

Abstract
Object
Brain injury from preterm birth predisposes children to cerebral palsy, epilepsy, cognitive delay, and behavioral abnormalities. The CNS injury often begins before the early birth, which hinders diagnosis and concurrent treatment. Safe, effective postnatal interventions are urgently needed to minimize these chronic neurological deficits. Erythropoietin (EPO) is a pleiotropic neuroprotective cytokine, but the biological basis of its efficacy in the damaged developing brain remains unclear. Coordinated expression of EPO ligand and receptor expression occurs during CNS development to promote neural cell survival. The authors propose that prenatal third trimester global hypoxia-ischemia disrupts the developmentally regulated expression of neural cell EPO signaling, and predisposes neural cells to death. Furthermore, the authors suggest that neonatal exogenous recombinant human EPO (rhEPO) administration can restore the mismatch of EPO ligand and receptor levels, and enhance neural cell survival.
Methods
Transient systemic hypoxia-ischemia (TSHI) on embryonic Day 18 in rats mimics human early-third-trimester placental insufficiency. This model was used to test the authors’ hypothesis using a novel clinically relevant paradigm of prenatal injury on embryonic Day 18, neonatal systemic rhEPO administration initiated 4 days after injury on postnatal Day 1, and histological, biochemical, and functional analyses in neonatal, juvenile, and adult rats.
Results
The results showed that prenatal TSHI upregulates brain EPO receptors, but not EPO ligand. Sustained EPO receptor upregulation was pronounced on oligodendroglial lineage cells and neurons, neural cell populations particularly prone to loss from CNS injury due to preterm birth. Postnatal rhEPO administration after prenatal TSHI minimized histological damage and rescued oligodendrocytes and γ-aminobutyric acidergic interneurons. Myelin basic protein expression in adult rats after insult was reduced compared with sham controls, but could be restored to near normal levels by neonatal rhEPO treatment. Erythropoietin-treated TSHI rats performed significantly better than their saline-treated peers as adults in motor skills tests, and showed significant seizure threshold restoration using a pentylenetetrazole increasing-dose paradigm.
Conclusions
These data demonstrate that neonatal rhEPO administration in a novel clinically relevant paradigm initiated 4 days after a global prenatal hypoxic-ischemic insult in rats rescues neural cells, and induces lasting histological and functional improvement in adult rats.
Keywords: cerebral palsy, epilepsy, oligodendrocyte, erythropoietin receptor, γ-aminobutyric acid, preterm brain injury
The incidence of preterm birth in the US continues to rise.32 Infants born before 28 weeks gestation are particularly vulnerable to CNS injury,60 including cerebral palsy, epilepsy, and cognitive and behavioral abnormalities.19 These deficits impart a significant burden to the children, their families, and society.39 Brain injury from preterm birth frequently begins prenatally.57 Current effective interventions to restore neurodevelopment after prenatal insults are limited. Multiple types of injury resulting from preterm birth, including systemic hypoxia-ischemia, converge to hinder neural cell survival, particularly for immature oligodendrocytes and cerebral neurons.57 Impaired neural cell survival and differentiation continues for a prolonged period after the initial injury.37,44
Signaling from the pleiotropic cytokine EPO through its cognate receptor EPOR is essential for CNS development. 9,62 Prenatal developmentally coordinated spatiotemporal expression of both EPO ligand and receptor regulates local neuronal survival after neurogenesis,25,26 while the expression of EPO and EPOR dramatically diminishes postnatally.31 Binding of EPO to EPOR activates cell survival pathways, while receptors without bound EPO ligand appear to promote cell death.13,21,46 After injury to the mature CNS, both EPO and EPOR expression are upregulated as part of an endogenous neuroprotective mechanism.5,6 In the developing brain, however, injuries appear to induce elevated EPOR expression without an apparent concomitant increase in EPO expression.24,51 This disruption of the coordinated expression of ligand and receptor potentially leaves neural cells vulnerable to cell death. Recombinant human EPO administration has been shown to improve markers of brain damage after multiple types of neonatal and adult CNS insults when initiated prior to or within 24 hours of the insult,41 but has never been investigated with a long, clinically relevant interval between the insult and treatment. We propose that the sustained mismatch of EPO ligand and receptor levels after prenatal injury creates a potential therapeutic window for treatment initiated well after the insult occurs.
Third trimester placental insufficiency injures the developing brain, including inducing persistent loss of oligodendroglial lineage cells and GABAergic neurons.43,57 To investigate whether a potential postnatal therapeutic window exists after a prenatal global hypoxic-ischemic insult, rhEPO treatment was initiated in newborn animals after embryonic Day 18 transient uterine artery occlusion. This model induces persistent and pervasive loss of oligodendrocytes and neurons with functional deficits in adult rats,44 similar to other rodent models,17 and consistent with findings observed in human infants with brain injury from early preterm birth.43 After prenatal TSHI on embryonic Day 18, EPOR transcription and expression is markedly upregulated on neural cells, whereas EPO ligand transcription fails to rise, suggesting a loss of the coordinated developmental regulation of ligand and receptor essential for cell survival. We propose that injury to the prenatal developing brain causes sustained elevated neural cell EPOR expression, and that addition of rhEPO can rescue these EPOR-positive neural cells from death. Prenatal TSHI also impairs motor performance and lowers the seizure threshold in adult rats, and these deficits improve after neonatal neuroprotective doses of rhEPO. Neonatal rhEPO after prenatal injury improves neural cell survival and functional outcomes in postinsult adult animals with restoration of myelin production, motor skills, and seizure threshold.
Methods
Prenatal TSHI Insult
Approval to perform the study was obtained from the Institutional Animal Care and Use Committee at Case Western Reserve University School of Medicine. Timed-pregnant Sprague-Dawley rats were purchased (Zivic Laboratories), and TSHI was induced on embryonic Day 18.44 Briefly, a laparotomy was performed on the rats under isoflurane inhalation anesthesia, uterine arteries were occluded for 60 minutes, and the laparotomy was closed. For sham control animals the laparotomy was performed without arterial occlusion. Pups were born at term (embryonic Day 22), except for those pups whose brains were harvested at embryonic Day 19. Fetal loss after the insult was 23%, and 0% for sham controls. Rats were killed and perfused with 4% paraformaldehyde, or tissue was collected and frozen at −80°C for biochemical analyses. All samples were coded for the performance of the histological and biochemical analyses. The experimental paradigm is illustrated in Fig. 1. Except where noted, consistent areas of frontal lobe were analyzed with anatomical and biochemical techniques to minimize potential differences due to regional variation.
Fig. 1.

Summary diagram illustrating the various experiments performed at each time point after TSHI on embryonic Day 18 (E18). Three neonatal rhEPO dosing regimens were used: low (L; 500 IU/kg intraperitoneally on postnatal Day 1 [P1]); moderate (MrEPO; 1000 IU/kg intraperitoneally on postnatal Days 1–3); and high (HighrEPO; 2000 IU/kg intraperitoneally on postnatal Days 1–5). Efforts were made to use as few litters as possible while generating adequate data, including using rats pups from more than 1 litter for every experiment. Two litters of TSHI and 2 sham control litters were used for studies at embryonic Day 19. Three insult and 3 sham control litters were used for PCR, ELISA, and Western blots (WESTERN) at postnatal Days 0, 2, and 5, and cell culture on postnatal Day 1 immunolabeled on postnatal Day 2 (immunocytochemistry [ICC]). For hematocrit (HCT) measurements on postnatal Day 5, 3 control and 3 insult litters were used to generate the pups with one-quarter of each litter receiving either saline or the low, moderate, or high EPO regimen; these brains were collected for other studies (immunohistochemistry [IHC], PCR, and Western blots). For immunohistochemistry at postnatal Days 5 and 9, 4 control and 4 insult litters were used to generate the pups, with one-half of each litter receiving saline or the moderate rhEPO regimen. For motor tests (MOTOR) at postnatal Day 15, 4 control and 4 insult litters were generated with one-quarter of each litter treated with saline or 1 of the 3 EPO regimens. Most saline-treated and high-dose rhEPO-treated rats from postnatal Day 15 were used at postnatal Day 24. For the adult studies, 6 control and 7 insult litters were generated with one-half of each litter treated with saline or the high-dose rhEPO regimen. Adult motor testing was completed first, followed by seizure threshold (SEIZURE). Brains from surviving adults were used for immunohistochemistry and biochemical analyses. The number of samples used for each experiment is listed with the data presentation.
Postnatal rhEPO Treatment
Recombinant human EPO (tissue culture grade 287-TC, R&D Systems) was administered to rat pups by intraperitoneal injection beginning on postnatal Day 1, with a portion of the pups in each litter receiving rhEPO, and an equivalent portion receiving saline. Each pup was tagged for future identification. Three rhEPO dosing regimens were used: low (500 IU/kg for 1 day), moderate (1000 IU/kg for 3 days), and high (2000 IU/kg for 5 days; Fig. 1). The dosing interval of 1 day was chosen based on pharmacokinetic studies.51,61 The low dose approximates the anemia dose currently used in human infants, and the moderate and high doses are neuroprotective doses previously used in young and mature rodents prior to or simultaneous with injury.33,49,50 All dose regimens were used for functional tests in juvenile rats. The moderate dose regimen was used to assess histological changes at postnatal Days 5 and 9. Only the high-dose regimen was used for adult outcome studies, as the objective was to determine whether neonatal rhEPO administered through a critical developmental window could affect the mature CNS after prenatal injury.
