and Jennett, 1974), which is the most widely used acute clinical measure of injury severity after pediatric TBI (Anderson et al., 2005; Ewing-Cobbs et al., 2006; Yeates et al., 1995), are developmentally inappropriate for infants and young children, and therefore can be unreliable in this age group (Durham et al., 2000; Shore et al., 2007; Simpson et al., 1991). Furthermore, it has been reported that young children with fatal TBI may present with an initial lucid period (no loss of consciousness), or a high GCS score (Arbogast et al., 2005).
Compared to age-matched uninjured animals, 3-mm depth of impact did not affect acquisition in the first week post-injury, an observation similar to that reported following diffuse brain trauma in the immature rat, for which learning deficits at lower levels of injury severity were either not present (Adelson et al., 1997), or were transient (Gurkoff et al., 2006). However, when the analysis was extended to either the third (this report) or the fifth week post-injury (Huh et al., 2007), 17-day-old rats injured at 3-mm depth of impact demonstrated acquisition deficits. These observations argue against the idea that the brain of the young child is more resistant to cerebral trauma (Kennard, 1938), but rather that children may “grow into their deficit” (Anderson et al., 2005, 2000). At more severe injury levels (4-mm depth of impact), acquisition deficits were observed as early as the first week post-trauma, and were sustained until the third week, thus lending support to reports that both volume of focal brain lesions, and tissue loss in brain-injured infants and children, were significantly correlated with worse cognitive and functional outcomes (Di Stefano et al., 2000; Wilde et al., 2005).
Spatial learning and memory deficits following brain trauma in adult rodents have been linked to loss of neurons in the dentate gyrus and pyramidal neurons of the CA3 area of the hippocampus (Baldwin et al., 1997; Colicos et al., 1996; Hicks et al., 1993; Smith et al., 1991). Together with previous reports (Huh et al., 2008; Tong et al., 2002), our data demonstrate that the dentate gyrus is vulnerable to traumatic insults regardless of age, injury type, or injury severity. However, it may be overly simplistic to argue that hippocampal cell death may be the anatomical substrate for post-traumatic learning deficits, given the observation that learning deficits after brain trauma can occur in the absence of overt cell death in the hippocampus (Lyeth et al., 1990). Fractin immunoreactivity, indicative of caspase activation, was predominantly seen in neurites within the molecular layer, an observation that supports earlier reports of a lack of immunoreactivity for active caspase-3 in the cell soma in any area of the hippocampus following closed head injury in immature rodents (Bayly et al., 2006; Dikranian et al., 2008).
Traumatic axonal injury is a pathological entity of TBI and is not restricted to diffuse axonal injury in infants and children (Wilde et al., 2005). Our observations of intra-axonal accumulations of APP in the white matter tracts immediately below and remote from the site of impact confirm our previous report of similar pathology following contusive trauma in the 11-day-old rat (Huh et al., 2006). Interestingly, in the current study, it appeared that the extent of APP-positive swollen (injured) axons in the white matter immediately below the impact site was greater at 24 h post-injury, while the APP-positive swellings in the corpus callosum were more extensive at 3 days. In part, this may reflect the fact that injury to axons below the impact site may result from a combination of primary (shearing forces) and secondary (impaired axonal transport, IAT) mechanisms, while axonal injury in regions remote from the impact site (corpus callosum) may predominantly reflect IAT. It must be noted here that as reported by Stone and colleagues in adult brain-injured rats (Stone et al., 2000), the antibody to the C-terminus of APP used in the present study appears to be more sensitive to detection of intra-axonal accumulation of APP than the N-terminus antibody used in our earlier study (Huh and Raghupathi, 2007). Caspase activation was observed in white matter tracts along with APP-positive axonal swellings; fractin immunoreactivity appeared as intensely-stained centrally-located cell bodies surrounded by diffusely-stained neuropil, suggestive of a glial phenotype. Fractin immunoreactivity in these non-neuronal cells was only observed at 24 h post-injury, and did not appear to be present to a greater extent when the depth of impact was increased, suggesting that TAI, which occurred to a greater extent at the more severe level of trauma, and glial apoptosis may not be causally related. Further, it appears that white matter injury in TBI may be very different from that observed in traumatic spinal cord injury, in which apoptotic oligodendrocytes were observed in areas remote from the contusion site, increased with time after trauma, and were temporally related to axonal degeneration (Crowe et al., 1997; Nottingham and Springer, 2003; Shuman et al., 1997).
