

Abstract
Formation of a transcriptionally competent open complex is a highly regulated multistep process involving at least two intermediates. The rate of formation and stability of the intermediate complexes often determine promoter strength. However, the detailed mechanism of formation of the open complex and the high resolution structures of these intermediates are not known. In this study the structures of the open and intermediate complexes formed on the lacUV5 promoter by Escherichia coli RNA polymerase were analyzed using ‘zero length’ DNA–protein cross-linking. In both the open and the intermediate complexes the core subunits (β′ and β) interact with lacUV5 DNA in a similar way, forming DNA–protein contacts flanking the initiation site. At the same time, the recognition (σ70) subunit closely interacts with the promoter only in the open complex. In combination with our previous results, the data suggest that during promoter recognition contacts of the σ subunit with core RNA polymerase and promoter DNA are rearranged in concert. These rearrangements constitute a landmark of transition from the intermediate to the open complex, identifying the σ subunit as a key player directing formation of the open complex.
INTRODUCTION
Escherichia coli RNA polymerase (RNAP) consists of a catalytic core (α2ββ′ subunits) and a σ70 subunit responsible for promoter recognition and melting. Formation of a transcriptionally competent open complex (RPo) between the RNA polymerase (R) and promoter (P) proceeds in three steps (1–5):
R + P ↔ RPc ↔ RPi ↔ RPo
1 2 3
First, RNAP contacts the surface of the promoter DNA helix forming a closed complex (RPc). Subsequently, the closed complex is transformed into an open complex (RPo) through formation of an intermediate closed complex (RPi). During the RPc→RPi transition there is a conformational change in RNAP (3,4). The RPi→RPo transition is characterized by a change in interaction of the σ subunit with the core subunits (6) and by melting of the DNA at the –10 promoter region (2,4,7–9). Regulation of transcription often occurs by changing the rate of formation and stability of RPc and RPi (10,11); therefore, analysis of the mechanism of the RPc→RPo transition is important for a better understanding of the regulation of gene expression.
The mechanism of the RPc→RPo transition is still not understood. Footprinting methods cannot distinguish contacts of different subunits of RNAP with DNA and therefore give a rather coarse picture of overall organization of promoter complexes. Considerable differences in the structures of RPi and RPo would be expected because DNA is melted during the RPi→RPo transition; however, the RPi- and RPo-specific footprints are very similar (1,4). At the same time, DNA–protein and RNA–protein cross-linking methods allow direct identification of the interacting proteins and detailed analysis of the structures of different transcriptional complexes (12–15).
In this study the structures of the open and intermediate complexes were investigated using a protein–DNA ‘zero length’ cross-linking method (16). This method allows detection of close contacts of RNAP subunits with DNA and therefore gives the overall topography of the DNA–protein interactions on the promoter DNA. As a model, we studied the strong constitutive promoter lacUV5, one of the best-characterized prokaryotic promoters (17). The data suggest that a rearrangement of the interactions of the σ subunit with the promoter DNA plays a central role in formation of the open complex.
MATERIALS AND METHODS
DNA fragments and RNAP
The 290 bp EcoRI–EcoRI fragment bearing two tandem lacUV5 promoters (18) was isolated from pOV10, a plasmid containing this 290 bp fragment cloned in the polylinker of pUC19. The 290 bp fragment was labeled either at the 5′-end using T4 phage polynucleotide kinase or 3′-end-labeled with [α-32P]dATP using the Klenow fragment of DNA polymerase I. The end-labeled 290 bp EcoRI–EcoRI fragment was digested with HaeIII and end-labeled 122 and 167 bp fragments, each containing the lacUV5 promoter, were isolated by PAGE.
The 114 bp randomly labeled fragment containing the lacUV5 promoter was obtained by PCR amplification of the 167 bp fragment using two oligonucleotide primers: (i) 5′-CGAGCATGCCGGAGAGCAGC-3′; (ii) 5′-CTGTTTCCTGTGTGAAATTG-3′.
The lacUV5 promoter start sites are located 62, 126 and 77 bp from the left ends of the 122, 167 and 114 bp fragments, respectively.
An aliquot of 25 ng of the 167 bp fragment and 100 pmol of each oligonucleotide were added on ice to a reaction mixture containing 10 mM KCl, 20 mM Tris–HCl pH 8.8, 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100, 60 µM each dNTP and 17 pM each [α-32P]dGTP and [α-32P]dATP in a final volume of 50 µl. To this was added 5 U Vent (exo–) DNA polymerase and amplification was carried out for 30 cycles in a Gene Amp System (Perkin Elmer). The 114 bp fragment was isolated by PAGE and its identity confirmed by analysis with restriction enzymes.
Escherichia coli RNAP (Eσ70) was isolated by the method of Burgess and Jendrisak (19) modified as described (20). SDS–PAGE showed that at least 80% of the polymerase contained the σ subunit and that the polymerase was at least 90% pure.
Cross-linking of RNA polymerase to the lacUV5-containing DNA fragment and determination of the positions of cross-links along the DNA
DNA–protein cross-linking was conducted as described (21), with the following modifications (see Fig. 1). End-labeled DNA fragments were methylated in the presence of 50 mM dimethylsulfate in 100 mM HEPES, pH 7.4, at 20°C for 3–5 min. The reaction was stopped by addition of 2-mercaptoethanol to 100 mM, DNA was precipitated by ethanol, dissolved in depurination buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, 0.1 mM EDTA) and incubated at 65°C for 60 min. Under these conditions all methylated bases were quantitatively depurinated (16). DNA was precipitated with ethanol and dissolved in 10 mM HEPES pH 7.4, 0.1 mM EDTA. In different experiments from 10 to 40% of DNA molecules contained a single depurinated nucleotide per DNA molecule, as determined by piperidine cleavage of DNA (22) and analysis of DNA by denaturing PAGE.