Erythropoietin and EPOR Real-Time RT-PCR
Total RNA was extracted from frontal lobe samples. For 18S rRNA positive controls, RNA was extracted from the samples, and for EPO pathway positive controls, kidney samples were used. Complementary DNA was synthesized from 2.5 μg of total RNA. Primers for EPO (GenBank No. EPO: NM_007942), EPOR (GenBank No. EPOR: NM_017002.2) and 18S rRNA were purchased (Integrated DNA Technologies). Real-time PCR was performed and analyzed using the BioRad iQ5 system (BioRad). The comparative cycle threshold (Ct) method was used.40 Standard curves were generated using a 2-fold dilution of the cDNA sample as the PCR template. The transcript level in each sample was normalized to the 18S mRNA (an endogenous reference gene used as an internal control) within that sample, named ΔCt. At least 3 independent experiments were performed and all reactions were performed with technical triplicates. The ratio of EPO ΔCt to EPOR ΔCt shows the relative discrepancy between EPO ligand and receptor transcript levels, with a higher ratio indicating less EPO mRNA present compared with EPOR mRNA.
In Situ Hybridization
An EPO receptor probe was made using complete rat EPOR cDNA as a template. The sequence of antisense primer was GTA ATA CGA CTC ACT ATA GGG AGA GGG GCA GGA AGA TGT TT and the sense primer was GAA TTA ACC CTC ACT AAA GGG TGA GTG TGT CCT GAG CAA CC. Polymerase chain reaction was used to generate EPOR primers with T7 and T3 promoter sequences. The PCR product was purified with the MinElute Gel Extraction Kit (Qiagen). Erythropoietin receptor RNA probes were synthesized using Digoxigenin Oligonucleotide Tailing Kit (Roche). Transcription reaction with T7 RNA polymerase (Digoxigenin RNA Labeling Kit, Roche) synthesized the antisense probe and the transcription with T3 RNA polymerase synthesized the sense probe. Probes were purified with ethanol and tested on agarose gel to confirm the appropriate band. Probes were aliquoted and stored at −80°C. Frozen coronal sections (20 μM) were cut from fixed embryonic Day 19 brains, treated with 1 μg/ml proteinase K, acetylated with triethanolamine buffer, hybridized with the EPOR probe (1:200), incubated with antidigoxigenin antibody (1:2500), equilibrated with buffer and levamisole (0.48 mg/ml), developed in nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate solution (Roche) for 6 hours, dehydrated, and mounted. The entire experiments were repeated 3 times.
Erythropoietin ELISA
Frontal lobe specimens were rinsed with phosphate-buffered saline, weighed, homogenized by ultrasonication in phosphate-buffered saline with protease inhibitor cocktail (Sigma), snap frozen, and stored at −80°C. Serum samples were also snap frozen after collection. Enzyme-linked immunosorbent assay was performed per the manufacturer’s directions using commercial rat and human EPO kits (R&D Systems). After 2 freeze-thaw cycles, homogenates were centrifuged at 8000 rpm for 5 minutes. Supernatants were collected, protein concentration was measured, and ELISA was performed in triplicate. Intensity was measured at 450 nm using a Kinetic microplate reader (Molecular Devices). At least 2 complete experiments were performed.
Western Blots
Frontal lobe samples were weighed, lysed in lysis buffer, centrifuged, pellet resuspended, and protein-concentration determined (Pierce). Equal amounts of protein were applied to 10% sodium dodecyl sulfate-polyacrylamide gels, and electrophoretically transferred onto a polyvinylidene difluoride membrane (Millipore). The membrane was incubated with EPOR or mouse anti-MBP antibody (Sternberg Monoclonals). Visualization was performed with horseradish peroxidase-conjugated goat anti–rabbit antibody (MPBiomedicals), and enhanced chemiluminescence (Pierce). After EPOR or MBP analysis, blots were stripped and reprobed with anti–β-actin (Santa Cruz Biotechnology) antibodies. Band intensity was quantified using a densitometer (Biorad). Three complete experiments were performed.
Immunohistochemical Analysis
Coronal sections (50 μm) were cut from anterior to posterior beginning at the ventral hippocampal commissure (bregma −1.4 mm) and ending at the anterior temporal horn (bregma −3.6 mm).38 Every fourth section was immunolabeled for a specific antigen to avoid any possibility of overlap from using adjacent sections. Sections were incubated with block, primary antibody, appropriate secondary antibody, Vectastain (Vector Laboratories), and diaminobenzidine, and dehydrated and mounted. Images were captured using a Leica microscope and Open Lab 3.17 (Improvision, PerkinElmer). For neonatal pups, images and labeled cell counts were obtained bilaterally at the level of the corpus callosum genu from the following areas: cleaved caspase-3 (Cell Signaling) at postnatal Days 5 and 9 in periventricular white matter; GAD-67 (Chemicon) at postnatal Day 9 in the cingulate region; O4-immunopositive oligodendrocytes44 at postnatal Day 9 in periventricular white matter and corpus callosum; and EPO (SC-7956, Santa Cruz Biotechnology) and EPOR (AF1390, R&D Systems; M-20, Santa Cruz Biotechnology) at postnatal Days 2, 5, and 9 in the subplate region superolateral to the lateral ventricle/hippocampus, and periventricular white matter. Both commercial EPOR antibodies produced the same pattern of immunolabeling. Sections incubated without primary antibodies were used to test the specificity of both EPOR antibodies. For GAD-67 or parvalbumin (Sigma)–labeled neurons at postnatal Day 24 and in adults, bilateral images were obtained from each section of the deep parietal trunk area (bregma −2.3 mm to −3 mm).38 To optimize comparisons between groups and minimize bias, nuclei of immunolabeled cells were counted in regions of interest from 3–5 fields (magnification 10) obtained from ≤ 3 sections per rat. For EPOR and O4 double-labeling in vivo, frozen floating sections were cut at 40 μm, blocked in 10% fetal calf serum, incubated sequentially with O4, fluorochrome Alexa564, anti–EPOR (R&D Systems), rabbit anti–goat immunoglobulin G–conjugated biotin antibodies, and streptavidin-Cy2, and mounted. Images were obtained using a Zeiss confocal microscope. For TUNEL staining to corroborate the activated caspase-3 immunohistochemistry at postnatal Day 9, perfused brains were immersed in 30% sucrose, coronal frozen sections were prepared, and the TUNEL reaction was performed using a commercial kit (Roche). Rats were obtained for each group from 3 separate litters.
Cell Culture
Mixed cerebral single-cell suspensions were plated at 50,000 cells/coverslip on poly-L-lysine-coated coverslips, and incubated overnight in supportive media (N2 supplement, 1% fetal bovine serum, and platelet-derived growth factor A (10 ng/ml)).42 The next day cells were incubated with monoclonal antibody for A2B5-immunopositive oligodendrocyte precursors, O4-immunopositive oligodendrocytes, and O1-immunopositive mature oligodendrocytes, 42 astrocytes (GFAP, Dako), neurons (β-tubulin, T8660 Sigma), and microglia (ED1 [CD68] no. 1435, Chemicon), incubated with rhodamine-conjugated secondary antibodies, anti–EPOR antibodies (R&D Systems), biotin-conjugated rabbit anti–goat antibodies, and Cy2-conjugated anti–rabbit tertiary antibodies. Nuclei were labeled with DAPI (Invitrogen). Controls without primary antibodies were investigated to confirm the specificity of the immunolabeling. Immunolabeling of cells (EPOR, cell type marker, DAPI) was photographed on a Leica microscope using software with consistent exposure settings. Erythropoietin receptor labeling on individual immunolabeled cells with round healthy DAPI-immunopositive nuclei was graded by an observer blinded to insult status semiquantitatively as 0 (no EPOR detected), 1 (faint EPOR immunolabeling), or 2 (strong EPOR-immunopositive) for > 50 cells per cell type per experiment, and 3 complete experiments were performed.
Motor Skills
The global insult produced by the prenatal TSHI results in bilateral damage, and consequently most skills tests designed for evaluation after a focal unilateral insult such as unilateral carotid occlusion are unsuitable. For motor tests, all groups of rats were tested on the same day under the same conditions. The bar hold test was used for juvenile rats,50 and the stride length44 and irregular horizontal ladder performance35 tests were used for adult rats (3–4 months) with the sex also recorded. For the bar hold test, the length of time each rat was able to support itself from a 12-mm diameter bar above a padded surface was recorded for 3 consecutive trials. The average ± SEM individual trial time for each group was calculated. For stride length, adult rats had nontoxic paint applied to their feet, and each rat ran once down a strip of paper covered by an open mesh wire tunnel into a home cage. The maximal unilateral stride length was measured for 5 steps for each rat, and the mean ± SEM for each group obtained.
For the irregular horizontal ladder performance test, a 1-m-long ladder with 27 irregularly spaced rungs, each 8-mm in diameter, was used.35 The ladder was suspended 15 cm above a mirrored surface to facilitate visualizing the rats’ feet. Rats ran toward a home cage and no other inducements were used. The day before testing, each rat had 1 trial in the reverse direction to habituate it to the apparatus. On the day of testing, rats were videotaped as they crossed the ladder. The videotape was reviewed by an observer blinded to the insult and treatment status to assess the time to cross the ladder, the number of times each foot stepped correctly on a rung (step), or dropped below a rung (error), similar to a score of 2 in the scale developed by Metz and Whishaw.35 The number of times both feet fell off simultaneously (drop) was also recorded. The mean number of rear leg errors and total errors per group was calculated. Rats for each group were obtained from at least 2 litters.
Pentylenetetrazole Seizure Threshold
An escalating dose paradigm with PTZ was used to define the average dose necessary to induce seizures, or seizure threshold.2 Each rat was uniquely marked, sex was recorded, and rats were weighed on each testing day. The first day rats were administered 20 mg/kg of PTZ intraperitoneally, and every third day afterward they received a PTZ dose increased by 10 mg/kg. The latency to the 3 seizure grades was recorded with a maximal observation period of 30 minutes. Grade I was head jerk only, Grade II was head-to-tail jerk, and Grade III was a generalized tonic-clonic seizure.15 A rat was removed from testing once it experienced a Grade III seizure. If a rat did not have a Grade III seizure, it was administered the increased dose on the next testing day. Rats for each group were obtained from at least 2 litters.