In addition to confirming our previous report that lateral impact at a 3-mm depth in 17-day-old rats resulted in a time-dependent increase in tissue loss (Huh and Raghupathi, 2007), the results in the present study suggest that the progression of cortical tissue loss appears to be dependent on the depth of the initial impact. First, a significant increase in loss of tissue was observed over the first 18 days following 4-mm depth of impact, but was only observed between 14 and 28 days following 3-mm depth of impact (Huh and Raghupathi, 2007). Second, the amount of cortical tissue lost after 18 days following a 4-mm depth of impact was quantitatively similar to that lost at 28 days following 3-mm depth of impact (Huh and Raghupathi, 2007). While longitudinal effects on brain morphology alterations following graded contusive trauma have not been evaluated in either adult or immature rodents, our data support previous reports of increased tissue loss and ventricular enlargement in adult rats over a 1-year period (Dixon et al., 1999; Pierce et al., 1998). In addition, at 8 days post-injury, there was no significant difference in tissue loss between the two depths of impact, an observation that is in contrast to adult mice, in which the loss of cortical tissue over the first week post-injury was significantly greater with increased depth of impact (Saatman et al., 2006). These observations underscore the importance of injury severity, time post-injury, and age at injury, in the evaluation of brain morphology following brain trauma.
The cellular basis for loss of cortical tissue may be related to neurodegeneration and/or cell volume. Unlike the loss of tissue, which increases with time after injury, Fluoro-Jade B labeling of neurons was most evident at 1 day post-injury, and was practically absent by 3 days. Second, caspase activation in the cortex in and around the lesion was predominantly in neuronal processes, suggestive of a loss of neurite length and dendrite arbor. Besides caspase activation leading to apoptotic cell death, it is well recognized that non-caspase-mediated apoptotic cell death and cellular calpain activation may contribute to cellular pathology, leading to cell loss (Borner and Monney, 1999; Huh et al., 2006; Raghupathi et al., 2000). While our observations of acute neuronal Fluoro-Jade B reactivity and cellular caspase activation support earlier reports of neurodegeneration and apoptosis following contusive trauma in the developing animal (Bittigau et al., 1999), to the best of our knowledge, the current study is the first to demonstrate neurodegeneration, caspase activation, and tissue loss in the ipsilateral cortex following graded brain trauma in the immature rat. Consistent with other rodent models of pediatric traumatic and hypoxic-ischemic brain injury (Bayly et al., 2006; Dikranian et al., 2008; Huh et al., 2008; Northington et al., 2001; Tong et al., 2002), we observed delayed neurodegeneration and caspase activation in neurons within the thalamus at both injury severities. In contrast, mild contusive trauma in the adult rat did not induce retrograde apoptosis of thalamic neurons (Raghupathi et al., 2002). Moreover, the thalamic nuclei containing degenerating cells are those that project to and from the lesioned areas of the cortex (Northington et al., 2001). Retrograde degeneration of neurons within these thalamic nuclei is further supported by the degeneration of axons within putative thalamocortical connections (Bayly et al., 2006; Dikranian et al., 2008; Iizuka et al., 1990; Pierce et al., 1998).
In summary, in the present study we present evidence of differential effects of graded contusion trauma on behavioral and pathological alterations in the immature rat. Clinically, injury severity influences the extent and types of acute supportive care used in the intensive care unit. Our data demonstrating that in the acute post-traumatic period, contusive trauma at both depths induces Fluoro-Jade B reactivity, caspase activation, and TAI, suggest that acute treatment strategies may need to be similar. However, the appearance of learning deficits only at the chronic time point, and the differential rate of tissue loss seen following a lower level of injury (3-mm depth of impact), imply that mechanisms (and treatment paradigms) in the chronic time period may be dependent on injury severity. These observations also raise important issues related to recovery of function, and future studies should focus on whether injury severity can influence the rate of return to baseline (uninjured) function. Importantly, these observations also underscore the importance and necessity of evaluating multiple outcome measures for a sustained period of time after trauma to the developing brain.