Figure 1.

Experimental approach for cross-linking and analysis of the cross-linked complexes. A DNA fragment containing two cloned tandem lacUV5 promoters was used in the experiments (18). DNA–protein ‘zero length’ cross-links are formed upon incubation of DNA-binding proteins with partly depurinated DNA (16). Arrows indicate the site of transcription initiation; black circles indicate positions of labeled nucleotides in the fragments; subunits of RNAP are shown as ovals; depurinated sites in DNA are shown as black rectangles. (Left) To map positions of cross-links along DNA, end-labeled DNA fragments were used. Cross-linking results in cleavage of one DNA strand such that the 3′-end of this strand is attached to the protein molecule. Thus, the length of the single-stranded DNA after cross-linking indicates the position of the cross-link with respect to the labeled DNA end and could be determined by analysis of end-labeled DNA in a sequencing gel. (Right) To analyze protein contents of different cross-linked products, the DNA fragment was uniformly labeled by PCR. Isolated cross-linked products were treated with nucleases so that only short, labeled, cross-linked DNA oligonucleotides remained attached to the proteins. Then molecular weights of oligonucleotide-labeled proteins were determined by SDS–PAGE.
Cross-linked complexes of RNAP (400 nmol) with depurinated DNA fragments (100 nmol) were obtained by incubating both at different temperatures for 5 (14°C) or 2 h (37°C) in 30–60 µl of cross-linking buffer (50 mM HEPES pH 7.5, 50 mM NaCl, 5 mM MgCl2, 5% glycerol). High concentrations of RNAP and DNA were used to increase the yield of the cross-linked complexes. Cross-linking was terminated by addition of SDS to a final concentration of 1%. The yield of cross-linked complexes was 2–5% of the total DNA.
Complexes were incubated for 1 h at 37°C and applied to a first dimension gradient SDS–PAGE (3.5–20%, 0.1% SDS, 20 mM Na phosphate, pH 7.0, at 5 V/cm, with buffer recirculation). The radioactive bands corresponding to different cross-linked complexes were cut out of the gel and cross-links were reversed by incubation of the first dimension gel for 2 h in 100 mM glycine–HCl, pH 3.0, at 20°C. DNA was eluted overnight in 0.1% SDS, 5 mM EDTA at 20°C, samples were phenol-extracted, DNA was precipitated by ethanol, washed with 70% ethanol and dissolved in formamide containing 10 mM EDTA. Samples were analyzed in 12% sequencing gels.
It has been found that DNA in cross-linked complexes shows more non-specific cleavage than non-cross-linked DNA (see Fig. 4). This ‘background’ cleavage is likely due to DNA fragmentation during isolation of the cross-linked complexes or during cross-link reversal at acidic pH. In order to determine the real sites of cross-linking the experiments were repeated several times and only strong reproducible cleavage sites were scored.
Figure 4.

Mapping of σ- and β′/β-specific cross-linking sites along lacUV5 promoter DNA. Different RPi (14°C)- or RPo (37°C)-specific cross-linked products were eluted from different β′/β- and σ-specific bands labeled as in Figure 3A. The cross-links were reversed and end-labeled DNA fragments were analyzed by denaturing PAGE. (A) DNA was 5′-labeled on the non-transcribed strand. Dots indicate positions of σ-specific cross-links (complex 4). M, sequencing markers (G→A reaction; 22); DNA, protein-free DNA eluted from the first dimension gel. (B) DNA was 3′-labeled on the transcribed strand. All σ-specific bands were eluted together.
DNase I and KMnO4 footprinting
Intermediate (RPi) and open (RPo) initiation complexes were formed on partially depurinated end-labeled DNA under the conditions used for cross-linking (see above).
DNase I (Boehringer) was added at the appropriate temperature to a final concentration of 1 U/ml, incubated for 1 min and the reaction terminated by addition of EDTA to 10 mM. Alternatively, KMnO4 was added to a final concentration of 10 mM, incubated for 2 min and the reaction terminated by addition of 1 µl of β-mercaptoethanol. The samples were phenol extracted, DNA was precipitated by ethanol, washed with 70% ethanol and dissolved in formamide containing 10 mM EDTA. Samples were analyzed in 10% sequencing gels.
Determination of the protein composition of the cross-linked complexes
Cross-linking of RNAP with the PCR labeled lacUV5 DNA fragment was carried out as described above. The cross-linked complexes were then either digested with nucleases in solution to determine the molecular weights of all proteins cross-linked to DNA or separated in a first dimension gel and gel slices digested to determine the protein composition of different cross-linked complexes (Fig. 1).
Nuclease digestion in solution. After cross-linking, 10 mM CaCl2 was added to a final concentration of 1 mM and DNA was digested in 30 µl with 18 U micrococcal nuclease and 25 U DNase I (Worthington) at 37°C for 1 h (open complexes) or 14°C for 4 h (intermediate complex). Then SDS was added to 1% and the samples were incubated for 1 h at 37°C and loaded on 8% SDS–PAGE gels.
Nuclease digestion in gel slices. After separation of the cross-linked complexes in a first dimension gel (as described above), gel slices were washed twice in 70 ml of 40 mM HEPES pH 7.4, 0.1 mM EDTA for 20 min each time. Then the complexes were digested in 1 ml of buffer (40 mM HEPES pH 7.4, 5 mM MgCl2, 1 mM CaCl2) containing 400 U micrococcal nuclease (Worthington) and 600 U DNase I for 1.5 h. For second dimension electrophoresis the gel slices were washed twice in Laemmli stacking buffer (20 min each time) and placed on top of 8% Laemmli SDS–PAGE gels. After electrophoresis the gels were dried and exposed to film or a PhosphorImager screen.