Quantification and Statistics
All observations and labeled cell counts were performed by an observer blinded to the insult and treatment status. Results were collected from rats from at least 2 litters for each study, and at least 3 independent experiments were performed (the exact number of experiments is noted in the figure legends), except for hematocrits (2 experiments). Results are expressed as the mean ± SEM. Statistical analyses were performed using SPSS version 11.5 (SPSS Inc.). Differences between the means for insult and sham control pups were compared using the Student 2-tailed t-test. Differences in the means between the multiple treatment groups were compared using 2-way ANOVA, with Bonferroni correction for post hoc analysis of ≥ 3 groups. Statistical significance was set at p ≤ 0.05.
Results
Prenatal Injury, EPO Ligand, and Receptor Transcription and Expression
Real-time RT-PCR with the comparative Ct technique was performed on perinatal frontal lobe samples to determine mRNA transcription, with ribosomal 18S used as an endogenous control in each sample (ΔCt = Ct(target gene)− Ct(18S)). For the EPO ligand more cycles were required to reach the fixed threshold in both sham controls and those animals with brain insult at postnatal Day 5, compared with postnatal Days 0 and 2 (4–7 cycles; p < 0.01, 2-way ANOVA), consistent with the postnatal downregulation of EPO signaling (Fig. 2a). No significant differences in EPO transcripts were detected between control and insult animals at any age. While EPOR transcript levels were relatively stable in sham controls from embryonic Day 19 to postnatal Day 5 (Fig. 2b), the embryonic Day 18 TSHI insult induced a marked upregulation of EPOR transcription on embryonic Day 19 (n = 9) compared with sham controls (n = 7; p = 0.04, 2-tailed t-test); that is, only approximately 10 cycles were required to reach the fixed threshold after the insult, while approximately 12.5 cycles were required in sham controls to reach the same threshold. The ratio of EPO ligand and receptor mRNA transcript levels in sham controls illustrates the gradual decrease in rate-limiting EPO ligand relative to EPOR transcripts during the perinatal period (Fig. 2c). Because a higher cycle threshold (ΔCt) reflects the presence of fewer target gene mRNA transcripts, a higher EPOΔCt/EPORΔCt ratio means a greater mismatch is likely with less EPO ligand mRNA present compared with receptor mRNA. After TSHI on embryonic Day 18, the insult ligand/receptor ΔCt ratio is about 30% higher on embryonic Day 19, indicating disruption of the developmentally coordinated regulation of ligand and receptor transcription. The ratio returns to the control level by postnatal Day 5. In situ hybridization performed on embryonic Day 19 localized the sites of increased EPOR mRNA in the cortex, subplate, and white matter after the TSHI on embryonic Day 18 (Fig. 2d). The increase in EPOR mRNA without concomitant rise in EPO ligand transcripts increases the discrepancy between ligand and receptor levels, and suggests that the potential response to rhEPO may be extended throughout a relatively long therapeutic window after the insult in the damaged developing brain.
Fig. 2.

Graphs and images showing that prenatal TSHI on embryonic day 18 disrupts the coordinated developmental transcription of EPO ligand and receptor in the cerebrum in vivo. a: In sham controls, RT-PCR using the Ct technique shows stable EPO ligand transcription from embryonic Day 19 (E19) through postnatal Day 2 (P2), and that more cycles are required to reach the threshold at postnatal Day 5, consistent with postnatal downregulation of EPO (*p < 0.01). Transcription levels appear unaffected by prenatal injury. A lower Ct for a target gene indicates fewer cycles are necessary to reach the threshold, and therefore that more mRNA transcripts are present in a sample, with each sample normalized to the endogenous ribosomal 18S mRNA (ΔCt). b: Reverse transcriptase PCR for EPOR demonstrates the TSHI insult induces more EPOR mRNA transcription at embryonic Day 19 compared with sham controls (*p = 0.043), disrupting the developmentally coordinated expression of ligand and receptor. c: During development the threshold cycle ratio (ΔCt EPO/ΔCt EPOR) for EPO ligand to receptor in controls gradually increases from embryonic Day 19 to postnatal Day 5, consistent with a rate-limiting amount of ligand regulating receptor-mediated signaling. Because a higher ΔCt means fewer mRNA transcripts are present, a higher EPO to EPOR ΔCt ratio indicates a greater discrepancy between the amount of EPO and EPOR transcription. After the TSHI insult on embryonic Day 18, the ratio is increased at embryonic Day 19 by about 30% compared with sham controls, and returns to control levels by postnatal Day 5. d: In situ hybridization with EPOR on embryonic Day 19 localized the marked increase in EPOR transcription after the insult compared with sham controls, particularly in the subplate (sp) and overlying cortex (ctx). Upper panels, Bar = 100 μm. Dotted line in higher magnification of boxed regions (lower panels) demarcates the gray matter-white matter junction just below the subplate. Bar = 10 μm. e: Enzyme-linked immunosorbent assay for EPO ligand shows developmental decrease in EPO expression from embryonic Day 19 through postnatal Day 5, with no significant difference induced by embryonic Day 18 injury (p = 0.003). f: Representative Western blot of EPOR and β-actin from postnatal Day 0 brain samples. g: Western blot EPOR levels at postnatal Days 0 and 5 from the frontal lobe brain, standardized to β-actin levels, showed a 2-fold increase in EPOR expression after the insult (n = 5–10; *p = 0.037, **p < 0.001).
Protein levels of EPO ligand and receptor followed similar trends to mRNA. A gradual decrease in EPO ligand was found in sham controls from embryonic Day 19 (n = 4) through postnatal Day 5 using ELISA (n = 8; p = 0.003, 2-way ANOVA; Fig. 2e), and this was not altered by injury on embryonic Day 18. In contrast to the lack of change in ligand levels after injury, frontal lobe Western blots showed that embryonic Day 18 TSHI induces an increase in EPOR expression at postnatal Day 0, which is sustained through postnatal Day 5 (Fig. 2f). Quantification of Western blots showed an increase in EPOR expression after prenatal insult at postnatal Days 0 (n = 5) and 5 (n = 10), compared with sham controls (postnatal Day 0: n = 7, p = 0.037, t-test; postnatal Day 5: n = 10, p = 0.03; Fig. 2g). Ligand and receptor expression was also assayed using immunohistochemistry. No significant difference was observed in the EPO ligand between postnatal Days 2 and 9 in sham controls or after embryonic Day 18 in the TSHI group (Fig. 3a). Unlike the lack of change in EPO ligand expression after injury, increased cerebral neural cell EPOR expression was observed diffusely in both the white and gray matter from postnatal Day 2 through postnatal Day 9, up to 13 days after the embryonic Day 18 insult (Fig. 3b), and in contrast to the relative progressive decrease in EPOR expression observed in sham controls during the same interval. Detailed examination of EPO ligand expression at postnatal Day 5 in the subplate and white matter did not reveal obvious differences between sham control and insult brains (Fig. 3c). Although fewer subplate cells were present at postnatal Day 5 after TSHI (control animals: 316 ± 27 cells/mm2; insult animals: 251 ± 13 cells/mm2; n = 3; p < 0.01), the percentage of postnatal Day 5 subplate cells immunolabeled by EPO antibodies was similar in control and insult rats (61 ± 4%). In contrast to the apparently stable EPO expression, increased EPOR expression was present in cortical cells with neuronal morphology in all cortical layers, but the increase was most prominent and persistent in the subplate (Fig. 3d). At postnatal Day 5 the proportion of subplate cells labeled with anti-EPOR antibodies was 72 ± 8% (n = 3; EPOR-immunopositive: 191 ± 18 cells/mm2; total: 265 ± 26 cells/mm2) after embryonic Day 18 TSHI, compared with 53 ± 9% in sham controls (n = 3; EPOR-immunopositive: 153 ± 27 cells/mm2; total: 289 ± 21 cells/mm2; p = 0.0001, t-test). Similarly, the number of EPOR-immunopositive cells in the white matter at postnatal Day 5 increased after the insult (n = 4; 250 ± 23 cells/mm2), compared with sham controls (n = 4; 171 ± 22 cells/mm2; p = 0.001). After prenatal TSHI, cerebral neural cell EPOR transcription and expression are increased in the developing CNS in both gray and white matter through postnatal Day 9, almost 2 weeks after the initial insult, whereas the insult does not markedly alter EPO ligand levels during the same period. The excess expression of EPOR after TSHI without a concomitant increase in EPO suggests that the mismatch between EPO ligand and receptor levels occurs after prenatal TSHI.
Fig. 3.

Images demonstrating that TSHI insult on embryonic Day 18 has minimal effect on postnatal EPO ligand expression, but alters expression of the EPOR. a: Erythropoietin ligand expression visualized with immunoperoxidase labeling gradually decreases postnatally from postnatal Day 2 through Day 9 in the cortex (ctx), subplate (sp), and white matter (wm) in sham controls, with minimal difference induced by the prenatal injury. Bar = 100 μm. b: Erythropoietin receptor immunolabeling also gradually decreases from postnatal Day 2 through Day 9 in sham controls, but remains elevated through postnatal Day 9 after the embryonic Day 18 insult, especially in the subplate. Bar = 100 μm c: Immunolabeling for the EPO ligand at postnatal Day 5 did not demonstrate any difference in the cortex or subplate after the embryonic Day 18 insult, and fewer cells appear immunolabeled with EPO ligand antibodies in the white matter after the embryonic Day 18 TSHI. Insets shown on right side for control (upper) and insult (lower). Bar = 100 μm. d: Erythropoietin receptor immunolabeling in the subplate and periventricular white matter at postnatal Day 5. Although fewer cells are present in the subplate after the insult, a greater proportion of the remaining subplate cells are labeled with EPORs by immunoperoxidase staining. More white matter cells express EPORs after the insult, in contrast to the lack of increased ligand expression observed after the insult. Bar = 10 μm.