Acknowledgments
These studies were supported in part by National Institute of Neurological Disorders and Stroke grants K08-NS053651 and R01-HD061963, and a VA merit review grant.
Author Disclosure Statement
No competing financial interests exist.
References
- Adelson P.D. Dixon C.E. Robichaud P. Kochanek P.M. Motor and cognitive functional deficits following diffuse traumatic brain injury in the immature rat. J. Neurotrauma. 1997;14:99–108. doi: 10.1089/neu.1997.14.99. [DOI] [PubMed] [Google Scholar]
- Adelson P.D. Whalen M.J. Kochanek P.M. Robichaud P. Carlos T.M. Blood-brain barrier permeability and acute inflammation in two models of traumatic brain injury in the immature rat: a preliminary report. Acta Neurochir. Suppl. 1998;71:104–106. doi: 10.1007/978-3-7091-6475-4_31. [DOI] [PubMed] [Google Scholar]
- Anderson V.A. Catroppa C. Rosenfeld J. Haritou F. Morse S.A. Recovery of memory function following traumatic brain injury in pre-school children. Brain Inj. 2000;14:679–692. doi: 10.1080/026990500413704. [DOI] [PubMed] [Google Scholar]
- Anderson V. Catroppa C. Morse S. Haritou F. Rosenfeld J. Functional plasticity or vulnerability after early brain injury? Pediatrics. 2005;116:1374–1382. doi: 10.1542/peds.2004-1728. [DOI] [PubMed] [Google Scholar]
- Arbogast K.B. Margulies S.S. Christian C.W. Initial neurologic presentation in young children sustaining inflicted and unintentional fatal head injuries. Pediatrics. 2005;116:180–184. doi: 10.1542/peds.2004-2671. [DOI] [PubMed] [Google Scholar]
- Baldwin S.A. Gibson T. Callihan C.T. Sullivan P.G. Palmer E. Scheff S.W. Neuronal cell loss in the CA3 subfield of the hippocampus following cortical contusion utilizing the optical dissector method for cell counting. J. Neurotrauma. 1997;14:385–398. doi: 10.1089/neu.1997.14.385. [DOI] [PubMed] [Google Scholar]
- Bayly P.V. Dikranian K.T. Black E.E. Young C. Qin Y.Q. Labruyere J. Olney J.W. Spatiotemporal evolution of apoptotic neurodegeneration following traumatic injury to the developing rat brain. Brain Res. 2006;1107:70–81. doi: 10.1016/j.brainres.2006.05.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittigau P. Sifringer M. Pohl D. Stadthaus D. Ishimaru M. Shimizu H. Ikeda M. Lang D. Speer A. Olney J.W. Ikonomidou C. Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann. Neurol. 1999;45:724–735. doi: 10.1002/1531-8249(199906)45:6<724::aid-ana6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Borner C. Monney L. Apoptosis without caspases: an inefficient molecular guillotine? Cell Death Differ. 1999;6:497–507. doi: 10.1038/sj.cdd.4400525. [DOI] [PubMed] [Google Scholar]
- Catroppa C. Anderson V.A. Morse S.A. Haritou F. Rosenfeld J.V. Children's attentional skills 5 years post-TBI. J. Pediatr. Psychol. 2007;32:354–369. doi: 10.1093/jpepsy/jsl019. [DOI] [PubMed] [Google Scholar]
- Catroppa C. Anderson V.A. Morse S.A. Haritou F. Rosenfeld J.V. Outcome and predictors of functional recovery 5 years following pediatric traumatic brain injury (TBI) J. Pediatr. Psychol. 2008;33:707–718. doi: 10.1093/jpepsy/jsn006. [DOI] [PubMed] [Google Scholar]
- Colicos M.A. Dixon C.E. Dash P.K. Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res. 1996;739:111–119. doi: 10.1016/s0006-8993(96)00819-0. [DOI] [PubMed] [Google Scholar]