RESULTS
Experimental approach
During initiation at physiological temperature (37°C) the closed complexes (RPc and RPi) are short-lived and are therefore difficult to study; however, they could be stabilized by formation of the complexes at lower temperatures (0–19°C; 2,3,9). Several studies have shown that these complexes are similar to the kinetic intermediates observed during initiation at 37°C (4,5,23,24). At the lacUV5 promoter, melting of the –10 promoter region (formation of the open complex) occurs between 16 and 24°C (2,8,9); complexes formed below 16°C represent closed intermediates (RPc and RPi) that can be completely converted into the open complex at higher temperature. It has also been shown that RPi is the predominant complex on the lacUV5 promoter at 14°C (1). Therefore, complexes formed at 14 and 37°C were analyzed as RPi and RPo, respectively. Since it has been shown that complexes formed at 14°C after temperature downshift from 37°C do not correspond to kinetically identified RPi (5), RPi was formed by mixing RNAP and promoter DNA at 0°C with a subsequent rise in the temperature to 14°C.
Our primary experimental approach was ‘zero length’ cross-linking of the proteins bound to DNA containing one depurinated base per molecule on average (Fig. 1). Aldehyde groups of deoxyriboses unmasked by depurination can react with ɛ-amino groups of lysines and imidazole rings of histidines with formation of a covalent cross-link between the protein and DNA (16). Cross-linking results in cleavage of one DNA strand such that the 3′-end of this strand is attached to the protein molecule.
In the cross-linking experiments ∼90% of the DNA was bound to RNAP, as determined by gel retardation experiments. The possibility of selective loss of a fraction of the complexes due to poor binding to the modified template is unlikely because it has been shown that the presence of a single depurinated base at many positions along λ prmup-1Δ265 promoter DNA only slightly changes the rate of formation of the open complex (25). In fact, depurination of the bases around the –10 consensus sequence strongly increased the rate of formation of the open complex (25). Similar data were obtained on the lacUV5 promoter using the hydroxyl radical-generated missing nucleosides approach (26). This technique introduces more dramatic changes in DNA structure, such as breakage of the phosphate backbone and addition of two negative charges to the backbone. In our experiments, 10–40% of the DNA contained a single depurinated base (see Materials and Methods).
Potassium permanganate and DNase I footprinting patterns of the complexes formed on partially depurinated and intact lacUV5 promoter DNA (Fig. 2) were very similar to each other and similar to published data (1,27). In particular, sites of preferential reactivity to potassium permanganate in the –10 to +1 region were detected only in RPo. We concluded that partial depurination of lacUV5 DNA does not induce strong changes in the structures of RPi and RPo.
Figure 2.

DNase I and KMnO4 footprinting of the RPi (14°C) and RPo (37°C) complexes formed on intact or partially depurinated (Depur.) lacUV5 promoter DNA. Either transcribed or non-transcribed DNA strands were end-labeled as indicated. Note that some depurinated sites (labeled with dots) are preferentially sensitive to DNase I (these bands are not detectable without DNase I digestion) and therefore are over-represented in the gel. Multiple bands present in the lanes containing KMnO4-treated depurinated DNA (beyond the –10 to +1 KMnO4-sensitive region) appear as a result of DNA cleavage at the depurinated sites during piperidine treatment (22). Some protection from DNase I close to DNA ends is likely to be explained by non-specific binding of the polymerase to DNA ends (28). D, DNA was treated with DNase I or KMnO4 at 37°C; M1, MspI digest of pBR322; M2, sequencing markers (G→A reaction; 22).
Protein composition of the cross-linked complexes
Complexes of RNAP with the partially depurinated lacUV5 promoter were formed at 37 (RPo) or 14°C (RPi) and the crosslinked products resolved by SDS–PAGE (Fig. 3A). Electrophoretic patterns of RPi and RPo were significantly different: while there were only three RPi-specific products, at least five different RPo-specific cross-linked products were resolved in the gel. They were identified as β/β′- or σ-specific in the experiments described below. Similar products were detected after cross-linking of lacUV5-containing DNA fragments of different lengths (114 and 167 bp; see Materials and Methods).
Figure 3.