Erythropoietin Receptor Presence on Immature Oligodendrocytes and Neurons
To determine the specific CNS cell types that expressed EPOR, double-labeling for EPOR and neural cells was performed. Erythropoietin receptors were present on O4-immunopositive oligodendrocytes in the white matter in sham controls in vivo (Fig. 4a). To allow more detailed evaluation of EPOR expression after prenatal TSHI on different neuronal and glial cell types, immunocytochemistry was performed on postnatal Day 2 equivalent (postnatal Day 1 + 1 day in vitro) cells. In sham controls, EPOR expression was found on all 3 stages of oligodendroglial cells examined. Indeed, 45 ± 3% of sham control O4-immunopositive immature oligodendrocytes had EPOR labeling present primarily on the cell body and proximal processes (Fig. 4b). In cultures derived from ischemic animals, the proportion of O4-immunopositive cells with EPOR expression increased 2-fold to 85 ± 5%. The intensity of EPOR labeling on each cell was estimated semiquantitatively (0 = none, 1 = faint, 2 = bright). The average intensity of EPOR expression per cell on O4-immunopositive cells increased markedly after embryonic Day 18 TSHI (n = 100) compared with sham controls (n = 86; p = 0.0001, t-test; Fig. 4c). The increase in EPOR on A2B5-immunopositive oligodendrocyte precursor cells after injury (99 cells) was also significant (116 controls; p = 0.02). No significant difference in EPOR expression was observed on mature O1-immunopositive oligodendrocytes (29 controls, 49 insult). Elevated EPOR expression was also found on other cell types in the brain after prenatal TSHI, including β-tubulin-positive neurons (713 controls, 432 insult; p < 0.0001), GFAP-positive astrocytes (340 in control group, 298 in insult group; p < 0.0003), and ED1-positive microglia (39 control, 51 insult; p < 0.0001; Fig. 4b and d). While the persistent excess expression of EPOR may contribute to neural cell loss if not adequately matched by sufficient ligand, the sustained EPOR expression also suggests a potential therapeutic window wherein the damaged developing CNS may potentially benefit from rhEPO administered postnatally.
Fig. 4.
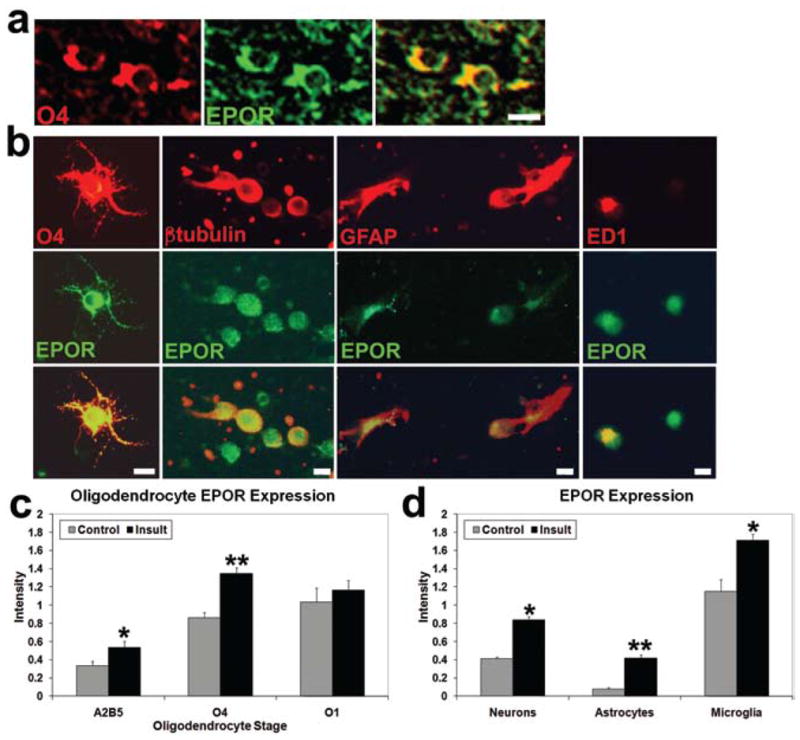
Images showing EPORs are present on neural cells in the developing brain, including oligodendroglial lineage cells. a: White matter (corpus callosum) at postnatal Day 5 double-labeled in vivo with O4 and EPOR antibodies show EPORs are present on O4-immunopositive oligodendrocytes. Bar = 10 μm. b: Frontal lobe cells from postnatal Day 1 and 1 day in vitro sham control pups double-labeled with O4 and EPOR antibodies in vitro show EPORs are present on oligodendroglial cell bodies and processes. Similar cultures from insult animals showed EPOR-immunopositive expression on β-tubulin–positive neurons, GFAP-positive astrocytes, and ED1-positive microglia. Bar = 10μm. c: A gradual increase in EPOR expression on individual postnatal Day 2 equivalent (postnatal Day 1 and 1 day in vitro) sham control cells in vitro visualized using double-labeling with immunofluorescence correlates with the maturation stage of cells in the oligodendroglial lineage, with more EPOR labeling visible on more mature oligodendroglial lineage cells (Semiquantitative Intensity Scale: 0 = none, 1 = faint, 2 = strong). After the embryonic Day 18 insult the average intensity of EPOR expression per cell is elevated compared with sham control cells (*p < 0.02, **p < 0.0001). d: The average intensity of EPOR expression per cell after the embryonic Day 18 insult was also increased in vitro at postnatal Day 2 on neurons, astrocytes, and microglia (*p < 0.0001, **p = 0.0003).
Exogenous EPO and Histological Signs of Damage in Vivo
To investigate the impact of neonatal rhEPO after TSHI on embryonic Day 18, 3 dosing regimens were used: low (500 IU/kg on postnatal Day 1), moderate (1000 IU/kg on postnatal Days 1–3), and high (2000 IU/kg on postnatal Days 1–5). The 3 dosing regimens were designed to identify an effective dose and define a therapeutic window, given the prolonged period of increased EPOR expression observed after the prenatal injury. Human EPO levels in rat serum were elevated after 2 days of systemic rhEPO administration, while human EPO levels were near zero without rhEPO administration (Fig. 5a). This suggests that intraperitoneal injection of rhEPO is readily absorbed and distributed within the vasculature. Hematocrits were measured on postnatal Day 5 after the different dosing regimens to assess the impact of systemic rhEPO administration. The TSHI on embryonic Day 18 lowered the postnatal Day 5 hematocrit of saline-treated TSHI pups (n = 6) compared with sham controls (n = 10; p = 0.003, 2-tailed t-test; Fig. 5b). The low (n = 5) and moderate (n = 5) dosing regimens did not alter the hematocrit, but the high-dose regimen increased the postnatal Day 5 hematocrit by 12% in both control and insult pups (n = 5; p < 0.001, 2-way ANOVA). The mild increase in hematocrit induced by the high-dose rhEPO regimen counterbalanced the injury-induced decrease, with the end result that the hematocrit of high-dose rhEPO-treated insult pups at postnatal Day 5 did not differ from the normal hematocrit of saline-treated sham control pups.
Fig. 5.
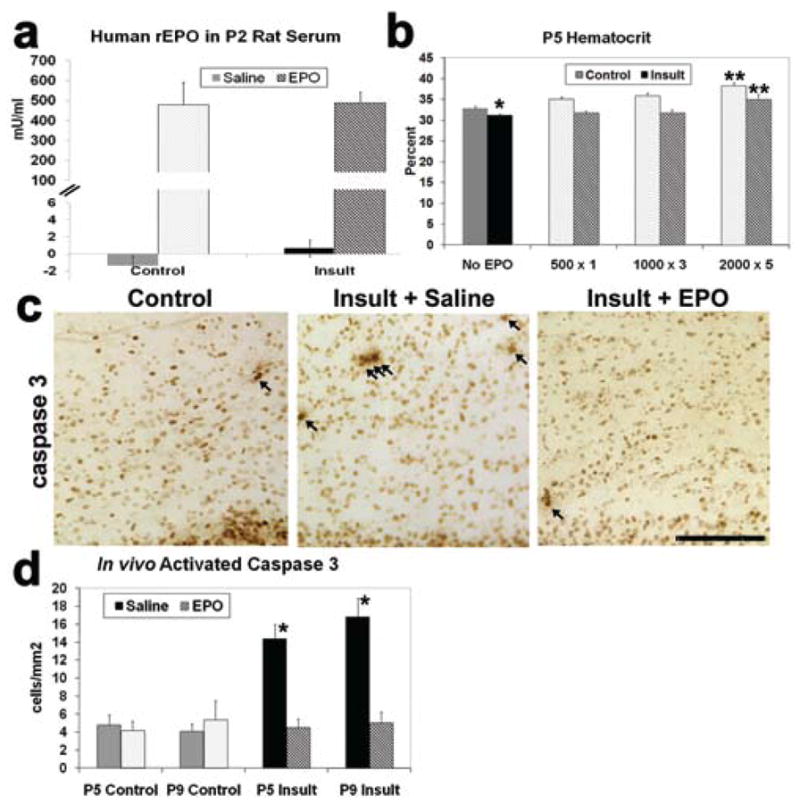
Neonatal intraperitoneal administration of exogenous rhEPO results in systemic absorption, influences the hematocrit, and decreases the number of activated caspase-3–positive cells in the brain. a: Serum human rhEPO levels collected on postnatal Day 2, 6 hours after rhEPO (2000U/kg intraperitoneally on postnatal Days 1–2) and measured by ELISA, were elevated from baseline of zero, with no difference noted between insult and control rats. b: Hematocrit comparison between control and insult rats at postnatal Day 5 after treatment with saline or 1 of the 3 rhEPO dosing regimens. The hematocrit of saline-treated insult pups is significantly lower than saline-treated sham controls (*p = 0.003), reflecting the impact of the systemic insult on embryonic Day 18. The high-dose rhEPO regimen (2000 IU/kg for postnatal Days 1–5) increases the hematocrit in both control and insult rats compared with the other 3 groups (saline, low, or moderate dose rhEPO regimens: **p < 0.0001). The hematocrit elevation induced by high-dose rhEPO in insult pups is counterbalanced by the insult-induced decrease in hematocrit. After high-dose rhEPO treatment, insult pup hematocrit does not differ from normal saline-treated sham control hematocrits. c: Anticleaved caspase-3 antibodies visualized with immunoperoxidase-labeled cells in the periventricular white matter undergoing cell death at postnatal Day 5. The number of cleaved caspase-3–positive cells is increased (arrows) after TSHI on embryonic Day 18 compared with sham controls. Neonatal EPO treatment (1000 U/kg on postnatal Days 1–3) causes a marked decrease in the number of activated caspase-3–positive cells. Bar = 100 μm. d: Quantification of cleaved caspase-3–positive expression in the periventricular white matter at postnatal Days 5 and 9 (*p < 0.0001).