- Conti A.C. Raghupathi R. Trojanowski J.Q. McIntosh T.K. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J. Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe M.J. Bresnahan J.C. Shuman S.L. Masters J.N. Beattie M.S. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- Di Stefano G. Bachevalier J. Levin H.S. Song J.X. Scheibel R.S. Fletcher J.M. Volume of focal brain lesions and hippocampal formation in relation to memory function after closed head injury in children. J. Neurol. Neurosurg. Psychiatry. 2000;69:210–216. doi: 10.1136/jnnp.69.2.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikranian K. Cohen R. Mac Donald C. Pan Y. Brakefield D. Bayly P. Parsadanian A. Mild traumatic brain injury to the infant mouse causes robust white matter axonal degeneration which precedes apoptotic death of cortical and thalamic neurons. Exp. Neurol. 2008;211:551–560. doi: 10.1016/j.expneurol.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiLeonardi A.M. Huh J.W. Raghupathi R. Impaired axonal transport and neurofilament compaction occur in separate populations of injured axons following diffuse brain injury in the immature rat. Brain Res. 2009;1263:174–182. doi: 10.1016/j.brainres.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon C.E. Clifton G.L. Lighthall J.W. Yaghmai A.A. Hayes R.L. A controlled cortical impact model of traumatic brain injury in the rat. J. Neurosci. Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- Dixon C.E. Kochanek P.M. Yan H.Q. Schiding J.K. Griffith R.G. Baum E. Marion D.W. DeKosky S.T. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J. Neurotrauma. 1999;16:109–122. doi: 10.1089/neu.1999.16.109. [DOI] [PubMed] [Google Scholar]
- Duhaime A.C. Margulies S.S. Durham S.R. O'Rourke M.M. Golden J.A. Marwaha S. Raghupathi R. Maturation-dependent response of the piglet brain to scaled cortical impact. J. Neurosurg. 2000;93:455–462. doi: 10.3171/jns.2000.93.3.0455. [DOI] [PubMed] [Google Scholar]
- Durham S.R. Clancy R.R. Leuthardt E. Sun P. Kamerling S. Dominguez T. Duhaime A.C. CHOP Infant Coma Scale (“Infant Face Scale”): a novel coma scale for children less than two years of age. J. Neurotrauma. 2000;17:729–737. doi: 10.1089/neu.2000.17.729. [DOI] [PubMed] [Google Scholar]
- Ewing-Cobbs L. Fletcher J.M. Levin H.S. Francis D.J. Davidson K. Miner M.E. Longitudinal neuropsychological outcome in infants and preschoolers with traumatic brain injury. J. Int. Neuropsychol. Soc. 1997;3:581–591. [PubMed] [Google Scholar]
- Ewing-Cobbs L. Prasad M.R. Kramer L. Cox C.S., Jr. Baumgartner J. Fletcher S. Mendez D. Barnes M. Zhang X. Swank P. Late intellectual and academic outcomes following traumatic brain injury sustained during early childhood. J. Neurosurg. 2006;105:287–296. doi: 10.3171/ped.2006.105.4.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox G.B. Fan L. Levasseur R.A. Faden A.I. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J. Neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- Goodman J.C. Cherian L. Bryan R.M., Jr. Robertson C.S. Lateral cortical impact injury in rats: pathologic effects of varying cortical compression and impact velocity. J. Neurotrauma. 1994;11:587–597. doi: 10.1089/neu.1994.11.587. [DOI] [PubMed] [Google Scholar]
- Grundl P.D. Biagas K.V. Kochanek P.M. Schiding J.K. Barmada M.A. Nemoto E.M. Early cerebrovascular response to head injury in immature and mature rats. J. Neurotrauma. 1994;11:135–148. doi: 10.1089/neu.1994.11.135. [DOI] [PubMed] [Google Scholar]