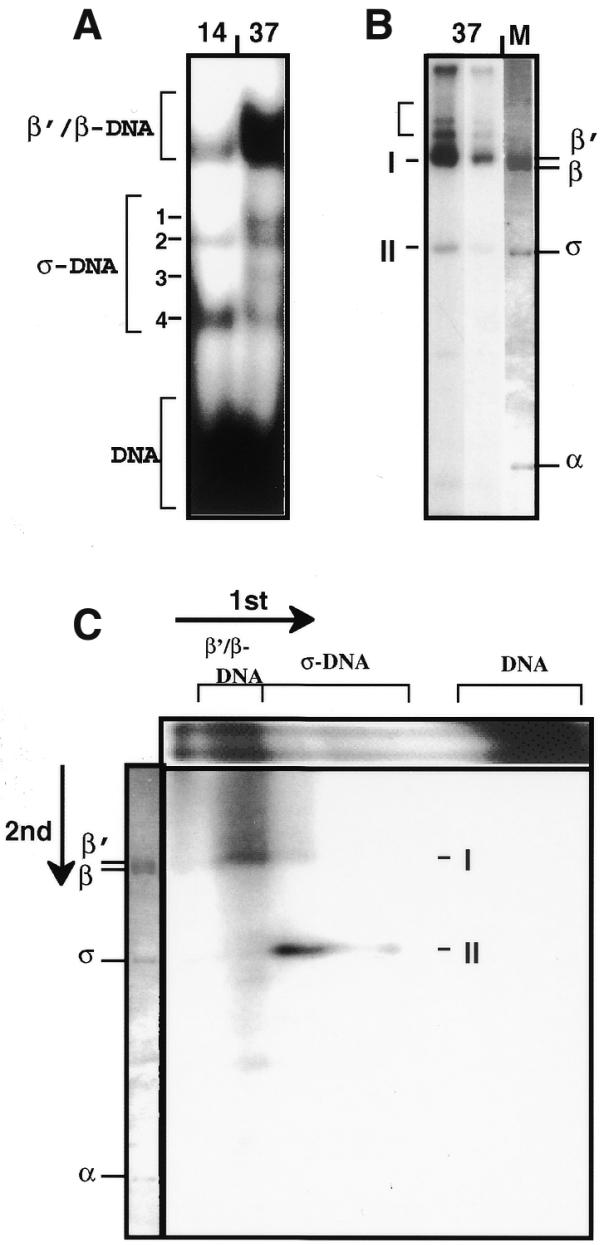
Analysis of protein composition of the cross-linked complexes. (A) 3.5–15% gradient SDS–PAGE of cross-linked products formed between RNAP and partially depurinated lacUV5 promoter DNA (end-labeled 122 bp fragment) at 37 (RPo) and 14°C (RPi). Brackets indicate positions of different β′/β- and σ-specific cross-linked products and protein-free DNA. Numbers 1–4 indicate the σ-specific products of different mobility. Different β′/β- and σ-specific cross-linked products have different mobilities due to cross-linking to different sites on the DNA (see text). (B) 8% SDS–PAGE of the cross-linked complexes formed at 37°C and digested with micrococcal nuclease and DNase I in solution. Two different exposures are shown. DNA was uniformly PCR-labeled. I and II, the final products of extensive digestion with nucleases. Intermediate products of digestion are indicated with a bracket. M, electrophoretic pattern of DNA-free RNAP in the same gel (Coomassie stain). (C) Two-dimensional SDS–PAGE of the cross-linked complexes formed at 37°C. The products resolved in the first dimension by 3.5–15% gradient SDS–PAGE (A) were digested with micrococcal nuclease and DNase I in the gel slice. Digestion products were separated in a second dimension 8% SDS–PAGE. Directions of separation and mobilities of the β′/β- and σ-specific cross-linked products in the first dimension are indicated. The electrophoretic pattern of free RNAP in the same gel (Coomassie stain) is shown on the left.
The relative intensities of β/β′- and σ-specific bands 1–3 were invariable within a range of concentration of RNAP. At the same time, the intensity of band 4 was disproportionally higher at higher concentrations of enzyme. Addition of heparin after formation of RPo caused disappearance of the band. This suggests that band 4 is a result of non-specific, heparin-sensitive interaction of RNAP with DNA ends through the σ subunit (28). This interpretation was supported by mapping of contact 4 close to DNA ends (Fig. 4A).
The protein composition of the cross-linked complexes was determined using a label transfer approach (Fig. 1). The 114 bp lacUV5-containing DNA fragment was PCR amplified/labeled, partly depurinated and cross-linked to RNAP at 37°C. Then the cross-linked products were digested with nucleases either in solution (Fig. 3B) or in gel after separation by SDS–PAGE (Fig. 3C). Digestion of DNA–protein cross-linked products with nucleases leaves short (10–20 bp) labeled oligonucleotides cross-linked to the protein and resistant to further digestion (29). Molecular weights of proteins cross-linked to labeled oligonucleotides were determined by SDS–PAGE.
After digestion of the cross-linked complexes in solution two products were detected (I and II, Fig. 3B). Analysis of the time course of digestion with nucleases indicated that these were the final products of digestion. Comparison of their mobilities with the mobilities of unlabeled RNAP subunits suggests that II contains the σ subunit. Product I contains the β′ and/or β subunit(s); the mobilities of the β′ and β subunits are very similar and therefore corresponding cross-linked complexes were not resolved by SDS–PAGE and were designated as β′/β complexes. There was no indication of a band containing the α subunit. The lack of α-specific cross-linked products could be explained by the absence of cross-linkable groups at the interacting surfaces of the protein and/or DNA (16).
Protein contents of different RPo-specific cross-linked products were analyzed by second dimension SDS–PAGE (Fig. 3C) after digestion of the DNA in gel slices excised from the first dimension gel (Fig. 3A). The same predominant products (I and II, β′/β- and σ-specific, respectively) were detected; they were separated from each other in the first dimension SDS–PAGE. Cross-linked products 1–4 having higher mobility in the first dimension gel (Fig. 3A) were identified as σ-specific. The σ-specific product 4 (Fig. 3A) was relatively under-represented in the second dimension gel, probably because the corresponding contact (localized at the end of the DNA fragment; see Fig. 4A) was less extensively labeled during amplification of the 114 bp fragment with the unlabeled PCR primers.
Arrangement of the RNAP subunits on lacUV5 promoter DNA in RPi and RPo
End-labeled DNA samples isolated from the RPi- and RPo-specific bands (Fig. 3A) were analyzed in a sequencing gel after reversal of the cross-links (Fig. 4). In RPo, strong σ-specific contacts were localized predominantly on the non-transcribed DNA strand and were clustered near the –35 and –10 promoter consensus regions (–40A, –39G, –38G, –17G and –16G) and near the start site (–2G and –1G). In σ-specific products having different mobilities in the first dimension gel (Fig. 3A) the σ subunit is cross-linked to different sites on the DNA fragment (Fig. 4A). Different mobilities of the cross-linking products could be explained by the presence of nicks in the DNA introduced during cross-linking. It has been shown that the electrophoretic mobility of protein–DNA complexes formed on nicked short linear DNA strongly depends on the position of the nick relative to the DNA ends (30).