Transient systemic hypoxia-ischemia on embryonic Day 18 causes increased cleaved caspase-3 expression, a marker of cell death, in white matter that persists for 2 weeks after the prenatal insult.44 To determine whether rhEPO treatment altered caspase-3 activation, the moderate rhEPO dosing regimen was administered (1000 U/kg on postnatal Days 1–3). Neonatal rhEPO markedly diminished cleaved caspase-3 expression in postinsult rat white matter at postnatal Day 5 (Figs. 5c and d) and postnatal Day 9 (Fig. 5d) to levels similar to sham controls, while saline showed no effect (n = 3–6; p < 0.0001, 2-way ANOVA). The lack of cell death at postnatal Day 9 was confirmed with TUNEL labeling (data not shown). In this model of prenatal TSHI, postnatal rhEPO administration initiated 4 days after the prenatal insult markedly reduced markers of cell death through postnatal Day 9, almost 2 weeks after the initial insult.
Presence of Neural Cells With Neonatal rhEPO After Prenatal TSHI
Significantly fewer O4-immunopositive oligodendrocytes were found in the periventricular white matter at postnatal Day 9 in saline-treated insult pups, compared with sham controls (n = 3–4; p < 0.0001; Fig. 6a and b). Neonatal rhEPO after prenatal TSHI restored the number of O4-immunopositive cells to near baseline (p < 0.0001; 2-way ANOVA). To determine whether myelin formation was influenced by neonatal rhEPO treatment, MBP expression in postnatal Day 24 and adult white matter was analyzed by Western blots (Fig. 6c and d). Less MBP was present at both ages in postinsult rats compared with sham controls (n = 4–6, adult; p = 0.005), and MBP levels were increased in EPO-treated insult animals (2000 IU/kg for 5 days) compared with saline-treated animals (adult: p = 0.002, 2-way ANOVA), suggesting that rhEPO after prenatal TSHI both restores oligodendrocyte numbers in the developing brain, and produces sustained improvement in adult myelination.
Fig. 6.
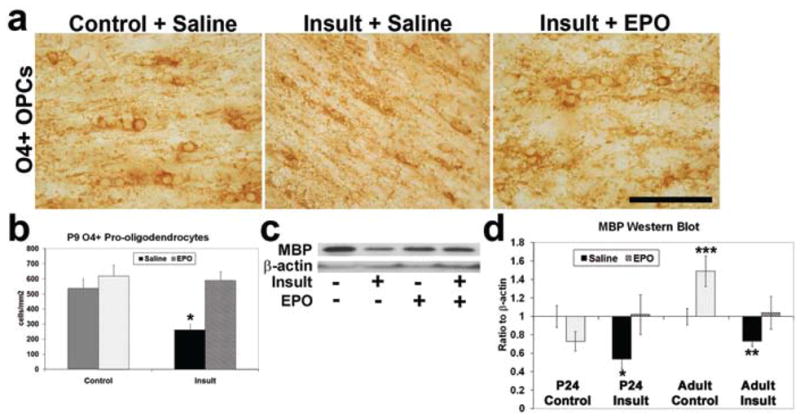
Neonatal rhEPO treatment after prenatal TSHI restores oligodendrocyte development and myelin production. a: Neonatal rhEPO treatment restores the number of O4-immunoperoxidase labeled oligodendrocyte lineage cells observed in the corpus callosum in vivo in postinsult rats at postnatal Day 9. Bar = 10 μm. OPCs = oligodendrocyte precursor cells. b: The number of O4-immunopositive oligodendrocytes immunolabeled at postnatal Day 9 is diminished in the periventricular white matter after prenatal TSHI, compared with sham controls, and restored to control density with rhEPO (*p < 0.0001). c: Western blot of MBP in juvenile (postnatal Day 24) periventricular white matter from saline-treated sham control and prenatal TSHI insult animals, and from EPO-treated controls and insults. d: Relative proportion of MBP to β-actin at postnatal Day 24 and in adults shows insult animals had significantly less MBP present (*p = 0.028). Myelin production in rhEPO-treated postinsult adults was restored (**p = 0.002), and supranormal MBP production appeared in adult EPO-treated sham controls (***p = 0.001, 2-way ANOVA).
The impact of neonatal rhEPO treatment on GABAergic cortical neurons was assayed, as perinatal brain damage from prematurity results in loss of GABAergic neurons in humans and rodents.17,43 In rhEPO-treated (1000 IU/kg for 3 days) postinsult rats, significantly more GAD-67–immunopositive neurons were present in the cingulate gyrus at postnatal Day 9 compared with saline-treated postinsult rats (n = 4; p < 0.0001, 2-way ANOVA; Fig. 7a). At postnatal Day 9, neurons in rhEPO-treated animals also appeared to be more robust and to have longer neurites. Gamma-aminobutyric acidergic neuronal development is primarily completed by 3 weeks in rats. Loss of GABAergic neurons was sustained at postnatal Day 24 in postinsult animals (n = 11) compared with sham controls (n = 10; p = 0.012; Fig. 7b), suggesting the loss of GAD-67–immunopositive cells found at postnatal Day 9 reflects a persistent loss of labeled cells, and not simply delayed appearance of this population. The loss was reversed by neonatal rhEPO (n = 6; p < 0.01, 2-way ANOVA). Fast-spiking parvalbumin cortical interneurons comprise a GABAergic subpopulation whose function is critical for inhibitory circuit formation,11 and are lost in postmortem preterm infants with white matter loss.20 Significant loss of parvalbumin-immunopositive neurons (n = 7) was present at postnatal Day 24 after embryonic Day 18 TSHI in deep parietal cortical layers (primary somatosensory area) compared with controls (n = 5; p < 0.0001; Fig. 7c and d), a cortical region known to be affected by perinatal brain injury.48 After neonatal rhEPO (2000 IU/kg from postnatal Days 1–5) the number of neurons present after the prenatal TSHI injury (n = 5) was restored to control levels (p < 0.0001, 2-way ANOVA). The loss of parvalbumin-immunopositive neurons persisted in adult insult rats (n = 5; control, n = 9; p = 0.0001), and improvement was observed after neonatal rhEPO (Fig. 7e). Together, these results show that neonatal rhEPO enhances the recovery of both GABAergic neurons and the oligodendrocyte lineage in both the developing and mature brain after a prenatal TSHI insult.
Fig. 7.
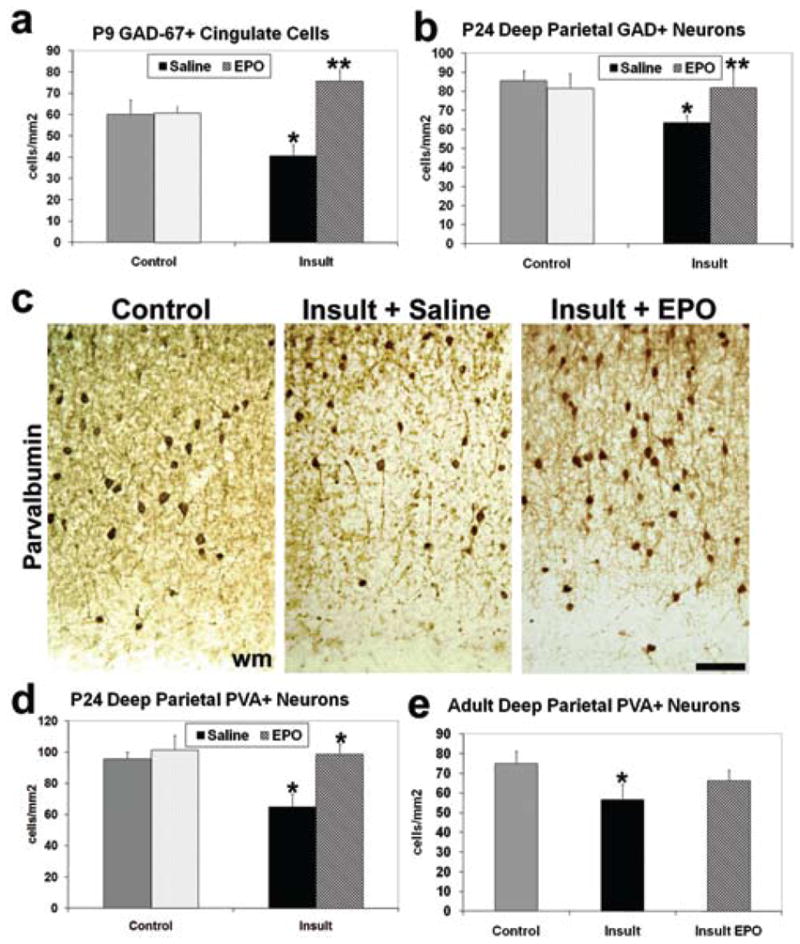
Exogenous neonatal EPO treatment restores GABAergic neuronal counts after prenatal insult. a: Loss of GAD-67 immunoperoxidase-labeled cells in layer IV of the cingulate gyrus in vivo occurs in insult rats at postnatal Day 9, compared with sham controls (*p < 0.0004), and is restored after neonatal rhEPO treatment (**p < 0.0001), comparable to sham controls. b: At postnatal Day 24, loss of GAD-67–immunopositive cells persists in the deep parietal cortex of postinsult rats compared with sham controls (*p < 0.0001), and is restored in rhEPO-treated postinsult rats compared with saline-treated insult rats (**p < 0.01). c: Parvalbumin (PVA) immunoperoxidase-labeling in postnatal Day 24 deep parietal cortex shows the loss of PVA-immunopositive neurons after the insult, and improvement with rhEPO treatment. Bar = 100 μm. d: Graph shows significant loss of PVA-immunopositive neurons at postnatal Day 24 after the embryonic Day 18 insult, and after rhEPO treatment in insult rats, numbers comparable to controls (*p < 0.0001). e: In adult rats, significantly fewer deep parietal PVA-immunopositive neurons are present after the embryonic Day 18 insult (*p < 0.0001). Improvement in PVA-immunopositive neuronal number occurs after neonatal EPO treatment.