- Gurkoff G.G. Giza C.C. Hovda D.A. Lateral fluid percussion injury in the developing rat causes an acute, mild behavioral dysfunction in the absence of significant cell death. Brain Res. 2006;1077:24–36. doi: 10.1016/j.brainres.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Hannay H.J. Feldman Z. Phan P. Keyani A. Panwar N. Goodman J.C. Robertson C.S. Validation of a controlled cortical impact model of head injury in mice. J. Neurotrauma. 1999;16:1103–1114. doi: 10.1089/neu.1999.16.1103. [DOI] [PubMed] [Google Scholar]
- Hickey R.W. Adelson P.D. Johnnides M.J. Davis D.S. Yu Z. Rose M.E. Chang Y.F. Graham S.H. Cyclooxygenase-2 activity following traumatic brain injury in the developing rat. Pediatr. Res. 2007;62:271–276. doi: 10.1203/PDR.0b013e3180db2902. [DOI] [PubMed] [Google Scholar]
- Hicks R.R. Baldwin S.A. Scheff S.W. Serum extravasation and cytoskeletal alterations following traumatic brain injury in rats. Comparison of lateral fluid percussion and cortical impact models. Mol. Chem. Neuropathol. 1997;32:1–16. doi: 10.1007/BF02815164. [DOI] [PubMed] [Google Scholar]
- Hicks R.R. Smith D.H. Lowenstein D.H. Saint Marie R. McIntosh T.K. Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J. Neurotrauma. 1993;10:405–414. doi: 10.1089/neu.1993.10.405. [DOI] [PubMed] [Google Scholar]
- Huh J.W. Raghupathi R. Chronic cognitive deficits and long-term histopathological alterations following contusive brain injury in the immature rat. J. Neurotrauma. 2007;24:1460–1474. doi: 10.1089/neu.2006.3787. [DOI] [PubMed] [Google Scholar]
- Huh J.W. Franklin M.A. Widing A.G. Raghupathi R. Regionally distinct patterns of calpain activation and traumatic axonal injury following contusive brain injury in immature rats. Dev. Neurosci. 2006;28:466–476. doi: 10.1159/000094172. [DOI] [PubMed] [Google Scholar]
- Huh J.W. Widing A.G. Raghupathi R. Midline brain injury in the immature rat induces sustained cognitive deficits, bihemispheric axonal injury and neurodegeneration. Exp. Neurol. 2008;213:84–92. doi: 10.1016/j.expneurol.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka H. Sakatani K. Young W. Neural damage in the rat thalamus after cortical infarcts. Stroke. 1990;21:790–794. doi: 10.1161/01.str.21.5.790. [DOI] [PubMed] [Google Scholar]
- Jenkins L.W. Peters G.W. Dixon C.E. Zhang X. Clark R.S. Skinner J.C. Marion D.W. Adelson P.D. Kochanek P.M. Conventional and functional proteomics using large format two-dimensional gel electrophoresis 24 hours after controlled cortical impact in postnatal day 17 rats. J. Neurotrauma. 2002;19:715–740. doi: 10.1089/08977150260139101. [DOI] [PubMed] [Google Scholar]
- Kennard M.A. Reorganization of motor function in the cerebral cortex of monkeys deprived of motor and premotor areas in infancy. J. Neurophysiol. 1938;1:477–496. [Google Scholar]
- Langlois J.A. Kegler S.R. Butler J.A. Gotsch K.E. Johnson R.L. Reichard A.A. Webb K.W. Coronado V.G. Selassie A.W. Thurman D.J. Traumatic brain injury-related hospital discharges. Results from a 14-state surveillance system. MMWR Surveill. Summ. 2003;1997;52:1–20. [PubMed] [Google Scholar]
- Levin H.S. Aldrich E.F. Saydjari C. Eisenberg H.M. Foulkes M.A. Bellefleur M. Luerssen T.G. Jane J.A. Marmarou A. Marshall L.F. Young H.F. Severe head injury in children: experience of the Traumatic Coma Data Bank. Neurosurgery. 1992;31:435–443. doi: 10.1227/00006123-199209000-00008. [DOI] [PubMed] [Google Scholar]