The majority of strong β′/β-specific cross-links were also localized on the non-transcribed DNA strand in the –10 to +20 promoter region around the transcription start site (–11A, –13G, –2G, –1G, +7G, +8A and +18A; Fig. 4A). β′/β-specific cross-linking at +30 probably occurs due to non-specific interaction of the polymerase with the DNA end because its intensity was directly proportional to concentration of the polymerase. Several less strong sites of cross-linking (–32G, –30G, +10G, +14A and +17G) and several weak but reproducible sites (–16G, –14G, –7A, +4A and +6A) were localized on the transcribed DNA strand in RPo (Fig. 4B). As expected from the specificity of the methylation reaction, the majority of cross-links occurred at adenine and guanine bases.
The distributions of β′/β-specific contacts along the DNA in RPi and RPo were very similar, with the exception of several weak RPo-specific cross-links on the transcribed DNA strand (–16G, –14G, –7A, +4A and +6A) that were not detectable in RPi. At the same time, none of the strong RPo-specific, σ-specific contacts with the non-transcribed DNA strand were observed in RPi. Thus, strong cross-links of the σ subunit at bases –40A, –39G, –38G, –18G, –17G, –2G and –1G constitute the structural landmark of the open complex.
There were also RPi-specific, β′/β- and σ-specific contacts at positions –49A and –48G (Fig. 4A). These contacts were clearly different from the non-specific contacts which the σ subunit makes with the ends of the DNA (Fig. 3A, band 4) because the –49A and –48G cross-linked products have different mobilities and were observed only in RPi and not in RPo.
The distribution of sites of cross-linking along the lacUV5 DNA is in agreement with data obtained by different techniques detecting close interactions of RNAP with the promoter (Fig. 5). Thus the positions of the RPo-specific cross-links correlate with the arrangement of the sites of differential sensitivity to modification by dimethylsulfate, which are localized at the positions of strong interactions of RNAP with the lacUV5 promoter (17). Like σ-specific cross-linking at the –35, –10 and +1 promoter regions, the sites of differential reactivity to dimethylsulfate were detected only in RPo (2). The majority of the cross-links were localized in positions protected from hydroxyl radical-induced DNA cleavage (data not shown).
Figure 5.

Positions of β′/β- and σ-specific cross-links in the open (RPo, 37°C) and closed intermediate (RPi, 14°C) complexes formed on lacUV5 promoter DNA. Weaker β′/β-specific cross-links are indicated by thinner bars. Sites protected from and hypersensitive to dimethylsulfate in the open complex (17) are also indicated. Note the ∼50% overlap between the sites of differential dimethylsulfate reactivity and the cross-linked sites in RPo. No strong cross-links were detected at several residues reactive to dimethylsulfate in RPo (–37G, –35A, –33A, –6G, –5A and –4G; Fig. 4). The likely explanation for this is absence of a cross-linkable residue on a protein interacting with DNA.
RNAP binds to the lacUV5 DNA in RPc (and presumably in RPi) specifically (27), implying that the specificity factor (σ) probably recognizes the promoter in these complexes. How could this be reconciled with the apparent absence of σ-specific cross-links and sites of differential reactivity to dimethylsulfate at the consensus regions in RPi? Our ‘zero length’ cross-linking and dimethylsulfate modification techniques detect only very close interactions, which could be absent or considerably different in less tight DNA–protein complexes. Indeed, RNAP binds much less tightly to lacUV5 RPc (and presumably to RPi) then to RPo (27). It is likely that longer DNA–protein cross-linking reagents would be able to detect more RPi-specific interactions of σ with lacUV5 DNA.
Many of the strong β′/β- and σ-specific contacts are localized near to and within the consensus promoter regions (–35 and –10) and the transcription start site in the RPo complex; these are mainly on the non-transcribed DNA strand, in agreement with laser UV cross-linking and dimethylsulfate protection studies of the lacUV5 promoter (5,17,31,32). The positions of the contacts are also consistent with cross-linking data obtained for lacUV5 RPo by a technique similar to that used in this work (21); in our work fewer contacts were detected, probably because of a more conservative interpretation of the cross-linking sites (see Materials and Methods). The data are also in good agreement with results obtained using longer (11 Å) cross-linking reagents (12). The agreement of the cross-linking data with the data obtained by other techniques strongly suggests that the cross-linking procedure does not change the structure of the initiation complexes.
The results of mapping σ-specific contacts are also in agreement with genetic data suggesting that the –35 consensus region interacts with the promoter recognition domain of σ (33,34), and that different functional regions of the σ subunit interact with lacUV5 at –33 to –37 (region 4.2), –12 to –19 (regions 2.5 and 3.1) and –4 to +2 (region 2.1) (35). Unfortunately, direct mapping of the regions of the proteins involved in zero length cross-linking to different promoter regions is extremely difficult because the proteins are cross-linked to multiple sites on the DNA and because the cross-links are unstable.
In summary, the strong β′/β-specific cross-links are rather similar in RPi and RPo (with the exception of the cross-link at position –50). In contrast, the σ-specific cross-links are dramatically different in RPi and RPo: the σ subunit closely interacts with DNA at the –10 and –35 consensus regions and at the transcription start site only in the RPo but not in the RPi complex.