Motor Skills Improvement in rhEPO-Treated Rats After Prenatal Insult
Prenatal TSHI also induces motor deficits in adult rats that were reversed with neonatal rhEPO. At postnatal Day 15 the length of time each rat held onto a bar during individual trials was shorter after prenatal TSHI (n = 24, 8.6 ± 0.7 seconds), compared with controls (n = 19, 12.7 ± 1.6 seconds; p < 0.0001; Fig. 8a). When the postinsult rats were treated with high-dose rhEPO (2000 IU/kg for 5 days, n = 26), the rats’ individual trial times improved to near control levels (p = 0.002, 2-way ANOVA), while a single low dose of rhEPO (500 IU/kg on postnatal Day 1, n = 10) or 3 moderate doses on postnatal Days 1–3 (1000 IU/kg, n = 9) were ineffective. The bar holding test may reflect endurance as well as coordination, but the changes clearly suggest that early improvement in motor performance is achievable after a prenatal insult with neonatal rhEPO in a dose-dependent manner.
Fig. 8.

Graphs showing motor skills and seizure threshold deficits from prenatal TSHI are improved by neonatal rhEPO treatment. a: Insult rats demonstrate dose-responsive improvement in bar holding with neonatal EPO treatment. While a single low nonneuroprotective dose (500 IU/kg) of rEPO on postnatal Day 1, or 3 moderate doses (1000 IU/kg on postnatal Days 1–3) failed to produce significant improvement in individual time trials at postnatal Day 15 on the bar test compared with saline-treated insult rats, a high neuroprotective dose (2000 IU/kg on postnatal Days 1–5) induced significant improvement in insult rats (*p = 0.002, 2-way ANOVA), such that the insult rats were indistinguishable from control rats treated with saline. b: Stride length in adult rats is decreased after the prenatal insult (*p < 0.0001) compared with sham controls, and improves after high-dose neonatal rhEPO (2000 IU/kg for postnatal Days 1–5; **p = 0.014, 2-way ANOVA). c: Adult rats after the prenatal insult made more total errors on the horizontal ladder with irregular rungs than sham controls. Insult rats with high-dose neonatal rhEPO made significantly fewer total errors than saline-treated rats (**p = 0.006, 2-way ANOVA). d–f: The seizure threshold was lowered using PTZ, a GABA antagonist, in an escalating dose paradigm. Seizures were evaluated on a scale with 3 grades. The dose required to induce each seizure grade was significantly lower in postinsult rats, compared with sham controls (*Grade 1: p = 0.012; Grade II: p = 0.02; Grade III: p = 0.03). With high-dose neonatal rhEPO, the PTZ dose necessary to induce each grade of seizure in adult rats after the prenatal insult was significantly higher than in saline-treated postinsult rats (**Grade I: p = 0.0001; Grade II: p = 0.006; Grade III: p = 0.006; 2-way ANOVA), and comparable to doses for control rats.
Motor skills in adult rats were assessed using stride length and the horizontal ladder test after high-dose neonatal rhEPO (2000 IU/kg from postnatal Days 1–5). Stride length was shorter in postinsult adults (n = 48) compared with sham controls (n = 49, p < 0.001). Neonatal rhEPO after a prenatal insult (n = 49) resulted in a longer mean stride length compared with saline-treated postinsult rats (p = 0.014, 2-way ANOVA; Fig. 8b). In the irregular rung horizontal ladder test, rhEPO-treated TSHI rats (n = 29) performed significantly better than their saline-treated peers (n = 28), with a lower number of rear leg (p = 0.005, 2-way ANOVA) and total errors (p = 0.006; Fig. 8c). These results suggest that TSHI-induced prenatal damage is mitigated by rhEPO given during neonatal development, allowing sustained improvement in motor function in adult rats.
Erythropoietin Treatment and Seizure Threshold After Prenatal Injury
The seizure threshold was tested in adult rats using a PTZ (GABA antagonist) dose escalation paradigm.2 For each seizure grade from I to III, a significantly lower PTZ dose was required to induce seizures in rats after prenatal TSHI (n = 34), compared with sham controls (n = 41; Grade I: p = 0.012, Grade II: p = 0.02, Grade III: p = 0.03), indicating that the prenatal insult lowers the seizure threshold (Fig. 8d–f). The lower seizure threshold was restored following high-dose neonatal rhEPO (n = 37) to near control levels, and was significantly higher than the threshold in saline-treated postinsult animals (Grade I: p = 0.0001; Grade II: p = 0.006; Grade III: p = 0.006, 2-way ANOVA). To our knowledge, this is the first demonstration that the seizure threshold in adults after a prenatal insult can be altered by postnatal treatment with rhEPO.
Discussion
Prenatal TSHI and EPO Ligand and Receptor Expression
Coordinated EPO signaling is essential for CNS development, 10,62 and exogenous rhEPO provides neuroprotection after a variety of insults in neonatal and mature animals.3,41 In this study we show that prenatal TSHI injury disrupts the developmentally regulated coordination of EPO ligand and receptor expression on multiple neural cell types, including oligodendroglial lineage cells. Erythropoietin ligand expression did not differ between insults and controls after TSHI on embryonic Day 18, and decreased postnatally. A similar lack of increased EPO expression after injury has been observed in prenatal hypoxia, neonatal excitotoxic (postnatal Day 5) or stroke (postnatal Day 7), or aged traumatic brain injury.1,24,51,53 By contrast, we found EPOR expression on neurons and white matter cells is elevated for almost 2 weeks after TSHI at embryonic Day 18. Elevated EPOR expression was found on oligodendrocytes and neurons, the cell populations most vulnerable to loss in perinatal brain injury. 4,44,58 This is the first demonstration that vulnerable O4-immunopositive oligodendrocytes alter EPOR expression in response to injury. Increased EPOR expression has also been shown in vivo after ischemia in neonatal rats,59 adult mice,5 and in adult human brains.47 The findings in this study suggest that injury to the developing brain increases the baseline level of EPOR expression for a sustained interval after the insult without a concomitant rise in EPO ligand production. Likewise, in adult rats with cerebral cortical contusion, brain EPOR expression was elevated for 7 days while EPO ligand was only elevated for 3 days,30 suggesting that the injury-induced mismatch of ligand and receptor may also occur in the mature brain in some instances. It may be that EPO levels are influenced by both age and injury type. For example, EPO ligand was markedly elevated in the CSF of term and older infants who suffered asphyxia,23 but CNS EPO levels have not been tested in very preterm infants after injury.
In the developing preterm brain, endogenous neuroprotective mechanisms that regulate EPO production may not yet be functionally mature. We are currently investigating if the transcription factor hypoxia-inducible factor 2, which stimulates astrocytes to secrete EPO in the mature brain during hypoxia,8 is functional after TSHI on embryonic Day 18. Erythropoietin signaling also does not occur in isolation, and complex interactions with other cytokines are emerging. For example, EPO acts synergistically with insulin-like growth factor-1 to enhance neuronal survival.14 Tumor necrosis factor-α signaling via tumor necrosis factor receptor-1 is also essential for EPO neuroprotection in the mature brain, as EPO fails to protect brains of tumor necrosis factor receptor-1–deficient mice from middle cerebral artery stroke as it does in wild type mice.52 The impact of these cytokine interactions in the damaged developing brain requires further study.
Neonatal rhEPO Restores Neural Cell Development and Function
The mismatch between EPO ligand and receptor levels after injury suggests a mechanism for neural cell loss after injury, as well as an opportunity to optimize survival of EPOR-immunopositive cells with rhEPO. The therapeutic window for rhEPO as defined by increased EPOR availability may extend days after the insult in the damaged developing brain. Thus, to mimic a typical clinical scenario, we examined whether neonatal rhEPO treatment administered with a significant interval between the insult and treatment could reverse brain damage from a prenatal global insult and result in lasting functional improvement in the mature CNS. Neonatal rhEPO administered after prenatal TSHI reduced signs of damage, including cleaved caspase-3 activation in developing white matter through postnatal Day 9, similar to the decrease in apoptosis found in other models using prior or concurrent rhEPO treatment with injury.27,29,30,36 Neonatal rhEPO after prenatal injury also restored the number of O4-immunopositive oligodendrocytes present at postnatal Day 9, and MBP levels present at postnatal Day 24 and in adult rats, the first demonstration of rhEPO-induced improvement of oligodendrocyte function in adults after prenatal brain injury. Improved myelin formation was also found with rhEPO after spinal cord injury in adult rats.56 We found treatment with neonatal rhEPO also improved survival of GAD-67–positive neurons at postnatal Day 9, and both GAD-67–positive neurons and parvalbumin-positive neurons in older rats. The ability of rhEPO to reverse neuronal damage after multiple types of injury in older animals, including neonatal and adult hypoxia-ischemia, adult concussive injury, and kainate toxicity, is a well-known phenomenon.6,12,54 In this study, neonatal rhEPO administered after prenatal TSHI insult enhanced the survival of oligodendrocytes and neurons with sustained histological improvement in adult rats.