- Levin H.S. Zhang L. Dennis M. Ewing-Cobbs L. Schachar R. Max J. Landis J.A. Roberson G. Scheibel R.S. Miller D.L. Hunter J.V. Psychosocial outcome of TBI in children with unilateral frontal lesions. J. Int. Neuropsychol. Soc. 2004;10:305–316. doi: 10.1017/S1355617704102129. [DOI] [PubMed] [Google Scholar]
- Lyeth B.G. Jenkins L.W. Hamm R.J. Dixon C.E. Phillips L.L. Clifton G.L. Young H.F. Hayes R.L. Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 1990;526:249–258. doi: 10.1016/0006-8993(90)91229-a. [DOI] [PubMed] [Google Scholar]
- Northington F.J. Ferriero D.M. Flock D.L. Martin L.J. Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. J. Neurosci. 2001;21:1931–1938. doi: 10.1523/JNEUROSCI.21-06-01931.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottingham S.A. Springer J.E. Temporal and spatial distribution of activated caspase-3 after subdural kainic acid infusions in rat spinal cord. J. Comp. Neurol. 2003;464:463–471. doi: 10.1002/cne.10806. [DOI] [PubMed] [Google Scholar]
- Ozdemir D. Tugyan K. Uysal N. Sonmez U. Sonmez A. Acikgoz O. Ozdemir N. Duman M. Ozkan H. Protective effect of melatonin against head trauma-induced hippocampal damage and spatial memory deficits in immature rats. Neurosci. Lett. 2005;385:234–239. doi: 10.1016/j.neulet.2005.05.055. [DOI] [PubMed] [Google Scholar]
- Pierce J.E. Smith D.H. Trojanowski J.Q. McIntosh T.K. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 1998;87:359–369. doi: 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- Prins M.L. Fujima L.S. Hovda D.A. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J. Neurosci. Res. 2005;82:413–420. doi: 10.1002/jnr.20633. [DOI] [PubMed] [Google Scholar]
- Prins M.L. Lee S.M. Cheng C.L.Y. Becker D.P. Hovda D.A. Fluid percussion brain injury in the developing and adult rat: a comparative study of mortality, morphology, intracranial pressure and mean arterial blood pressure. Dev. Brain Res. 1996;95:272–282. doi: 10.1016/0165-3806(96)00098-3. [DOI] [PubMed] [Google Scholar]
- Pullela R. Raber J. Pfankuch T. Ferriero D.M. Claus C.P. Koh S.E. Yamauchi T. Rola R. Fike J.R. Noble-Haeusslein L.J. Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. Dev. Neurosci. 2006;28:396–409. doi: 10.1159/000094166. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Huh J.W. Diffuse brain injury in the immature rat: evidence for an age-at-injury effect on cognitive function and histopathologic damage. J. Neurotrauma. 2007;24:1596–1608. doi: 10.1089/neu.2007.3790. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Conti A.C. Graham D.I. Krajewski S. Reed J.C. Grady M.S. Trojanowski J.Q. McIntosh T.K. Mild traumatic brain injury induces apoptotic cell death in the cortex that is preceded by decreases in Bcl-2 immunoreactivity. Neuroscience. 2002;110:605–616. doi: 10.1016/s0306-4522(01)00461-4. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Graham D.I. McIntosh T.K. Apoptosis after traumatic brain injury. J. Neurotrauma. 2000;17:927–938. doi: 10.1089/neu.2000.17.927. [DOI] [PubMed] [Google Scholar]
- Rasband W.S. In: Image J., editor. U.S. National Institutes of Health; Bethesda, MD: 2007. [Google Scholar]