DISCUSSION
The principal result of this work is the demonstration of a close interaction of the σ70 recognition subunit with functionally important regions (the –10 and –35 consensus regions and the transcription start site) of the lacUV5 promoter only in the open (RPo) but not in the closed intermediate (RPi) complex. In contrast, close contacts of the catalytic β′/β subunit(s) with the promoter are much more similar in RPi and RPo. The data are consistent with results obtained by other laboratories (see Results) and suggest that contacts of the β′/β core RNAP subunit(s) with DNA are mainly established in RPi, while formation of close σ–promoter contacts is fully completed only after formation of RPo. Thus the σ subunit is likely to be the key player in the RPi→RPo transition.
The contacts of the core subunit(s) at the –10 to +20 region of lacUV5 are very similar in RPi and RPo (Fig. 4). This observation is consistent with the footprinting data, suggesting that the RPi- and RPo-specific footprints are very similar (36–38). At the same time, at least 10 bp of DNA are melted in the –10 to +3 region in RPo but not in RPi (2,8,9,39). There are at least two possible explanations for the lack of changes in DNA–protein contacts of the core subunits with the –10 to +20 promoter region during DNA melting. One is that during the RPi→RPo transition the DNA is untwisted by precisely one helical turn; this untwisting is relaxed by DNA rotation downstream of the –10 to +5 region and the contact pattern is restored when rotation is completed (37). Alternatively, DNA could already be unwound in RPi; during the RPi→RPo transition the bases in the unwound region are ‘flipped out’ and become accessible to the catalytic region of RNAP (36,38). The data on the high similarity of the β′/β-specific cross-linking patterns in RPi and RPo (Figs 4 and 5) are more consistent with the base flipping model because DNA untwisting by one helical turn would be expected to result in a more pronounced change in the cross-linking pattern due to the different geometry of the DNA after melting. Base flipping has recently been shown to occur in many systems where enzymes need to gain access to a DNA base (40). In fact, it has been proposed that base flipping could nucleate opening of the DNA helix during initiation of transcription (40).
It has been proposed that the region –40 to –50 of the lacUV promoter can interact with the C-terminal domain of the α subunit of RNAP (41–43). A set of strong β′/β- and σ-specific, RPi-specific contacts was detected around position –50 (Figs 4 and 5). It is possible that the α-specific, RPo-specific contacts can be substituted by σ- and β′/β-specific contacts in RPi.
Our previous data on analysis of the RPc→RPo transition using formaldehyde-induced protein–protein cross-linking suggest that the interaction(s) between the β′ and σ subunits characteristic of the DNA-free holoenzyme in solution are disrupted in the RPc and RPi complexes and recovered in the open complex (6). These data, in combination with the DNA–protein cross-linking data described here, suggest that during promoter recognition interactions of the σ subunit with DNA and with core polymerase are rearranged in concert. This suggests the following mechanism of lacUV5 promoter recognition (Fig. 6).
Figure 6.
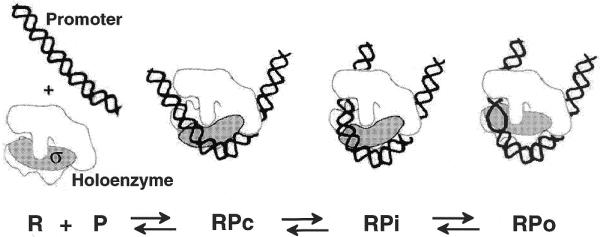
Proposed structures of the closed (RPc), closed intermediate (RPi) and open (RPo) complexes formed on the lacUV5 promoter. The structure of the polymerase holoenzyme in different complexes is modeled after Zhang et al. (45) (viewed from the flexible ‘flap’ side, roughly parallel with the main axis of the RNAP channel). The polymerase ‘jaws’ are likely to be closed around the DNA in RPi and RPo, but not in RPc (4,44; not shown).
Initial binding of the holoenzyme to lacUV5 (RPc formation) probably destabilizes a β′–σ interaction(s) in the holoenzyme (detected as disappearance of the β′–σ protein–protein cross-links), perhaps inducing a ‘core-like’ conformation of RNAP (Fig. 6). Destabilization of the β′–σ interaction(s) could in turn induce closing of the ‘jaws’ of the polymerase (RPi formation) because closed jaws are a structural landmark of the core enzyme (44). At this stage, contacts of the β′/β subunit(s) with the promoter are mainly established, but formation of strong contacts of σ with the DNA is not completed and the DNA is not yet melted. Then the σ subunit completes DNA recognition (RPo formation); this coincides with DNA melting and restoration of the holoenzyme-specific β′–σ contact. The contacts of σ with the core enzyme in RPo are likely to be less stable than in the holoenzyme because the enzyme probably has the ‘closed jaws’ conformation characteristic of the core enzyme free of the σ subunit (4). This might help the core enzyme to break the σ–core interactions and escape to the elongation stage of transcription. While speculative, this model offers a molecular logic for the complex conformational transitions observed during transcription initiation.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr L. Savochkina and J. Becker for providing the preparations of the RNA polymerase, Dr O. N. Voloshin for providing the pOV10 plasmid, Dr A. Chenchick for advice on the cross-linking and Drs M. Kashlev, R. Needleman, M. T. Record and O. V. Tsodikov for critical reading of the manuscript. The V.M.S. laboratory is funded by NIH grant GM58650-02.