The enhanced neural cell survival observed after neonatal rhEPO translated into functional neurological improvement in juvenile pups in a dose-dependent manner, similar to others using rhEPO for neonatal focal stroke.7,27 The functional benefit for motor skills in this model with diffuse global prenatal injury from a systemic insult was sustained in adult rats after neonatal rhEPO treatment. Other studies that investigated long-term rhEPO effects in rodents after neonatal insults also found lasting improvement at 6 weeks and in adults when EPO was administered coincident with or immediately after injury.18,28,33,50 In our paradigm the extended interval between the embryonic Day 18 TSHI insult and postnatal rhEPO treatment may more accurately reflect the likely clinical scenario for infants born very preterm in which the brain injury likely begins prenatally and is typically diagnosed postnatally.
Children with perinatal brain damage are prone to develop epilepsy.45 In a study of PTZ-induced seizures in previously healthy rats, rhEPO administered prior to PTZ challenge minimized alterations in seizure threshold.55 In this study, neonatal rhEPO elevated the adult PTZ seizure threshold back to normal levels after embryonic Day 18 TSHI, suggesting that the alterations in neuronal development and circuit formation that occur after a prenatal insult and cause epilepsy are potentially reversible. For several reasons related to the anatomical simplicity of the rat compared with humans, the impact of rhEPO in rodents may be muted compared with that possible in humans. To our knowledge this is the first study to demonstrate that a neuroprotective agent administered several days after a prenatal insult can normalize the seizure threshold in the mature brain.
Clinical Implications
Erythropoietin shows particular promise for neuroprotection after perinatal brain injury. Neuroprotective EPO doses do not cause harmful side effects when administered to neonatal rodents.33 Erythropoietin neuroprotection may be particularly useful in neonates because it could enhance oligodendroglial and neuronal maturation during a critical developmental window, decrease injury-induced inflammation, and minimize apoptosis.34 Recent initial Phase I/II studies to test the safety of neuroprotective doses in human preterm infants showed no adverse events occurred.16,22 The safety and efficacy of neuroprotective EPO dosing regimens in neonates born preterm should be specifically evaluated.
Conclusions
Using a novel, clinically relevant treatment paradigm, we found neonatal rhEPO treatment initiated 4 days after TSHI on embryonic Day 18 resulted in histological and functional improvement in adult rats. As EPO binding of EPOR enhances neuronal and oligodendroglial survival and maturation, rhEPO has significant potential to safely improve the neurodevelopmental outcome of high-risk infants born preterm. In addition, EPO signaling will be useful for further investigation of how the developing brain undergoes repair after insults.
Acknowledgments
The authors thank Anne DeChant, Stephanie Eaton, Qing Li, and Elizabeth Shick for their excellent technical assistance with the preparation of this manuscript.
Abbreviations used in this paper
- DAPI
4,6′-diamino-2-phenylindole-dihydrochloride
- ELISA
enzyme-linked immunosorbent assay
- EPO
erythropoietin
- EPOR
EPO receptor
- GABA
γ-aminobutyric acid
- GAD
glutamic acid decarboxylase
- GFAP
glial fibrillary acidic protein
- MBP
myelin basic protein
- PTZ
pentylenetetrazole
- rhEPO
recombinant human EPO
- RT-PCR
reverse transcriptase polymerase chain reaction
- TSHI
transient systemic hypoxia-ischemia
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling
Footnotes
Portions of this work were previously presented in abstract form at the 2006 American Society of Clinical Investigators and Association of American Physicians Joint Meeting, Chicago, Illinois; the 2006 CNS Annual Meeting, Chicago, Illinois; the 2006 AANS Annual Meeting, Greensboro, Georgia; the 2007 American Society of Pediatric Neurosurgeons Annual Meeting, Lani’i, Hawaii; and the 2008 American Society of Clinical Investigators and Association of American Physicians Joint Meeting, Chicago, Illinois.
Disclosure
This work was supported by grants to Dr. Robinson from the National Institute of Neurological Disorders and Stroke, National Institutes of Health, No. K08 NS46486 and R01 NS060765, and from the Rainbow Babies and Children’s Hospital Board of Trustees.
References
- 1.Anderson J, Sandhir R, Hamilton ES, Berman NE. Impaired expression of neuroprotective molecules in the HIF-1alpha pathway following traumatic brain injury in aged mice. J Neurotrauma. 2009;26:1557–1566. doi: 10.1089/neu.2008.0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arrieta O, Palencia G, García-Arenas G, Morales-Espinosa D, Hernández-Pedro N, Sotelo J. Prolonged exposure to lead lowers the threshold of pentylenetetrazole-induced seizures in rats. Epilepsia. 2005;46:1599–1602. doi: 10.1111/j.1528-1167.2005.00267.x. [DOI] [PubMed] [Google Scholar]
- 3.Aydin A, Genç K, Akhisaroglu M, Yorukoglu K, Gokmen N, Gonullu E. Erythropoietin exerts neuroprotective effect in neonatal rat model of hypoxic-ischemic brain injury. Brain Dev. 2003;25:494–498. doi: 10.1016/s0387-7604(03)00039-1. [DOI] [PubMed] [Google Scholar]
- 4.Back SA, Luo NL, Borenstein NS, Volpe JJ, Kinney HC. Arrested oligodendrocyte lineage progression during human cerebral white matter development: dissociation between the timing of progenitor differentiation and myelinogenesis. J Neuropathol Exp Neurol. 2002;61:197–211. doi: 10.1093/jnen/61.2.197. [DOI] [PubMed] [Google Scholar]
- 5.Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, et al. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19:643–651. doi: 10.1097/00004647-199906000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci U S A. 2000;97:10526–10531. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang YS, Mu D, Wendland M, Sheldon RA, Vexler ZS, Mc-Quillen PS, et al. Erythropoietin improves functional and histological outcome in neonatal stroke. Pediatr Res. 2005;58:106–111. doi: 10.1203/01.PDR.0000163616.89767.69. [DOI] [PubMed] [Google Scholar]
- 8.Chavez JC, Baranova O, Lin J, Pichiule P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J Neurosci. 2006;26:9471–9481. doi: 10.1523/JNEUROSCI.2838-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen G, Shi JX, Hang CH, Xie W, Liu J, Liu X. Inhibitory effect on cerebral inflammatory agents that accompany traumatic brain injury in a rat model: a potential neuroprotective mechanism of recombinant human erythropoietin (rhEPO) Neurosci Lett. 2007;425:177–182. doi: 10.1016/j.neulet.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 10.Chen ZY, Asavaritikrai P, Prchal JT, Noguchi CT. Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J Biol Chem. 2007;282:25875–25883. doi: 10.1074/jbc.M701988200. [DOI] [PubMed] [Google Scholar]
- 11.Daw MI, Ashby MC, Isaac JT. Coordinated developmental recruitment of latent fast spiking interneurons in layer IV barrel cortex. Nat Neurosci. 2007;10:453–461. doi: 10.1038/nn1866. [DOI] [PubMed] [Google Scholar]
- 12.Demers EJ, McPherson RJ, Juul SE. Erythropoietin protects dopaminergic neurons and improves neurobehavioral outcomes in juvenile rats after neonatal hypoxia-ischemia. Pediatr Res. 2005;58:297–301. doi: 10.1203/01.PDR.0000169971.64558.5A. [DOI] [PubMed] [Google Scholar]
- 13.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 14.Digicaylioglu M, Garden G, Timberlake S, Fletcher L, Lipton SA. Acute neuroprotective synergy of erythropoietin and insulin-like growth factor I. Proc Natl Acad Sci U S A. 2004;101:9855–9860. doi: 10.1073/pnas.0403172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–421. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 16.Fauchère J-C, Dame C, Vonthein R, Koller B, Arri S, Wolf M, et al. An approach to using recombinant erythropoietin for neuroprotection in very preterm infants. Pediatrics. 2008;122:375–382. doi: 10.1542/peds.2007-2591. [DOI] [PubMed] [Google Scholar]
- 17.Golan H, Huleihel M. The effect of prenatal hypoxia on brain development: short- and long-term consequences demonstrated in rodent models. Dev Sci. 2006;9:338–349. doi: 10.1111/j.1467-7687.2006.00498.x. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez FF, McQuillen P, Mu D, Chang Y, Wendland M, Vexler Z, et al. Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Dev Neurosci. 2007;29:321–330. doi: 10.1159/000105473. [DOI] [PubMed] [Google Scholar]
- 19.Hack M, Taylor HG, Drotar D, Schluchter M, Cartar L, Andreias L, et al. Chronic conditions, functional limitations, and special health care needs of school-aged children born with extremely low-birth-weight in the 1990s. JAMA. 2005;294:318–325. doi: 10.1001/jama.294.3.318. [DOI] [PubMed] [Google Scholar]
- 20.Iai M, Takashima S. Thalamocortical development of parvalbumin neurons in normal and periventricular leukomalacia brains. Neuropediatrics. 1999;30:14–18. doi: 10.1055/s-2007-973450. [DOI] [PubMed] [Google Scholar]
- 21.Junk AK, Mammis A, Savitz SI, Singh M, Roth S, Malhotra S, et al. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2002;99:10659–10664. doi: 10.1073/pnas.152321399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juul SE, McPherson RJ, Bauer LA, Ledbetter KJ, Gleason CA, Mayock DE. A phase I/II trial of high-dose erythropoietin in extremely low birth weight infants: pharmacokinetics and safety. Pediatrics. 2008;122:383–391. doi: 10.1542/peds.2007-2711. [DOI] [PubMed] [Google Scholar]