- Saatman K.E. Feeko K.J. Pape R.L. Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J. Neurotrauma. 2006;23:1241–1253. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- Saatman K.E. Graham D.I. McIntosh T.K. The neuronal cytoskeleton is at risk after mild and moderate brain injury. J. Neurotrauma. 1998;15:1047–1058. doi: 10.1089/neu.1998.15.1047. [DOI] [PubMed] [Google Scholar]
- Scheff S.W. Baldwin S.A. Brown R.W. Kraemer P.J. Morris water maze deficits in rats following traumatic brain injury: lateral controlled cortical impact. J. Neurotrauma. 1997;14:615–627. doi: 10.1089/neu.1997.14.615. [DOI] [PubMed] [Google Scholar]
- Shore P.M. Berger R.P. Varma S. Janesko K.L. Wisniewski S.R. Clark R.S. Adelson P.D. Thomas N.J. Lai Y.C. Bayir H. Kochanek P.M. Cerebrospinal fluid biomarkers versus Glasgow Coma Scale and Glasgow Outcome Scale in pediatric traumatic brain injury: the role of young age and inflicted injury. J. Neurotrauma. 2007;24:75–86. doi: 10.1089/neu.2006.0062. [DOI] [PubMed] [Google Scholar]
- Shuman S.L. Bresnahan J.C. Beattie M.S. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J. Neurosci. Res. 1997;50:798–808. doi: 10.1002/(SICI)1097-4547(19971201)50:5<798::AID-JNR16>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Simpson D.A. Cockington R.A. Hanieh A. Raftos J. Reilly P.L. Head injuries in infants and young children: the value of the Paediatric Coma Scale. Review of literature and report on a study. Childs Nerv. Syst. 1991;7:183–190. doi: 10.1007/BF00249393. [DOI] [PubMed] [Google Scholar]
- Smith D.H. Okiyama K. Thomas M.J. Claussen B. McIntosh T.K. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J. Neurotrauma. 1991;8:259–269. doi: 10.1089/neu.1991.8.259. [DOI] [PubMed] [Google Scholar]
- Stone J.R. Singleton R.H. Povlishock J.T. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res. 2000;871:288–302. doi: 10.1016/s0006-8993(00)02485-9. [DOI] [PubMed] [Google Scholar]
- Sutton R.L. Lescaudron L. Stein D.G. Unilateral cortical contusion injury in the rat: vascular disruption and temporal development of cortical necrosis. J. Neurotrauma. 1993;10:135–149. doi: 10.1089/neu.1993.10.135. [DOI] [PubMed] [Google Scholar]
- Teasdale G. Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81–84. doi: 10.1016/s0140-6736(74)91639-0. [DOI] [PubMed] [Google Scholar]
- Tong K.A. Ashwal S. Holshouser B.A. Nickerson J.P. Wall C.J. Shutter L.A. Osterdock R.J. Haacke E.M. Kido D. Diffuse axonal injury in children: clinical correlation with hemorrhagic lesions. Ann. Neurol. 2004;56:36–50. doi: 10.1002/ana.20123. [DOI] [PubMed] [Google Scholar]
- Tong W. Igarashi T. Ferriero D.M. Noble L.J. Traumatic brain injury in the immature mouse brain: characterization of regional vulnerability. Exp. Neurol. 2002;176:105–116. doi: 10.1006/exnr.2002.7941. [DOI] [PubMed] [Google Scholar]
- Wilde E.A. Hunter J.V. Newsome M.R. Scheibel R.S. Bigler E.D. Johnson J.L. Fearing M.A. Cleavinger H.B. Li X. Swank P.R. Pedroza C. Roberson G.S. Bachevalier J. Levin H.S. Frontal and temporal morphometric findings on MRI in children after moderate to severe traumatic brain injury. J. Neurotrauma. 2005;22:333–344. doi: 10.1089/neu.2005.22.333. [DOI] [PubMed] [Google Scholar]
- Yeates K.O. Blumenstein E. Patterson C.M. Delis D.C. Verbal learning and memory following pediatric closed-head injury. J. Int. Neuropsychol. Soc. 1995;1:78–87. doi: 10.1017/s1355617700000138. [DOI] [PubMed] [Google Scholar]