References
- 1.Buc H. and McClure,W.R. (1985) Kinetics of open complex formation between Escherichia coli RNA polymerase and the lac UV5 promoter. Evidence for a sequential mechanism involving three steps. Biochemistry, 24, 2712–2723. [DOI] [PubMed] [Google Scholar]
- 2.Spassky A., Kirkegaard,K. and Buc,H. (1985) Changes in the DNA structure of the lac UV5 promoter during formation of an open complex with Escherichia coli RNA polymerase. Biochemistry, 24, 2723–2731. [DOI] [PubMed] [Google Scholar]
- 3.Roe J.H., Burgess,R.R. and Record,M.T.,Jr (1985) Temperature dependence of the rate constants of the Escherichia coli RNA polymerase-lambda PR promoter interaction. Assignment of the kinetic steps corresponding to protein conformational change and DNA opening. J. Mol. Biol., 184, 441–453. [DOI] [PubMed] [Google Scholar]
- 4.Craig M.L., Tsodikov,O.V., McQuade,K.L., Schlax,P.E.,Jr, Capp,M.W., Saecker,R.M. and Record,M.T.,Jr (1998) DNA footprints of the two kinetically significant intermediates in formation of an RNA polymerase-promoter open complex: evidence that interactions with start site and downstream DNA induce sequential conformational changes in polymerase and DNA. J. Mol. Biol., 283, 741–756. [DOI] [PubMed] [Google Scholar]
- 5.Buckle M., Pemberton,I.K., Jacquet,M.A. and Buc,H. (1999) The kinetics of sigma subunit directed promoter recognition by E. coli RNA polymerase. J. Mol. Biol., 285, 955–964. [DOI] [PubMed] [Google Scholar]
- 6.Brodolin K.L., Studitsky,V.M. and Mirzabekov,A.D. (1993) Conformational changes in E. coli RNA polymerase during promoter recognition. Nucleic Acids Res., 21, 5748–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsodikov O.V., Craig,M.L., Saecker,R.M. and Record,M.T.,Jr (1998) Quantitative analysis of multiple-hit footprinting studies to characterize DNA conformational changes in protein-DNA complexes: application to DNA opening by E sigma-70 RNA polymerase. J. Mol. Biol., 283, 757–769. [DOI] [PubMed] [Google Scholar]
- 8.Kirkegaard K., Buc,H., Spassky,A. and Wang,J.C. (1983) Mapping of single-stranded regions in duplex DNA at the sequence level: single-strand-specific cytosine methylation in RNA polymerase-promoter complexes. Proc. Natl Acad. Sci. USA, 80, 2544–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Straney D.C. and Crothers,D.M. (1985) Intermediates in transcription initiation from the E. coli lac UV5 promoter. Cell, 43, 449–459. [DOI] [PubMed] [Google Scholar]
- 10.Roy S., Garges,S. and Adhya,S. (1998) Activation and repression of transcription by differential contact: two sides of a coin. J. Biol. Chem., 273, 14059–14062. [DOI] [PubMed] [Google Scholar]
- 11.Helmann J.D. and deHaseth,P.L. (1999) Protein-nucleic acid interactions during open complex formation investigated by systematic alteration of the protein and DNA binding partners. Biochemistry, 38, 5959–5967. [DOI] [PubMed] [Google Scholar]
- 12.Naryshkin N., Revyakin,A., Kim,Y., Mekler,V. and Ebright,R.H. (2000) Structural organization of the RNA polymerase-promoter open complex. Cell, 101, 601–611. [DOI] [PubMed] [Google Scholar]
- 13.Owens J.T., Miyake,R., Murakami,K., Chmura,A.J., Fujita,N., Ishihama,A. and Meares,C.F. (1998) Mapping the sigma 70 subunit contact sites on Escherichia coli RNA polymerase with a sigma 70-conjugated chemical protease. Proc. Natl Acad. Sci. USA, 95, 6021–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brodolin K., Mustaev,A., Severinov,K. and Nikiforov,V. (2000) Identification of RNA polymerase beta′ subunit segment contacting the melted region of the lacUV5 promoter. J. Biol. Chem., 275, 3661–3666. [DOI] [PubMed] [Google Scholar]
- 15.Korzheva N., Mustaev,A., Kozlov,M., Malhotra,A., Nikiforov,V., Goldfarb,A. and Darst,S.A. (2000) A structural model of transcription elongation. Science, 289, 619–625. [DOI] [PubMed] [Google Scholar]
- 16.Mirzabekov A.D., Bavykin,S.G., Belyavsky,A.V., Karpov,V.L., Preobrazhenskaya,O.V., Shick,V.V. and Ebralidse,K.K. (1989) Mapping DNA-protein interactions by cross-linking. Methods Enzymol., 170, 386–408. [DOI] [PubMed] [Google Scholar]
- 17.Siebenlist U., Simpson,R.B. and Gilbert,W. (1980) E. coli RNA polymerase interacts homologously with two different promoters. Cell, 20, 269–281. [DOI] [PubMed] [Google Scholar]
- 18.Johnsrud L. (1978) Contacts between Escherichia coli RNA polymerase and a lac operon promoter. Proc. Natl Acad. Sci. USA, 75, 5314–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burgess R.R. and Jendrisak,J.J. (1975) A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving Polymin P precipitation and DNA-cellulose chromatography. Biochemistry, 14, 4634–4638. [DOI] [PubMed] [Google Scholar]
- 20.Lowe P.A., Hager,D.A. and Burgess,R.R. (1979) Purification and properties of the sigma subunit of Escherichia coli DNA-dependent RNA polymerase. Biochemistry, 18, 1344–1352. [DOI] [PubMed] [Google Scholar]
- 21.Chenchick A., Beabealashvilli,R. and Mirzabekov,A. (1981) Topography of interaction of Escherichia coli RNA polymerase subunits with lac UV5 promoter. FEBS Lett., 128, 46–50. [DOI] [PubMed] [Google Scholar]
- 22.Maxam A.M. and Gilbert,W. (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol., 65, 499–560. [DOI] [PubMed] [Google Scholar]