- 23.Juul SE, Stallings SA, Christensen RD. Erythropoietin in the cerebrospinal fluid of neonates who sustained CNS injury. Pediatr Res. 1999;46:543–547. doi: 10.1203/00006450-199911000-00009. [DOI] [PubMed] [Google Scholar]
- 24.Keller M, Yang J, Griesmaier E, Gorna A, Sarkozy G, Urbanek M, et al. Erythropoietin is neuroprotective against NMDA-receptor-mediated excitotoxic brain injury in newborn mice. Neurobiol Dis. 2006;24:357–366. doi: 10.1016/j.nbd.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Knabe W, Knerlich F, Washausen S, Kietzmann T, Sirén AL, Brunnett G, et al. Expression patterns of erythropoietin and its receptor in the developing midbrain. Anat Embryol (Berl) 2004;207:503–512. doi: 10.1007/s00429-003-0365-y. [DOI] [PubMed] [Google Scholar]
- 26.Knabe W, Sirén AL, Ehrenreich H, Kuhn HJ. Expression patterns of erythropoietin and its receptor in the developing spinal cord and dorsal root ganglia. Anat Embryol (Berl) 2005;210:209–219. doi: 10.1007/s00429-005-0019-3. [DOI] [PubMed] [Google Scholar]
- 27.Kumral A, Ozer E, Yilmaz O, Akhisaroglu M, Gokmen N, Duman N, et al. Neuroprotective effect of erythropoietin on hypoxic-ischemic brain injury in neonatal rats. Biol Neonate. 2003;83:224–228. doi: 10.1159/000068926. [DOI] [PubMed] [Google Scholar]
- 28.Kumral A, Uysal N, Tugyan K, Sonmez A, Yilmaz O, Gokmen N, et al. Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hypoxia-ischemia in rats. Behav Brain Res. 2004;153:77–86. doi: 10.1016/j.bbr.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Lu Z, Keogh C, Yu SP, Wei L. Erythropoietin-induced neurovascular protection, angiogenesis, and cerebral blow flow restoration after focal ischemia in mice. J Cereb Blood Flow Metab. 2007;27:1043–1054. doi: 10.1038/sj.jcbfm.9600417. [DOI] [PubMed] [Google Scholar]
- 30.Liao ZB, Zhi XG, Shi QH, He ZH. Recombinant human erythropoietin administration protects cortical neurons from traumatic brain injury in rats. Eur J Neurol. 2008;15:140–149. doi: 10.1111/j.1468-1331.2007.02013.x. [DOI] [PubMed] [Google Scholar]
- 31.Liu C, Shen K, Liu Z, Noguchi CT. Regulated human erythropoietin receptor expression in mouse brain. J Biol Chem. 1997;272:32395–32400. doi: 10.1074/jbc.272.51.32395. [DOI] [PubMed] [Google Scholar]
- 32.Martin JA, Hamilton BE, Sutton PD, Ventura SJ, Menacker F, Kirmeyer S, et al. Births: final data for 2005. Natl Vital Stat Rep. 2007;56:1–103. [PubMed] [Google Scholar]
- 33.McPherson RJ, Demers EJ, Juul SE. Safety of high-dose recombinant erythropoietin in a neonatal rat model. Neonatology. 2007;91:36–43. doi: 10.1159/000096969. [DOI] [PubMed] [Google Scholar]
- 34.McPherson RJ, Juul SE. Recent trends in erythropoietin-mediated neuroprotection. Int J Dev Neurosci. 2008;26:103–111. doi: 10.1016/j.ijdevneu.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metz GA, Whishaw IQ. Cortical and subcortical lesions impair skilled walking in the ladder rung walking test: a new task to evaluate fore- and hindlimb stepping, placing, and coordination. J Neurosci Methods. 2002;115:169–179. doi: 10.1016/s0165-0270(02)00012-2. [DOI] [PubMed] [Google Scholar]
- 36.Mizuno K, Hida H, Masuda T, Nishino H, Togari H. Pre-treatment with low doses of erythropoietin ameliorates brain damage in periventricular leukomalacia by targeting late oligodendrocyte progenitors: a rat model. Neonatology. 2008;94:255–266. doi: 10.1159/000151644. [DOI] [PubMed] [Google Scholar]
- 37.Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, et al. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci. 2000;20:7994–8004. doi: 10.1523/JNEUROSCI.20-21-07994.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 4. San Diego: Academic Press; 1998. [Google Scholar]
- 39.Petrou S. The economic consequences of preterm birth during the first 10 years of life. BJOG. 2005;112 (Suppl 1):10–15. doi: 10.1111/j.1471-0528.2005.00577.x. [DOI] [PubMed] [Google Scholar]
- 40.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rabie T, Marti HH. Brain protection by erythropoietin: a manifold task. Physiology (Bethesda) 2008;23:263–274. doi: 10.1152/physiol.00016.2008. [DOI] [PubMed] [Google Scholar]
- 42.Robinson S. Systemic prenatal insults disrupt telencephalon development: implications for potential interventions. Epilepsy Behav. 2005;7:345–363. doi: 10.1016/j.yebeh.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson S, Li Q, Dechant A, Cohen ML. Neonatal loss of γ-amino butyric acid pathway expression after human perinatal brain injury. J Neurosurg. 2006;104 (6 Suppl):396–408. doi: 10.3171/ped.2006.104.6.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson S, Petelenz K, Li Q, Cohen ML, Dechant A, Tabrizi N, et al. Developmental changes induced by graded prenatal hypoxic-ischemic insults in rats. Neurobiol Dis. 2005;18:568–581. doi: 10.1016/j.nbd.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 45.Ronen GM, Buckley D, Penny S, Streiner DL. Long-term prognosis in children with neonatal seizures: a population-based study. Neurology. 2007;69:1816–1822. doi: 10.1212/01.wnl.0000279335.85797.2c. [DOI] [PubMed] [Google Scholar]
- 46.Sirén AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci U S A. 2001;98:4044–4049. doi: 10.1073/pnas.051606598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sirén AL, Knerlich F, Poser W, Gleiter CH, Brück W, Ehrenreich H. Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol. 2001;101:271–276. doi: 10.1007/s004010000297. [DOI] [PubMed] [Google Scholar]
- 48.Sizonenko SV, Kiss JZ, Inder T, Gluckman PD, Williams CE. Distinctive neuropathologic alterations in the deep layers of the parietal cortex after moderate ischemic-hypoxic injury in the P3 immature rat brain. Pediatr Res. 2005;57:865–872. doi: 10.1203/01.PDR.0000157673.36848.67. [DOI] [PubMed] [Google Scholar]
- 49.Solaroglu I, Solaroglu A, Kaptanoglu E, Dede S, Haberal A, Beskonakli E, et al. Erythropoietin prevents ischemia-reperfusion from inducing oxidative damage in fetal rat brain. Childs Nerv Syst. 2003;19:19–22. doi: 10.1007/s00381-002-0680-2. [DOI] [PubMed] [Google Scholar]
- 50.Spandou E, Papadopoulou Z, Soubasi V, Karkavelas G, Simeonidou C, Pazaiti A, et al. Erythropoietin prevents long-term sensorimotor deficits and brain injury following neonatal hypoxia-ischemia in rats. Brain Res. 2005;1045:22–30. doi: 10.1016/j.brainres.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 51.Statler PA, McPherson RJ, Bauer LA, Kellert BA, Juul SE. Pharmacokinetics of high-dose recombinant erythropoietin in plasma and brain of neonatal rats. Pediatr Res. 2007;61:671–675. doi: 10.1203/pdr.0b013e31805341dc. [DOI] [PubMed] [Google Scholar]
- 52.Taoufik E, Petit E, Divoux D, Tseveleki V, Mengozzi M, Roberts ML, et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc Natl Acad Sci U S A. 2008;105:6185–6190. doi: 10.1073/pnas.0801447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trollmann R, Strasser K, Keller S, Antoniou X, Grenacher B, Ogunshola OO, et al. Placental HIFs as markers of cerebral hypoxic distress in fetal mice. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1973–R1981. doi: 10.1152/ajpregu.00053.2008. [DOI] [PubMed] [Google Scholar]
- 54.Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J, et al. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosci. 2006;26:1269–1274. doi: 10.1523/JNEUROSCI.4480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uzüm G, Sarper Diler A, Bahçekapili N, Ziya Ziylan Y. Erythropoietin prevents the increase in blood-brain barrier permeability during pentylentetrazol induced seizures. Life Sci. 2006;78:2571–2576. doi: 10.1016/j.lfs.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 56.Vitellaro-Zuccarello L, Mazzetti S, Madaschi L, Bosisio P, Gorio A, De Biasi S. Erythropoietin-mediated preservation of the white matter in rat spinal cord injury. Neuroscience. 2007;144:865–877. doi: 10.1016/j.neuroscience.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 57.Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110–124. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Volpe JJ. Encephalopathy of prematurity includes neuronal abnormalities. Pediatrics. 2005;116:221–225. doi: 10.1542/peds.2005-0191. [DOI] [PubMed] [Google Scholar]
- 59.Wen TC, Rogido M, Genetta T, Sola A. Permanent focal cerebral ischemia activates erythropoietin receptor in the neonatal rat brain. Neurosci Lett. 2004;355:165–168. doi: 10.1016/j.neulet.2003.10.078. [DOI] [PubMed] [Google Scholar]
- 60.Wilson-Costello D, Friedman H, Minich N, Fanaroff AA, Hack M. Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics. 2005;115:997–1003. doi: 10.1542/peds.2004-0221. [DOI] [PubMed] [Google Scholar]
- 61.Xenocostas A, Cheung WK, Farrell F, Zakszewski C, Kelley M, Lutynski A, et al. The pharmacokinetics of erythropoietin in the cerebrospinal fluid after intravenous administration of recombinant human erythropoietin. Eur J Clin Pharmacol. 2005;61:189–195. doi: 10.1007/s00228-005-0896-7. [DOI] [PubMed] [Google Scholar]
- 62.Yu X, Shacka JJ, Eells JB, Suarez-Quian C, Przygodzki RM, Beleslin-Cokic B, et al. Erythropoietin receptor signalling is required for normal brain development. Development. 2002;129:505–516. doi: 10.1242/dev.129.2.505. [DOI] [PubMed] [Google Scholar]