- 23.Li X.Y. and McClure,W.R. (1998) Characterization of the closed complex intermediate formed during transcription initiation by Escherichia coli RNA polymerase. J. Biol. Chem., 273, 23549–23557. [DOI] [PubMed] [Google Scholar]
- 24.Wilson C. and Dombroski,A.J. (1997) Region 1 of sigma 70 is required for efficient isomerization and initiation of transcription by Escherichia coli RNA polymerase. J. Mol. Biol., 267, 60–74. [DOI] [PubMed] [Google Scholar]
- 25.Li X.Y. and McClure,W.R. (1998) Stimulation of open complex formation by nicks and apurinic sites suggests a role for nucleation of DNA melting in Escherichia coli promoter function. J. Biol. Chem., 273, 23558–23566. [DOI] [PubMed] [Google Scholar]
- 26.Noel R.J.,Jr and Reznikoff,W.S. (2000) Structural studies of lacUV5-RNA polymerase interactions in vitro. Ethylation interference and missing nucleoside analysis. J. Biol. Chem., 275, 7708–7712. [DOI] [PubMed] [Google Scholar]
- 27.Kovacic R.T. (1987) The 0 degree C closed complexes between Escherichia coli RNA polymerase and two promoters, T7-A3 and lacUV5. J. Biol. Chem., 262, 13654–13661. [PubMed] [Google Scholar]
- 28.Melancon P., Burgess,R.R. and Record,M.T.,Jr (1983) Direct evidence for the preferential binding of Escherichia coli RNA polymerase holoenzyme to the ends of deoxyribonucleic acid restriction fragments. Biochemistry, 22, 5169–5176. [DOI] [PubMed] [Google Scholar]
- 29.Chodosh L.A., Carthew,R.W. and Sharp,P.A. (1986) A single polypeptide possesses the binding and transcription activities of the adenovirus major late transcription factor. Mol. Cell. Biol., 6, 4723–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Werel W., Schickor,P. and Heumann,H. (1991) Flexibility of the DNA enhances promoter affinity of Escherichia coli RNA polymerase. EMBO J., 10, 2589–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borowiec J.A. and Gralla,J.D. (1986) High-resolution analysis of lac transcription complexes inside cells. Biochemistry, 25, 5051–5057. [DOI] [PubMed] [Google Scholar]
- 32.Buckle M., Geiselmann,J., Kolb,A. and Buc,H. (1991) Protein-DNA cross-linking at the lac promoter. Nucleic Acids Res., 19, 833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siegele D.A., Hu,J.C., Walter,W.A. and Gross,C.A. (1989) Altered promoter recognition by mutant forms of the sigma 70 subunit of Escherichia coli RNA polymerase. J. Mol. Biol., 206, 591–603. [DOI] [PubMed] [Google Scholar]
- 34.Gardella T., Moyle,H. and Susskind,M.M. (1989) A mutant Escherichia coli sigma 70 subunit of RNA polymerase with altered promoter specificity. J. Mol. Biol., 206, 579–590. [DOI] [PubMed] [Google Scholar]
- 35.Owens J.T., Chmura,A.J., Murakami,K., Fujita,N., Ishihama,A. and Meares,C.F. (1998) Mapping the promoter DNA sites proximal to conserved regions of sigma 70 in an Escherichia coli RNA polymerase-lacUV5 open promoter complex. Biochemistry, 37, 7670–7675. [DOI] [PubMed] [Google Scholar]
- 36.Jeppesen C. and Nielsen,P.E. (1989) Uranyl mediated photofootprinting reveals strong E.coli RNA polymerase–DNA backbone contacts in the +10 region of the DeoP1 promoter open complex. Nucleic Acids Res., 17, 4947–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schickor P., Metzger,W., Werel,W., Lederer,H. and Heumann,H. (1990) Topography of intermediates in transcription initiation of E.coli. EMBO J., 9, 2215–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mecsas J., Cowing,D.W. and Gross,C.A. (1991) Development of RNA polymerase-promoter contacts during open complex formation. J. Mol. Biol., 220, 585–597. [DOI] [PubMed] [Google Scholar]
- 39.Cowing D.W., Mecsas,J., Record,M.T.,Jr and Gross,C.A. (1989) Intermediates in the formation of the open complex by RNA polymerase holoenzyme containing the sigma factor sigma 32 at the groE promoter. J. Mol. Biol., 210, 521–530. [DOI] [PubMed] [Google Scholar]
- 40.Roberts R.J. and Cheng,X. (1998) Base flipping. Annu. Rev. Biochem., 67, 181–198. [DOI] [PubMed] [Google Scholar]
- 41.Kolb A., Igarashi,K., Ishihama,A., Lavigne,M., Buckle,M. and Buc,H. (1993) Escherichia coli RNA polymerase, deleted in the C-terminal part of its alpha-subunit, interacts differently with the cAMP–CRP complex at the lacP1 and at the galP1 promoter. Nucleic Acids Res., 21, 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross W., Gosink,K.K., Salomon,J., Igarashi,K., Zou,C., Ishihama,A., Severinov,K. and Gourse,R.L. (1993) A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science, 262, 1407–1413. [DOI] [PubMed] [Google Scholar]
- 43.Burns H.D., Ishihama,A. and Minchin,S.D. (1999) Open complex formation during transcription initiation at the Escherichia coli gal P1 promoter: the role of the RNA polymerase alpha subunit at promoters lacking an UP-element. Nucleic Acids Res., 27, 2051–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polyakov A., Severinova,E. and Darst,S.A. (1995) Three-dimensional structure of E. coli core RNA polymerase: promoter binding and elongation conformations of the enzyme. Cell, 83, 365–373. [DOI] [PubMed] [Google Scholar]
- 45.Zhang G., Campbell,E.A., Minakhin,L., Richter,C., Severinov,K. and Darst,S.A. (1999) Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell, 98, 811–824. [DOI] [PubMed] [Google Scholar]